

Abstract
BACKGROUND
Ruxolitinib, a selective JAK1 and JAK2 inhibitor, has clinically significant activity in myelofibrosis.
METHODS
In a double-blind trial, patients with intermediate-2 or high-risk myelofibrosis were randomized to twice-daily oral ruxolitinib (n=155) or placebo (n=154). The primary endpoint was the proportion of patients with ≥35% spleen volume reduction at 24 weeks assessed by magnetic resonance imaging. Secondary endpoints included durability of response, changes in symptom burden (assessed by Total Symptom Score [TSS]), and overall survival.
RESULTS
In the ruxolitinib group, 41.9% achieved the primary endpoint versus 0.7% in the placebo group (P<0.001). Spleen response was maintained while taking ruxolitinib: 67% of responding patients maintained response for ≥48 weeks. A ≥50% improvement in TSS at 24 weeks was achieved by 45.9% of ruxolitinib-treated versus 5.3% of placebo-treated patients (P<0.001). Thirteen deaths occurred in the ruxolitinib and 24 in the placebo group (hazard ratio, 0.50; 95% CI, 0.25–0.98; P=0.04). Discontinuations for adverse events were similar between groups (11% each). Among ruxolitinib-treated patients, anemia and thrombocytopenia were the most common adverse events, but rarely led to discontinuation (1 patient for each event). Two patients underwent transformation to acute myeloid leukemia (AML), both in the ruxolitinib group.
CONCLUSIONS
Ruxolitinib provided significant clinical benefits in patients with myelofibrosis by reducing spleen size, improving debilitating myelofibrosis-related symptoms, and improving overall survival. Improvement came at a cost of more frequent anemia and thrombocytopenia in the early part of the treatment period. The imbalance in AML transformation requires attention in further studies. (Funded by Incyte Corporation; ClinicalTrials.gov, NCT00952289)
INTRODUCTION
Myelofibrosis, a myeloproliferative neoplasm, presents with abnormal blood cell counts (anemia, thrombocytosis or thrombocytopenia, and leukocytosis or leukopenia); splenomegaly; and debilitating symptoms (eg, fatigue, weakness, abdominal pain, cachexia, weight loss, pruritus, night sweats, and bone pain) thought to be driven by the combined effects of massive splenomegaly and elevated proinflammatory cytokines.1 Survival ranges from approximately 2 to 11 years, depending on defined prognostic factors.2 Traditional therapeutic options, including splenectomy, have limited benefit.3 Although allogeneic stem-cell transplantation may cure myelofibrosis, few patients are eligible.
While the JAK2V617F gain-of-function mutation is present in approximately 50% of patients with primary myelofibrosis, other mechanisms of direct or indirect activation of the intracellular JAK-STAT pathway are known,4 suggesting that dysregulation of this pathway is a central pathogenic component in myelofibrosis regardless of the mutational status of JAK2. Also, proinflammatory cytokines that play an important role in myelofibrosis signal through JAK1 and JAK2.5 In a phase 1/2 trial with ruxolitinib (INCB018424), a potent inhibitor of JAK1 and JAK2,6,7 patients with myelofibrosis experienced durable reductions in splenomegaly and improvements in myelofibrosis-related symptoms regardless of JAK2V617F mutation status. To further evaluate the efficacy and safety of ruxolitinib, we conducted a randomized, double-blind, placebo-controlled trial in patients with advanced myelofibrosis.
METHODS
PATIENTS
Patients were ≥18 years of age with primary (PMF), post–polycythemia vera (PPV-MF), or post–essential thrombocythemia myelofibrosis (PET-MF) based on 2008 World Health Organization criteria,8 with life expectancy ≥6 months, International Prognostic Scoring System (IPSS) score2 (Appendix Table S1) of 2 (intermediate-2 risk) or ≥3 (high risk), Eastern Cooperative Oncology Group (ECOG) performance status9 ≤3 (on a scale from 0 to 5, with higher scores indicating greater disability; Appendix), peripheral blood blasts <10%, absolute peripheral blood CD34+ cell count >20×106/l, platelets ≥100×109/l, and palpable splenomegaly (≥5 cm below left costal margin). Patients were resistant or refractory to, intolerant of, or not candidates for available therapies and had disease requiring treatment (inclusion and exclusion criteria are listed in the protocol posted on NEJM.org).
The protocol was approved by an institutional review board of each site. The study was conducted in accordance with Good Clinical Practice guidelines per the International Conference on Harmonisation. All patients provided written informed consent.
STUDY DESIGN AND TREATMENT
This randomized, double-blind, placebo-controlled phase 3 trial was conducted at 89 sites in the United States, Australia, and Canada. Patients were randomized 1:1 to receive oral ruxolitinib phosphate tablets or matched placebo. The starting dose of ruxolitinib was 15 mg or 20 mg twice daily, depending on baseline platelet count (100 to 200×109/l or >200×109/l, respectively). The dose was adjusted for lack of efficacy or excess toxicity per protocol (Appendix). Unblinding of therapy and crossover from placebo to ruxolitinib was permitted for protocol-defined worsening splenomegaly (Appendix). The prospectively defined data cutoff occurred when half the patients remaining in the study completed the week 36 visit, and all completed the week 24 evaluation or discontinued treatment. Data for placebo-treated patients after crossover are not included in these analyses, except for the intent-to-treat (ITT) analysis of overall survival.
The primary endpoint was the proportion of patients achieving a ≥35% reduction in spleen volume from baseline to week 24, measured by magnetic resonance imaging (MRI) or computed tomography. Secondary endpoints included duration of maintenance of spleen volume reduction, proportion of patients with ≥50% reduction in Total Symptom Score (TSS) from baseline to week 24 using the modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.0 diary (Appendix),10,11 change in TSS from baseline to week 24, and overall survival. The overall survival analysis was updated at the time of a planned data cutoff 4 months after the primary analysis. Patients completed the MFSAF every night; this electronic diary evaluated, on a scale of 0 (absent) to 10 (worst imaginable), night sweats, itching, abdominal discomfort, pain under the ribs on the left side, feeling of fullness (early satiety), muscle/bone pain, and inactivity. TSS was the sum of individual symptom scores, excluding inactivity. Exploratory endpoints included changes in body weight and JAK2V617F allele burden, achievement of transfusion independence,12 and additional patient-reported outcomes (Appendix).
The study was designed to enroll 240 patients, providing 97% power to detect a treatment difference in spleen volume response at a 2-sided alpha level of 0.05 assuming ≥30% response rate for ruxolitinib and ≤10% response rate for placebo. Analyses were conducted in accordance with intent-to-treat (ITT) principles. For all applicable variables, however, patients with missing baseline values were excluded from analyses of change and percent change from baseline. In analyses of change from baseline to week 24, patients who discontinued or crossed over before week 24 were counted as nonresponders (for response measures of spleen volume reduction and symptom improvement). Comparative secondary efficacy variables were tested in a fixed-sequence-testing procedure at an alpha level of 0.05. Durability of spleen response and survival were analyzed using the Kaplan-Meier method. The statistical analysis plan is posted on NEJM.org.
This study was funded by Incyte Corporation. The first author (S.V.) and a coauthor (V.S.) wrote the initial draft of the manuscript. Medical writing assistance with an early draft was provided by Daniel Hutta, Ph.D., of Articulate Science, LLC, and funded by Incyte Corporation. All coauthors contributed to subsequent drafts and decided to submit for publication. Data were analyzed at Incyte Corporation (W.S.). All authors vouch for the accuracy and completeness of reported data and for fidelity of this report to the protocol.
RESULTS
PATIENTS
From September 2009 through April 2010, 309 patients were enrolled: 155 randomized to ruxolitinib, and 154 to placebo. Baseline characteristics were similar between groups (Table 1). Median spleen volume was >2500 cm3 (>10 times normal spleen volume of 150–200 cm3).13–15 Both IPSS intermediate-2–risk (38%) and high-risk categories (61%) were well represented.
Table 1.
Baseline Demographics and Disease Characteristics.
| Parameter | Ruxolitinib (N=155) | Placebo (N=154) |
|---|---|---|
| Median age (range) – yr | 66 (43–91) | 70 (40–86) |
| Proportion of male patients – % | 51 | 57 |
| Myelofibrosis subtype – % of patients | ||
| PMF | 45 | 55 |
| PPV-MF | 32 | 31 |
| PET-MF | 23 | 14 |
| IPSS risk status | ||
| High-risk disease – % of patients | 58 | 64 |
| Intermediate-2 – % of patients | 41 | 35 |
| Previous hydroxyurea use – % of patients | 67 | 57 |
| Median baseline platelet count (range) – ×109/l | 262 (81–984) | 238 (100–887) |
| Median baseline hemoglobin (range) – g/l | 105 (66–170) | 105 (35–173) |
| Median palpable spleen length (range) – cm/spleen volume (range) – cm3 | 16 (0–33)*/2598 (478–7462) | 16 (5–34)/2566 (521–8881) |
| JAK2V617F–positive – % of patients | 73 | 80 |
One patient had a baseline spleen length recorded as nonpalpable in error, but had a prior measurement of 16 cm and a baseline spleen volume of 2449.6 cm3. IPSS denotes International Prognosis Scoring System; PET-MF, post–essential thrombocythemia myelofibrosis; PMF, primary myelofibrosis; and PPV-MF, post–polycythemia vera myelofibrosis.
At the time of the prospectively defined data cutoff (median follow-up, 32 weeks), 134 patients (87%) in the ruxolitinib and 78 (52%) in the placebo group were receiving randomized treatment. Thirty-six patients (24%) in the placebo group crossed over to ruxolitinib (16 before and 20 after week 24; Appendix.)
EFFICACY
Spleen Size
For the primary endpoint, 41.9% of ruxolitinib-treated patients achieved a ≥35% reduction in spleen volume at week 24 compared with 0.7% of placebo-treated patients (odds ratio [OR], 134.4; 95% confidence interval [CI], 18.0–1005; P<0.001; Fig. 1A). Additional prespecified analyses showed that, for patients with baseline and week 24 data, patients receiving ruxolitinib (n=139) had a mean 31.6% (median 33.0%) reduction in spleen volume at week 24; patients on placebo (n=106) had a mean 8.1% (median 8.5%) increase. Almost all patients receiving ruxolitinib experienced some degree of spleen volume reduction (Fig. 1B). The majority of patients receiving placebo experienced spleen growth. Palpable spleen length changes in the ruxolitinib and placebo groups mirrored changes in spleen volume. Spleen volume response was durable with continued therapy (Fig. 1C). For this secondary endpoint, among patients who had a ≥35% reduction in spleen volume, 67% (95% CI, 0.46–0.81) maintained a spleen volume reduction for ≥48 weeks (loss of response defined as <35% reduction from baseline and ≥25% increase from nadir).
Figure 1. Assessment of Spleen Volume Reduction.
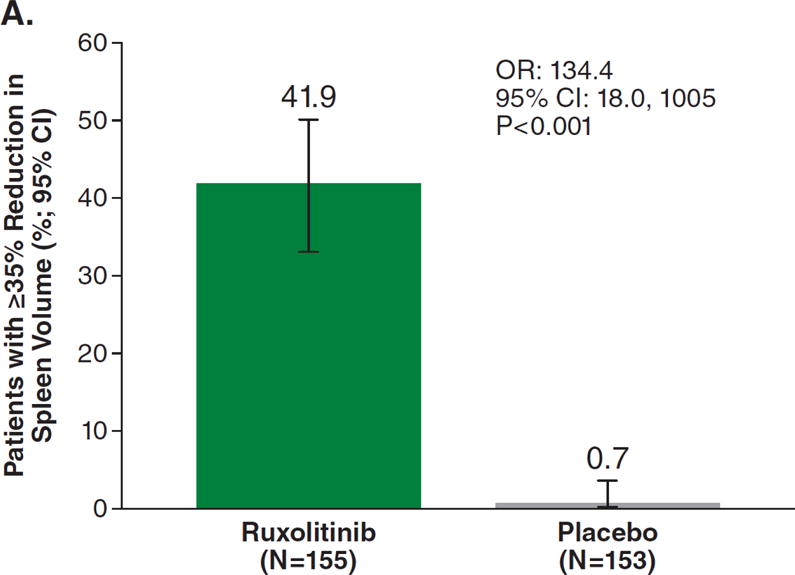
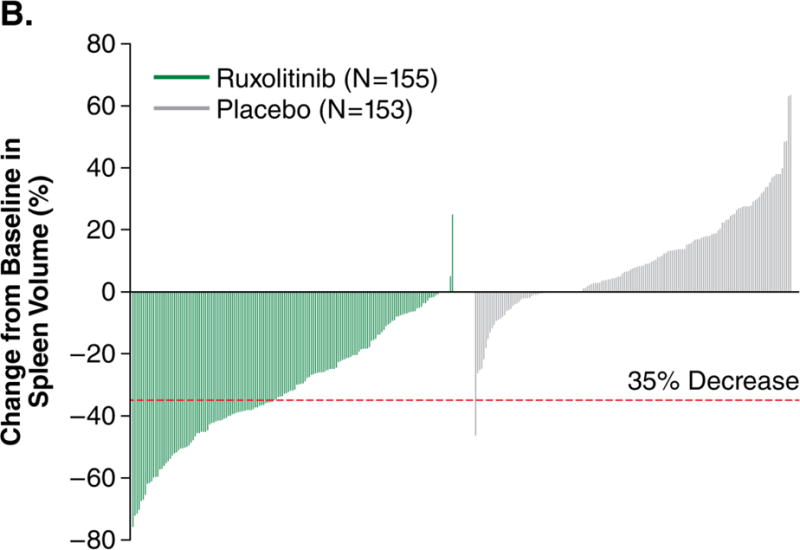
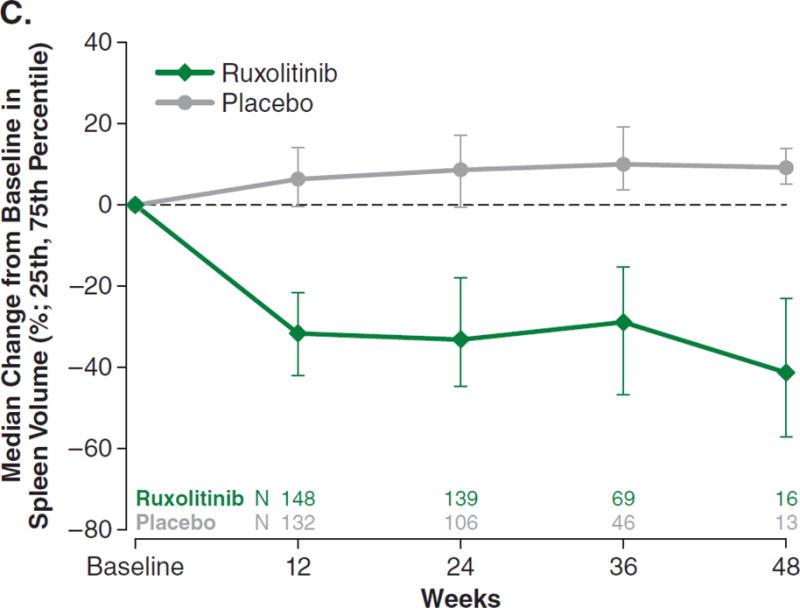
Figure 1A represents the intent-to-treat analysis of the percentage of patients in each treatment group achieving the primary endpoint of a ≥35% reduction in spleen volume by magnetic resonance imaging or computed tomography. Patients who discontinued before week 24 or crossed over before week 24 were counted as nonresponders. Only patients with baseline data were included in this analysis. Figure 1B depicts a waterfall plot of percent change in spleen volume from baseline for patients in the ruxolitinib and placebo groups at week 24 (n=139 and n=106, respectively) or the last evaluation before week 24 (n=16 and n=47). One patient with a missing baseline value is not included on the graph. Most patients in the ruxolitinib group (150/155) experienced a reduction in spleen volume, whereas most patients in the placebo group exhibited either an increase (102/153) or no change in spleen volume (15/153). Figure 1C shows the median percent change in spleen size, as measured by volume assessed by magnetic resonance imaging or computed tomography over time. Reductions in spleen volume were apparent at the first on-study measurement at 12 weeks and were maintained over the course of the study. OR denotes odds ratio. CI denotes confidence interval.
Symptoms and Other Patient-Reported Outcomes
The proportion of patients achieving a ≥50% reduction in TSS from baseline to week 24, a prespecified secondary endpoint, was significantly higher in the ruxolitinib than in the placebo group (45.9% vs. 5.3%, respectively; OR, 15.3; 95% CI, 6.9–33.7; P<0.001). Additional prespecified analyses showed that, for patients with baseline and week 24 data, those receiving ruxolitinib (n=129) had a mean 46.1% (median 56.2%) improvement in TSS at week 24; those receiving placebo (n=103) had a mean 41.8% (median 14.6%) worsening (P<0.001). The improvement was rapid and maintained over the 24 weeks symptom data were collected (Fig. 2A). Most ruxolitinib-treated patients experienced improvement; the majority of placebo-treated patients had worsening of symptoms (Fig. 2B).
Figure 2. Assessment of Symptom Improvement.
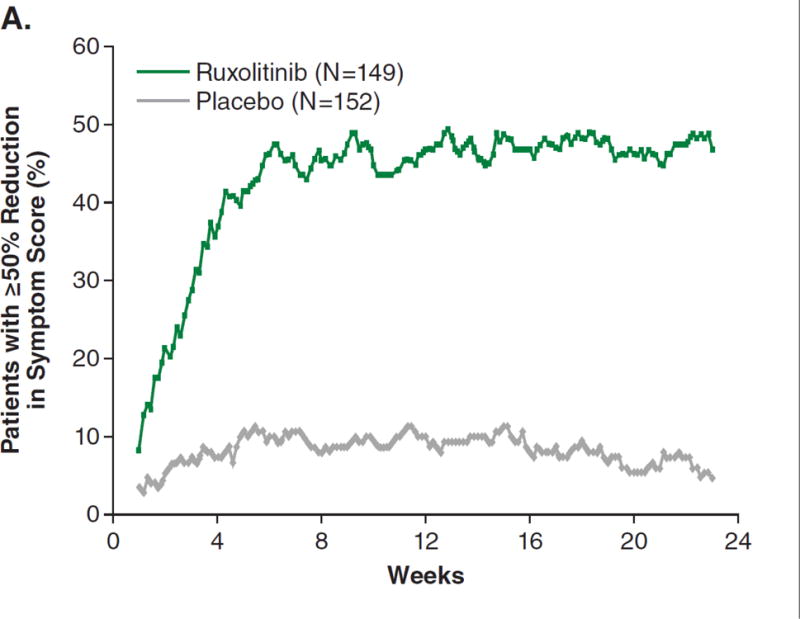
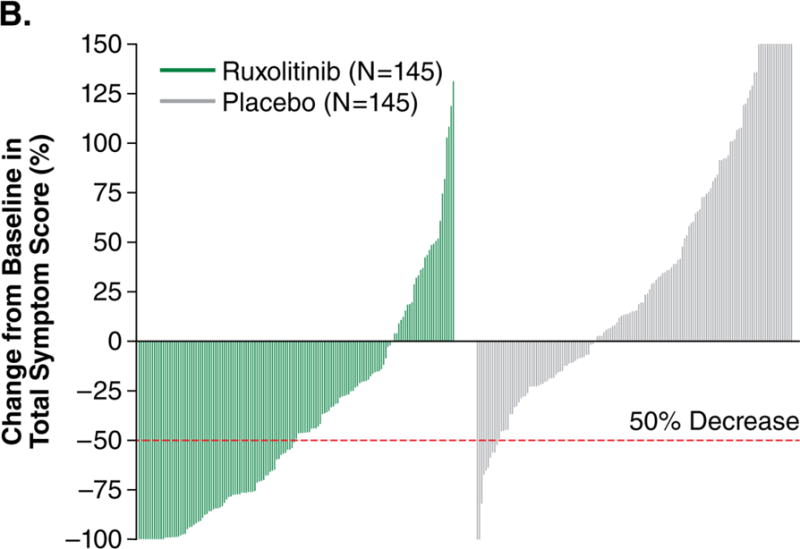
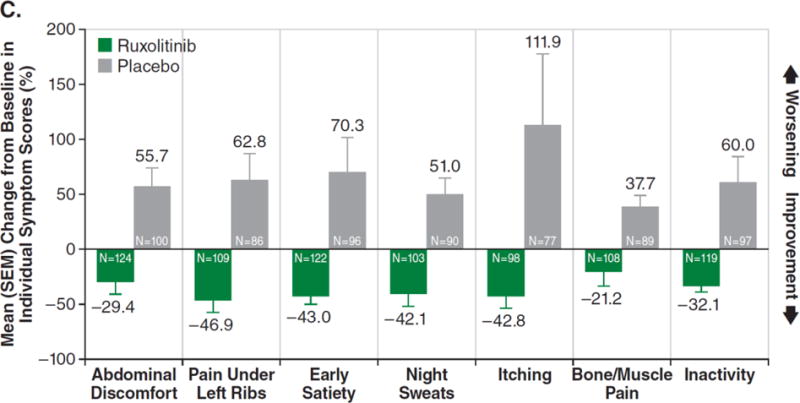
Figure 2A shows the intent-to-treat analysis of the proportion of patients achieving at least a 50% reduction in symptom score over time (each value plotted represents the moving average of the prior 7 days). Patients who discontinued or had missing data were considered nonresponders. The majority of responses occurred rapidly, within the first 4 weeks of treatment. Only patients with baseline data were included in this analysis. Figure 2B depicts a waterfall plot of percent change in TSS from baseline for patients in the ruxolitinib and placebo groups at week 24 (n=129 and n=103, respectively) or the last evaluation on randomized therapy (n=16 and n=42). Patients with a baseline score of 0 (n=5), missing baseline values (n=8), and insufficient postbaseline data (n=6) are not included on the graph. Whereas most ruxolitinib-treated patients experienced a reduction in TSS, the majority of placebo-treated patients had a worsening of symptoms (all TSS worsening ≥150% are shown as 150%). Figure 2C illustrates the mean percent change in score for each individual symptom that comprises the modified Myelofibrosis Symptom Assessment Form v2.0. All symptoms improved in the ruxolitinib group and worsened in the placebo group (P<0.01 for ruxolitinib vs. placebo). SEM denotes standard error of the mean.
A post hoc analysis showed ruxolitinib-treated patients improving in each individual symptom of the MFSAF (Fig. 2C), whereas symptoms worsened in the placebo group (P<0.01 for each).
Prespecified analyses were conducted to cross validate the modified MFSAF v2.0. The Patient Global Impression of Change and other patient-reported outcomes mirrored changes in symptom scores (Appendix Figs. S3A–3C). Ruxolitinib-treated patients experienced weight gain, whereas those receiving placebo experienced weight loss (Appendix Fig. S4). In the ruxolitinib group, 63% of patients with a ≥35% reduction in spleen volume achieved ≥50% improvement in spleen-related symptoms (sum of MFSAF scores for abdominal discomfort, pain under ribs on the left side, and feeling of fullness [early satiety]); however, this level of improvement also occurred in 47% with <35% spleen volume reduction. An additional post hoc analysis showed a ≥50% improvement in nonabdominal symptoms (night sweats, bone/muscle pain, and pruritus) in 59% and 54% of patients with ≥35% and <35% reductions in spleen volume, respectively.
Subgroups
In a post hoc analysis of subgroups, mean changes in spleen volume in patients with and without the JAK2V617F mutation (ruxolitinib vs. placebo) were −34.6% vs. +8.1% and −23.8% vs. +8.4%, respectively (p-value for interaction = 0.07). The respective changes in TSS were −52.6% (improvement) vs. +42.8% (worsening) and −28.1% vs. +37.2% (p-value for interaction = 0.11). Across MF subtypes (PMF, PPV-MF, and PET-MF), ruxolitinib-treated patients showed a decrease in spleen volume and improvement in TSS; patients receiving placebo had increases in spleen volume and worsening of TSS (p-value for interaction = 0.52 for spleen volume and 0.46 for TSS). See Appendix Figs. S5A–5B).
Biomarkers
In a prespecified analysis of biomarkers, ruxolitinib-treated patients experienced mean reductions in JAK2V617F allele burden of 10.9% and 21.5%; patients receiving placebo had a mean increase of 3.5% and 6.3% at weeks 24 and 48, respectively (Appendix Fig. S6). Furthermore, patients receiving ruxolitinib experienced reductions in plasma levels of C-reactive protein, and the proinflammatory cytokines tumor necrosis factor-α and interleukin-6, and increases in plasma leptin and erythropoietin (Appendix Fig. S7).
Overall Survival
For the secondary endpoint of overall survival, at the time of data cutoff, 10 deaths (6.5%) were reported in the ruxolitinib and 14 (9.1%) in the placebo group (hazard ratio [HR], 0.67; 95% CI, 0.30–1.50; P=0.33). Subsequently, a survival analysis based on a planned data cutoff with 4 additional months of follow-up revealed a significant survival advantage for ruxolitinib-treated patients, with 13 (8.4%) deaths in the ruxolitinib and 24 (15.7%) in the placebo group (median follow-up, 51 weeks; HR, 0.50; 95% CI, 0.25–0.98; P=0.04) (Fig. 3).
Figure 3. Overall Survival.
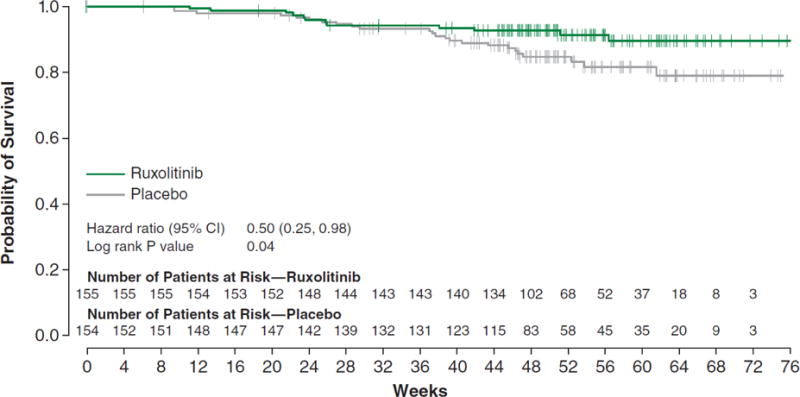
Figure 3 shows the Kaplan-Meier plot of overall survival, including 4 months of additional follow-up after the primary analysis. There were 13 (8.4%) deaths in the ruxolitinib and 24 (15.7%) in the placebo group (median follow-up, 51 weeks). CI denotes confidence interval.
SAFETY
The number of patient-years of exposure was 105 in the ruxolitinib and 87 in the placebo group; study discontinuation and crossover to ruxolitinib accounted for lower exposure in the placebo group. Seventeen (11%) ruxolitinib-treated and 16 (11%) placebo-treated patients discontinued treatment for adverse events (AEs; any grade). There were 20 deaths on study or within 28 days of the last dose (9 in the ruxolitinib and 11 in the placebo group, including 1 after crossover; Appendix, patient disposition). Principal causes of death in the ruxolitinib group were muscle weakness, subdural hematoma, renal failure, non-small cell lung cancer, acute myeloid leukemia (AML), pneumonia (2), sepsis (2); principal causes of death in the placebo group were staphylococcal infection, gastrointestinal hemorrhage, intestinal perforation, multi-organ failure, pneumonia, sepsis (2), and disease progression (4).
Overall, nonhematologic AEs occurred at a similar rate in each group. Events that occurred more frequently in the ruxolitinib group were ecchymosis, dizziness, and headache (predominantly grade 1 or 2; Table 2). The most common grade 3 or 4 nonhematologic AEs (abdominal pain, fatigue, and dyspnea) occurred more frequently in the placebo group.
Table 2.
Adverse Events Observed in ≥10% of Ruxolitinib-Treated Patients.
| Adverse Event | Ruxolitinib (N=155) % of patients |
Placebo (N=151) % of patients |
||
|---|---|---|---|---|
|
| ||||
| All Grades | Grade 3 or 4 | All Grades | Grade 3 or 4 | |
| Nonhematologic | ||||
| Fatigue | 25.2 | 5.2 | 33.8 | 6.6 |
| Diarrhea | 23.2 | 1.9 | 21.2 | 0 |
| Peripheral edema | 18.7 | 0 | 22.5 | 1.3 |
| Ecchymosis | 18.7 | 0 | 9.3 | 0 |
| Dyspnea | 17.4 | 1.3 | 17.2 | 4.0 |
| Dizziness | 14.8 | 0.6 | 6.6 | 0 |
| Nausea | 14.8 | 0 | 19.2 | 0.7 |
| Headache | 14.8 | 0 | 5.3 | 0 |
| Constipation | 12.9 | 0 | 11.9 | 0 |
| Vomiting | 12.3 | 0.6 | 9.9 | 0.7 |
| Pain in extremity | 12.3 | 1.3 | 9.9 | 0 |
| Insomnia | 11.6 | 0 | 9.9 | 0 |
| Arthralgia | 11.0 | 1.9 | 8.6 | 0.7 |
| Pyrexia | 11.0 | 0.6 | 7.3 | 0.7 |
| Abdominal pain | 10.3 | 2.6 | 41.1 | 11.3 |
| Hematologic (laboratory values)* | ||||
| Hemoglobin | 96.1 | 45.2 | 86.8 | 19.2 |
| Platelets | 69.7 | 12.9 | 30.5 | 1.3 |
| Neutrophils | 18.7 | 7.1 | 4.0 | 2.0 |
Patients are included at their worst grade on study regardless of whether this represents a change from their baseline.
Anemia and thrombocytopenia were the most frequent hematologic AEs (overall and grade 3 or 4; Table 2), and a cause for treatment discontinuation in 1 patient in each group for each event. About half of all grade 3 or 4 AEs of anemia in the ruxolitinib group occurred during the first 8 weeks of therapy. The mean hemoglobin level in ruxolitinib-treated patients reached a nadir of 95 g/l after approximately 8 to 12 weeks of therapy (Appendix Fig. S8), then recovered by week 24 to a new steady state (101 g/l). The monthly prevalence of grade 3 or 4 anemia and the proportion of patients requiring transfusions (1 or more units of red blood cells) also followed this pattern (Appendix Figs. S9A–9B). Using International Working Group for Myelofibrosis Research and Treatment response criteria, 41.2% of ruxolitinib-treated and 46.9% of placebo-treated patients who were transfusion-dependent at baseline changed their classification to transfusion-independent on study (Appendix Table S4). Importantly, ruxolitinib-treated patients with new-onset grade 3 or 4 anemia experienced improvements in symptoms and reductions in spleen volume similar to ruxolitinib-treated patients without anemia (Appendix Figs. 9C–9D).
Approximately half the grade 3 or 4 thrombocytopenia events (11 of 20) occurred during the first 8 weeks of treatment (Appendix Fig. S10), and led to dose adjustments or brief treatment interruptions. Five patients experienced more than 1 episode of grade 3 or 4 thrombocytopenia. Grade 3 or 4 episodes of bleeding (terms listed in the Appendix) occurred in 2.6% and 1.3%, respectively, of ruxolitinib-treated and 2.0% and 1.3%, respectively, of placebo-treated patients. Bruising, as described by bleeding events related to skin and subcutaneous tissue (Appendix), was explored separately; 23.2% of ruxolitinib- and 14.6% of placebo-treated patients had a bruising event; all were grade 1 or 2 except for 1 grade 3 event in the ruxolitinib group.
In patients with dosing interruption, symptoms (TSS) gradually returned to baseline levels over approximately 1 week (Appendix Fig. S11). AEs of grade ≥3 in the ruxolitinib and placebo groups, respectively, developed in 8/49 (16.3%) and 7/54 (13.0%) patients after study drug interruption and in 12/21 (57.1%) and 17/37 (45.9%) after discontinuation. There was no clear pattern in these events to suggest a specific withdrawal effect (Appendix Tables S5–S6).
Two patients in the ruxolitinib group experienced transformation to AML during the study: 1 patient with 7% bone marrow blasts at baseline and a history of breast cancer transformed after 8 months on study; the second patient entered the study with 2% baseline marrow blasts and a trisomy 8 chromosomal abnormality, and transformed after 5 months on study. There were no transformations in the placebo group.
DISCUSSION
In this study, ruxolitinib therapy was significantly better than placebo for all primary and secondary endpoints, as well as an updated analysis of overall survival. In addition to the proportion of patients meeting the defined response threshold of a ≥35% reduction in spleen volume, nearly all ruxolitinib-treated patients achieved some reduction in spleen volume. Spleen volume responses were durable, with 67% of responding patients maintaining response for ≥48 weeks with continued therapy.
Improvements in symptoms were measured using the modified MFSAF v2.0 diary, a tool designed specifically to assess symptoms of myelofibrosis. The majority of patients experienced improvements in symptoms which occurred even in patients not achieving a ≥35% reduction in spleen volume. In contrast, most patients receiving placebo experienced progressive splenomegaly and worsening of myelofibrosis-related symptoms. Changes in symptoms recorded with the MFSAF were directionally consistent with other validated and common patient-reported outcome instruments used in this study.
Anemia and thrombocytopenia were more common in ruxolitinib-treated patients than in patients treated with placebo. These AEs were manageable, as evidenced by the low discontinuation rate (1 patient in each group for each event). Thrombocytopenia rarely recurred at a grade 3 or 4 level after appropriate dose modifications and was not associated with an increase in bleeding events, although events of bruising were more common in the ruxolitinib group. The prevalence of grade 3 or 4 anemia peaked after approximately 8 to 12 weeks of ruxolitinib therapy, subsequently decreasing to levels similar to placebo-treated patients. Importantly, patients on ruxolitinib who developed new-onset grade 3 or 4 anemia demonstrated symptomatic improvement similar to those without anemia. In comparison, patients receiving placebo (with and without grade 3 or 4 anemia) had worsening of symptoms, which were greater in patients with anemia.
There were 2 transformations to AML in this study, both in ruxolitinib-treated patients who had baseline characteristics putting them at higher risk for transformation. Although there were no transformations to AML in 146 patients treated with ruxolitinib and 2 transformations in 73 patients treated with best available therapy in another phase 3 trial with a median follow-up of 61 weeks,16 longer-term follow-up will be required to better define rates of AML transformation.
After interruption of ruxolitinib therapy, myelofibrosis-related symptoms gradually returned to baseline levels. A comparison of AEs in the ruxolitinib and placebo groups reported after treatment interruption or permanent discontinuation showed no clear pattern of a specific withdrawal effect.
Ruxolitinib reduced the splenomegaly and symptoms that are prominent manifestations of myelofibrosis and appeared to improve overall survival. Toxicities were generally managed with dose modification. These findings demonstrate that ruxolitinib is an effective therapy for myelofibrosis.
Supplementary Material
Acknowledgments
This study was supported by Incyte Corporation. Editorial assistance was provided by Kelly Reith of Incyte Corporation.
Footnotes
All investigators and participating centers are listed in the Supplementary Appendix.
References
- 1.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29:1356–63. doi: 10.1200/JCO.2010.32.9490. [DOI] [PubMed] [Google Scholar]
- 2.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895–901. doi: 10.1182/blood-2008-07-170449. [DOI] [PubMed] [Google Scholar]
- 3.Abdel-Wahab OI, Levine RL. Primary myelofibrosis: update on definition, pathogenesis, and treatment. Annu Rev Med. 2009;60:233–45. doi: 10.1146/annurev.med.60.041707.160528. [DOI] [PubMed] [Google Scholar]
- 4.Delhommeau F, Jeziorowska D, Marzac C, Casadevall N. Molecular aspects of myeloproliferative neoplasms. Int J Hematol. 2010;91:165–73. doi: 10.1007/s12185-010-0530-z. [DOI] [PubMed] [Google Scholar]
- 5.Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19:385–93. doi: 10.1016/j.semcdb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–17. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–27. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. 2008;22:14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 9.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–55. [PubMed] [Google Scholar]
- 10.Mesa RA, Kantarjian H, Tefferi A, et al. Evaluating the serial use of the Myelofibrosis Symptom Assessment Form for measuring symptomatic improvement: performance in 87 myelofibrosis patients on a JAK1 and JAK2 inhibitor (INCB018424) clinical trial. Cancer. 2011 Apr 8; doi: 10.1002/cncr.26129. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesa RA, Kantarjian H, Tefferi A, et al. Validation of the serial use of the Myelofibrosis Symptom Assessment Form (MF-SAF) for measuring symptomatic improvement: performance in 86 myelofibrosis patients on INCB018424 clinical trial. Blood. 2009;114 doi: 10.1002/cncr.26129. Abstract 3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT) Blood. 2006;108:1497–503. doi: 10.1182/blood-2006-03-009746. [DOI] [PubMed] [Google Scholar]
- 13.Harris A, Kamishima T, Hao HY, et al. Splenic volume measurements on computed tomography utilizing automatically contouring software and its relationship with age, gender, and anthropometric parameters. Eur J Radiol. 2010;75:e97–101. doi: 10.1016/j.ejrad.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Mazonakis M, Damilakis J, Maris T, Prassopoulos P, Gourtsoyiannis N. Estimation of spleen volume using MR imaging and a random marking technique. Eur Radiol. 2000;10:1899–903. doi: 10.1007/s003300000551. [DOI] [PubMed] [Google Scholar]
- 15.Prassopoulos P, Daskalogiannaki M, Raissaki M, Hatjidakis A, Gourtsoyiannis N. Determination of normal splenic volume on computed tomography in relation to age, gender and body habitus. Eur Radiol. 1997;7:246–8. doi: 10.1007/s003300050145. [DOI] [PubMed] [Google Scholar]
- 16.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib vs best available therapy in myelofibrosis. N Engl J Med. 2011 doi: 10.1056/NEJMoa1110556. (in press) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.