

Abstract
After crossing the cellular membrane barrier during cell entry, most animal viruses must undergo further disassembly before initiating viral gene expression. In many cases, these disassembly mechanisms remain poorly defined. For this report, we examined a final step in disassembly of the mammalian reovirus outer capsid: cytoplasmic release of the central, δ fragment of membrane penetration protein μ1 to yield the transcriptionally active viral core particle. An in vitro assay with reticulocyte lysate recapitulated the release of intact δ molecules. Requirements for activity in this system were shown to include a protein factor, ATP, and Mg2+ and K+ ions, consistent with involvement of a molecular chaperone such as Hsc70. Immunodepletion of Hsc70 and Hsp70 impaired δ release, which was then rescued by addition of purified Hsc70. Hsc70 was associated with released δ molecules not only in the lysate but also during cell entry. We conclude that Hsc70 plays a defined role in reovirus outer capsid disassembly, during or soon after membrane penetration, to prepare the entering particle for gene expression and replication.
After crossing the cellular membrane barrier during cell entry, a virus must initiate its own gene expression. In many cases, one or more intervening steps are also required, including further disassembly of the virus particle and/or transport to an appropriate intracellular location. For example, with human immunodeficiency virus 1, translocation of virus into the cytoplasm upon membrane fusion is followed by partial disassembly of the viral nucleocapsid. This allows reverse transcription of the RNA genome, which is in turn followed by translocation of these “pre-integration complexes” into the cell nucleus. Only after the viral DNA is integrated into the host DNA does viral gene expression commence.
Cell entry by mammalian orthoreovirus (reovirus), a nonenveloped double strand RNA virus of the family Reoviridae, is also accompanied by stepwise partial uncoating of the virus particle. Host proteases degrade the stabilizing outer capsid protein σ3 and cleave the autolytic outer capsid protein μ1 (76 kDa) near residue 580, generating a particle termed the infectious subvirion particle (ISVP)5 (1, 2). This cleavage of μ1 yields the myristoylated N-terminal fragment μ1δ (63 kDa) and the C-terminal fragment ϕ (13 kDa) (Fig. 1A), both of which remain particle associated (3). Because a portion of the μ1 autocleavage between residues 42 and 43 may take place during reovirus assembly, a portion of the ISVP-associated μ1δ may consist of the myristoylated N-terminal peptide μ1N (4 kDa) and the large central fragment δ (59 kDa) (Fig. 1A) (4, 5). ISVPs can be generated in vitro by protease treatment of intact virions (3, 6, 7). The ISVP is metastable and can spontaneously convert to a particle termed the ISVP*, a transition associated with perforation of target membranes (8, 9). ISVP → ISVP* conversion is characterized by μ1 rearrangement to a hydrophobic and protease-sensitive conformer, release of the adhesion fiber σ1, and derepression of the transcriptase activity of the particle (9). Moreover, further μ1 autocleavage to yield the μ1N peptide is completed during this transition (4), and μ1N and ϕ are largely released from the ISVP* (8, 10)6. The δ fragment, in contrast, remains largely particle associated (8–10). μ1 conformational rearrangement and σ1 release also occur during cell entry by reovirus and are associated with membrane penetration and productive infection (11). In contrast to the stable association between δ and the particle observed in vitro, however, the ISVP*-like particle that is formed during productive cell entry appears to exist only transiently; its formation is rapidly followed by separation of δ from the core particle (11), which can then mediate viral transcription in the cytoplasm.
FIGURE 1. δ molecules are released from reovirus particles entering Mv1Lu cells.

A, a line diagram of μ1 and its various cleavage fragments is shown. B, T2J and T3c9 ISVPs were attached to cycloheximide-pretreated Mv1Lu cells at 4 °C. Cells were then fixed either immediately (0 h post-infection (p.i.)) or after a 2-h incubation at 37 °C in the continued presence of cycloheximide (2 h p.i.). Entering viral components were visualized by immunofluorescence using μ1-specific mAb 4A3 (green) and anti-core serum antibodies (red). C, [35S]Met/Cys-labeled T1L ISVPs were attached to Mv1Lu cells at 4 °C. Cells were then either kept on ice (0 h p.i.) or incubated at 37 °C for 2 h (2 h p.i.), before being lysed in buffer A with protease inhibitors. The postnuclear supernatants were cleared of viral particles by pelleting. Both pellet and supernatant fractions were analyzed by SDS-PAGE and phosphorimaging.
Both released δ molecules and core particles appear to be cytoplasmically localized after membrane penetration by reovirus, with δ immunostaining in a diffuse distribution and core particles in a more punctate pattern that does not colocalize with membrane markers (11). If infecting particles contain mutant μ1 protein N42A, which is defective at membrane penetration, δ and core immunostaining continue to colocalize in a punctate distribution, consistent with ISVP*-like particle trapping within intracellular vacuoles (10). Furthermore, new protein synthesis is not required for removal of δ from ISVP*-like particles during infection (11). Together, these observations have led to the hypothesis that constitutively expressed host cytoplasmic factors play a role in removing δ from ISVP*-like particles either during or soon after membrane penetration. We now further hypothesize that the hydrophobic conformer of δ present in the ISVP* (9) is a substrate for cellular molecular chaperones, which effect its removal from the particle.
Molecular chaperones are proteins that facilitate protein folding and unfolding and prevent aggregation of misfolded or denatured proteins during cellular stress (12). They aid in assembly and disassembly of protein complexes, protein degradation, cellular transport, translocation of proteins across cellular membranes, and signal transduction (13). One conserved family of molecular chaperones with a molecular weight near 70,000 is termed Hsp70/DnaK (14). Hsp70 family members are present in prokaryotes and in most compartments of eukaryotic cells. Examples include Hsc70 and Hsp70, which reside in the eukaryotic cytoplasm and are expressed either constitutively (Hsc70) or in response to cellular stress (Hsp70). Proteins of the Hsp70 family are weak ATPases, which bind and release their substrates through regulated cycles of ATP binding and hydrolysis. Targeting to their substrates, as well as ATP hydrolysis, is stimulated by other molecular chaperones (cochaperones) that contain a DnaJ-like domain, such as Hsp40 and auxilin (13). Additional families include Hsp90 molecular chaperones and chaperonins (12).
Given the diverse functions of molecular chaperones, it is not surprising that they play numerous roles in the life cycles of different viruses. Viral infection can induce endogenous molecular chaperone expression and/or relocalization, and in some cases, this induction has been shown to be essential for virus replication. For example, avian adenovirus CELO encodes a protein that increases expression of both Hsp70 and Hsp40, as well as their relocalization to the nucleus; viruses unable to express this protein are replication defective, and this defect is partially rescued when molecular chaperone expression is elevated by other means (15). Some viruses encode their own molecular chaperones. For example, polyomavirus expresses T antigens, which are DnaJ-containing proteins that act as cochaperones for Hsc70 (16). Specific roles ascribed to molecular chaperones during viral infections include regulation of viral and host gene expression, assembly and disassembly of viral replication complexes, and virus particle assembly (17).
Roles of molecular chaperones in virus particle disassembly are less well appreciated, but recently it has been shown that Hsp70 family members can disassemble polyoma- and papillomavirus capsids in vitro (18). A role for Hsc70 in one or more early postattachment steps in rotavirus infection has also been demonstrated (19–21). Several viruses incorporate cellular molecular chaperones within mature virions. For example, Hsc70, Hsp70, and Hsp60 have been found in the virion of human immunodeficiency virus 1 (22), and Hsc70 has been found in the virions of rabies, vesicular stomatitis, Newcastle disease, and influenza A viruses (23). Although incorporation of molecular chaperones might be a nonspecific by-product of virus assembly, one can speculate that these proteins may play specific roles in virus disassembly. Thus, molecular chaperones may be more widely involved in virus disassembly than has so far been understood.
For the current study, we examined the newly hypothesized role of molecular chaperones in δ release to yield cytoplasmic viral cores, a final step in reovirus outer capsid disassembly. Our results both in vitro and in cells identify a role for Hsc70 in this process. We conclude that Hsc70 contributes in a defined manner to reovirus disassembly, during or soon after membrane penetration, to prepare the entering particle for viral gene expression and replication.
EXPERIMENTAL PROCEDURES
Reagents, Antibodies, and Cells
Rabbit reticulocyte lysate (RRL) was from Green Hectares. ATP, ATPγS, apyrase, glucose, hexokinase, creatine phosphate, creatine kinase, phenylmethyl sulfonyl fluoride, Nα-p-tosyl-l-lysine chloromethyl ketone-treated α-chymotrypsin, trypsin, and cycloheximide were from Sigma-Aldrich. Protein A and G beads were from Dynal. Recombinant bovine Hsc70 protein was from Stressgen (product SPP-751). Monoclonal antibodies (mAbs) against Hsc70 (clone 1B5) and Hsp70 (clone C92F3A-5) were from MBL. mAbs against λ2 (7F4) and μ1 (4A3) and serum antibodies against μNS, Type 1 Lang (T1L) virions, and T1L cores have been described previously (11). Alexa-488- and Alexa-594-conjugated goat anti-mouse and goat anti-rabbit immunoglobulin G antibodies were from Molecular Probes. Spinner-adapted L929 and Mv1Lu cells were grown as described previously (11).
Buffers
Buffers used were as follows: virion-Na+ buffer (150 mM NaCl, 10 mM MgCl2, 10 mM Tris-HCl, pH 7.5); virion-K+buffer (150 mM KCl in place of NaCl); buffer A (150 mM NaCl, 1% Nonidet P-40, 10 mM Tris-HCl, pH 7.9); buffer E (80 mM potassium acetate, 0.5 mM magnesium acetate, 10 mM Hepes-KOH, pH 7.4); dialysis buffer (10 or 50 mM Hepes-NaOH, pH 7.5, 2 mM β-mercaptoethanol); conversion buffer (300 mM CsCl, 50 mM Tris-HCl, pH 7.5); Hsc70 buffer (150 mM NaCl, 1 mM dithiothreitol, 0.1 mM PMSF, 10 mM Tris-HCl, pH 7.5); attachment buffer (phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.5) with 2 mM MgCl2).
Virions and ISVPs
Virions of reoviruses T1L, Type 2 Jones (T2J), and Type 3 clone 9 (T3c9) were grown and purified by the standard protocol (24) and stored in virion-Na+ buffer. To generate T1L virions containing [35S]Met/Cys-labeled proteins, 7 mCi of Tran35S label (ICN) was added at the onset of infection. ISVPs were obtained by digesting virions in virion-Na+ buffer at concentrations of 1 × 1013 particles/ml with 200 μg/ml chymotrypsin for 10–20 min at 32 or 37 °C. Digestion was stopped with 2–5 mM phenylmethyl sulfonyl fluoride on ice. To attach particles to protein A beads, beads were washed with buffer A and incubated with an equal volume of mAb 7F4 (1:2–1:4 dilution in buffer A) for 1 h at room temperature or overnight at 4 °C. The 7F4-bead conjugates were then washed with buffer A and incubated, with rotary mixing, with particles in buffer A at 5 × 109 ISVPs or 1 × 1010 virions per μl of beads for 1–5 h at 4 °C. Approximately 5 μl of the original bead volume were used per δ release reaction (see “RRL δ Release Reactions”).
Infections, Fractionation, Immunofluorescence
Mv1Lu cells (~1–2 × 106 cells per 60-mm dish) were left untreated (see Fig. 1C) or were pretreated with 100 μg/ml cycloheximide (see Fig. 6B) for 30 min at 37 °C. Cells were washed with attachment buffer and then incubated with [35S]Met/Cys-labeled T1L ISVPs in that buffer (see Fig. 1C), unlabeled T1L ISVPs in that buffer (see Fig. 6B), or that buffer alone (see Fig. 6B, uninfected) for 1 h at 4 °C (~2.5 ×105 particles/cell). After washing with attachment buffer, cells were either left in that buffer on ice (time zero) or incubated in the presence (see Fig. 6B) or absence (see Fig. 1C) of 100 μg/ml cycloheximide for 2 h at 37 °C. After washing with phosphate-buffered saline, cells were lysed in buffer A with protease inhibitors (Roche) for 30 min at 4 °C and centrifuged at 8000 × g for 10 min to remove nuclei and cell debris. Particles were then separated from soluble protein fraction by pelleting at 16,000 × g for 1 h at 4 °C. If pellets were used in analysis (see Fig. 1C), they were washed once with buffer A, resuspended in gel sample buffer, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and phosphorimaging using a Typhoon system (GE Healthcare). Supernatants were analyzed directly by SDS-PAGE and phosphorimaging (see Fig. 1C) or were subjected to immunoprecipitation using anti-virion serum antibodies (see Fig. 6B). Infection of Mv1Lu cells and immunofluorescence analysis shown in Fig. 1B were performed as described previously (11).
FIGURE 6. Hsc70 associates with δ molecules released from ISVP*s in RRL as well as ones entering Mv1Lu cells.

A, SDS-PAGE and either immunoblotting using anti-Hsc70 mAb (top) or phosphorimaging of a paired sample (bottom): [35S]Met/Cys-labeled T1L ISVP*s were incubated with RRL, and reaction supernatants were treated with apyrase or ARS. Hsc70 was immunoprecipitated from each reaction. Immunoprecipitated protein (beads) as well as that remaining in the supernatant under each condition (sup) was analyzed. B, SDS-PAGE and immunblotting: T1L ISVPs (infected +) or buffer alone (infected −) were attached to cycloheximide-pretreated Mv1Lu cells at 4 °C. Cells were then incubated at 37 °C for 2 h in the continued presence of cycloheximide (CHX), before being lysed in buffer A with protease inhibitors. The postnuclear supernatants were cleared of viral particles by pelleting and subjected to immunoprecipitation using anti-virion serum antibodies (α-virion) or buffer alone (Mock). Immunoprecipitated proteins (beads) and those remaining in the supernatant (sup) were detected using anti-Hsc70 mAb, anti-virion serum antibodies, or anti-μNS serum antibodies.
Chymotrypsin Treatment of RRL
RRL was treated with 280 μg/ml chymotrypsin or an equivalent amount of chymotrypsin previously inactivated with 30 mM phenylmethyl sulfonyl fluoride (final concentration in RRL, 3 mM) for 2 h on ice. Chymotrypsin digestion was stopped with 3 mM phenylmethyl sulfonyl fluoride.
Dialysis of RRL
RRL was dialyzed twice against 10 mM dialysis buffer and twice against 50 mM dialysis buffer. Following dialysis, the volume of RRL increased and was concentrated to half the original volume using Microcon-YM10 centrifugal device (Millipore) according to manufacturer’s recommendations.
RRL δRelease Reactions
Standard assay conditions consisted of 60% RRL, 80 mM potassium acetate, 0.5 mM magnesium acetate, 2 mM ATP, and 2 mM dithiothreitol. If apyrase or ATPγS was used to pretreat RRL, ATP was omitted. If RRL was dialyzed prior to the reaction, salt concentrations were adjusted to 90 mM monovalent salts and 1.5 mM divalent salts. A slightly different percentage of RRL was used in some reactions (see below). In the experiment in which reactions were performed in the absence of RRL, 10 mM dialysis buffer was used in its place (Fig. 2C). Three general variations of the protocol were used.
FIGURE 2. RRL supports release of intact δ molecules from ISVP*s.

A, experimental design: V, virion; I, ISVP; I*, ISVP*; and C, core; 1, chymotrypsin treatment; 2, attachment of particles to beads (omitted in protocol 2); 3, incubation with CsCl; 4, addition to RRL; 5, incubation at 32 °C; 6, separation of reaction supernatants from bead-bound particles. B, SDS-PAGE and immunoblotting with anti-virion serum antibodies: T1L, T2J, and T3c9 ISVP*s prior to incubation with RRL (RRL−) were compared with particles recovered after incubation with RRL (RRL+). C, [35S]Met/Cys-labeled T1L ISVP*s were either left on ice (lane 1) or incubated at 32 °C in the presence of RRL (lanes 3 and 4) or in its absence (lane 2). Reaction supernatant was then in one case treated with chymotrypsin (lane 4). Particles and reaction supernatants were analyzed by SDS-PAGE and phosphorimaging. δ and λ bands were quantified, and the δ/λ ratio of output particles or that of output particles and supernatant together was expressed relative to that of particles incubated on ice (input particles), which was set to 100%. Data are presented as means ± S.D. for three replicate experiments.
Protocol 1
Bead-bound particles were washed with buffer A, then buffer A with no Nonidet P-40, and resuspended in gel sample buffer (input particles) or an equal volume of ice-cold conversion buffer. To make ISVP*s, bead-bound ISVPs in conversion buffer were incubated at 32 °C for 20 min. Bead-bound particles in conversion buffer comprised 5% of the 100-μl reaction volume (~5 μl of the original bead volume). Incubations were 30 min long unless otherwise indicated. Beads were immobilized on a magnet (Dynal) to separate reaction supernatants, washed four times with 200 μl of buffer A, and boiled in gel sample buffer (output particles). δ release from particles of different strains (see Fig. 2B) was performed using this protocol, and input and output particles were detected by SDS-PAGE and immunoblotting using anti-virion serum antibodies. δ-release reactions with [35S]Met/Cys-labeled T1L particles shown in Fig. 3A were performed using this method, and input and output particles were analyzed by SDS-PAGE and phosphorimaging.
FIGURE 3. Release of δ molecules in RRL is dependent on ISVP → ISVP* conversion.

A, SDS-PAGE and phosphorimaging: [35S]Met/Cys-labeled T1L virions, ISVPs, and ISVP*s prior to incubation with RRL reaction mixtures (RRL−) were compared with particles recovered after incubation with RRL (RRL+). B, SDS-PAGE and immunoblotting with anti-virion serum antibodies: T1L ISVPs were incubated at 37 °C in the presence of CsCl and at the indicated times, one aliquot was transferred to a tube with trypsin on ice and the other to a tube with RRL at 32 °C. Both incubations were for 45 min.
Protocol 2
ISVP → ISVP* conversion of free particles was attained by incubation of 5 × 1012 particles/ml in virion-Na+ buffer with 300 mM CsCl at 37 °C for 10 min or as indicated. 10-μ1 aliquots of the reaction were assayed for conformational status of the μ1 protein by trypsin digestion (100 μg/ml trypsin for 45 min on ice) and/or for δ release in 200-μ1 reactions (45 min at 32 °C). Particles were separated from the reaction supernatants by pelleting at 16,000 × g for 30 min and washed with buffer A. Input and output particles, as well as trypsin-treated particles, were analyzed by SDS-PAGE and immunoblotting using anti-virion serum antibodies (see Fig. 3B).
Protocol 3
Bead-bound ISVP*s were obtained as in protocol 1, then washed with virion-K+buffer (or buffer A with no Nonidet P-40 for dialyzed RRL experiment, see Fig. 4C) and resuspended in an equal volume of virion-K+buffer (or buffer A with no Nonidet P-40). One 5-μ1 aliquot of beads was saved (input particles) and the rest were resuspended in 20-μ1 reaction mixtures following removal of buffer. Incubations were 0.5–1 h long. Supernatant was separated from bead-bound particles as in protocol 1. In the experiments in which reaction supernatants were directly analyzed (Figs. 2C, 4C, and 5B), two initial 6-μ1 washes with buffer A were pooled with each respective supernatant. Beads were then washed twice more with 200 μl of buffer A prior to boiling in sample buffer. Input and output particles and if applicable, reaction supernatants were analyzed by SDS-PAGE and phosphorimaging. This protocol was used in the following experiments. 1) To detect the released δ directly (see Fig. 2C), reaction supernatant was left untreated or was treated with chymotrypsin before analysis. 2) To assay for chymotrypsin sensitivity of δ release in RRL (see Fig. 4A), RRL was pretreated with chymotrypsin or inactivated chymotrypsin prior to the reaction. 3) To assay for ATP requirement (see Fig. 4B), RRL was preincubated for 10 min at 32 °C with 2 mM ATP, 10 units/ml apyrase, or 10 mM ATPγS. 4) To test dialyzed RRL, see Fig. 4C. 5) For co-immunoprecipitation between Hsc70 and released δ (50% RRL) (see Fig. 6A), 30-μl aliquots of pooled reaction supernatants were incubated with 3 μl of 10× ATP-regenerating system (ARS) (300 mM creatine phosphate, 4 mg/ml creatine kinase), 100 units/ml apyrase, or 50 mM glucose plus 500 units/ml hexokinase, for 20 min at 32 °C. 6) To test immunodepleted RRL (64% RRL) (see Fig. 5B), the reaction also contained 10% Hsc70 buffer or 10% Hsc70 protein (1.8 μg) in Hsc70 buffer.
FIGURE 4. Release of δ molecules in RRL is protease-sensitive, ATP-dependent, and cation-dependent.

[35S]Met/Cys-labeled T1L ISVP*s were used in each panel, and samples were analyzed by SDS-PAGE and phosphorimaging. Amount of δ release was quantified as described in the legend to Fig. 2. The means ± S.D. of particle-associated δ content from three (A and B) or five (C) replicate experiments were plotted. A, ISVP*s prior to reaction (input *) were compared with particles recovered after incubation with RRL, which I was left untreated (untr.) or was pretreated either with chymotrypsin (CHT) or with inactivated chymotrypsin (PMSF-CHT). B, ISVP*s prior to incubation with RRL (input I*) were compared with particles recovered after incubation with RRL containing ATP, ATPγS, or apyrase. C, particles recovered after incubation with RRL dialyzed against buffer lacking salts and ATP, then supplemented with ATP and/or the indicated salts. Either the monovalent cation or the divalent cation was varied. Salts contained either chloride or acetate anions, which did not differently affect the amount of δ release (data not shown). Bottom panel shows representative gels of particles and corresponding reaction supernatants from one of the replicate experiments.
FIGURE 5. Hsc70 contributes to δ release in RRL.

A, SDS-PAGE and immunblotting: RRL was treated with ARS and then either left untreated (ARS) or incubated with a combination of anti-Hsc70 and anti-Hsp70 mAbs (ARS/α-Hscp70) or buffer alone (ARS/mock). Protein complexes removed with three sets of beads (beads, lanes 7–9 and 10 –12), the resulting supernatant fractions (RRL, lanes 3–6), and RRL treated only with ARS (RRL, lanes 1 and 2) were assayed for Hsc70 (top panel) and Hsp70 (bottom panel) protein content by immunoblotting with the preceding mAbs. B, SDS-PAGE and phosphorimaging: [35S]Met/Cys-labeled T1L ISVP*s prior to incubation with RRL (input ISVP*, lane 1) were compared with particles recovered after incubation with RRL treated as indicated on top (lanes 2–5). Purified Hsc70 was added to antibody-treated sample in one case (lane 6). Particle-associated δ content was quantified as described in the legend to Fig. 2 and presented as means ± S.D. for five replicate experiments. A statistical test was performed between the paired values for mock- and antibody-treated samples as well as between the paired values for antibody-treated samples to which purified Hsc70 was or was not added. The resulting p values (highly significant in both cases) are shown above lines connecting the corresponding pairs of bars. Bottom panel shows representative gels of particles and corresponding reaction supernatants from one of the replicate experiments.
Immunodepletion of RRL
Anti-Hsc70 and anti-Hsp70 mAbs (20 μl each) were separately dialyzed against buffer E (three times) and concentrated to one-half to one-third of the original volume. RRL was treated with ARS (ARS-RRL). ARS-RRL (24 μl) was incubated with 3 μl of each mAb (ARS/anti-Hscp70-RRL) or with 6 μl of buffer E (ARS-RRL and ARS/mock-RRL) for 2–3 h. mAbs and associated proteins were removed by three rounds of 1–1.5 h incubations with 20 μl of protein G beads (ARS/anti-Hscp70-RRL and ARS/mock-RRL). After washing with buffer E, one-fifth of total protein recovered from beads by boiling in gel sample buffer, along with aliquots of differently treated RRL, was analyzed for Hsc70 and Hsp70 protein content by SDS-PAGE and immunoblotting (see Fig. 5A).
Immunoprecipitations
RRL Reaction Supernatants
RRL reaction supernatants treated with apyrase, ARS (see Fig. 6A), or glucose/hexokinase (not shown) were each split into two 10- or 15-μl aliquots and incubated with 4 or 6 μl of anti-Hsc70 mAb (~0.3 μg/μl final concentration) or phosphate-buffered saline overnight at 4 °C. Antibody and associated proteins were removed by three rounds of 4- to 12-h incubations with 10 or 15 μl of protein G beads, which effectively depleted the input antibody (data not shown) and significantly reduced the target protein levels (see Fig. 6A). Supernatants were split into four aliquots (supernatant). Beads were washed with buffer A, boiled in gel sample buffer, pooled, and then split into four equal aliquots (beads). One aliquot of supernatant and one aliquot of beads were analyzed by SDS-PAGE and phosphorimaging to detect associated δ molecules (see Fig. 6A, bottom panel). The second aliquot of each was analyzed by SDS-PAGE and immunoblotting using anti-Hsc70 mAb (see Fig. 6A, top panel).
Mv1Lu Cell Infections
Soluble protein cytoplasmic extracts corresponding to two 60-mm dishes, each of infected and uninfected cells were pooled in a final volume of 300 μl of buffer A with protease inhibitors and incubated with 25 μl of anti-virion serum antibodies (1/12 dilution) or buffer alone (phosphate-buffered saline, 50% glycerol) overnight at 4 °C. Antibody complexes were removed with four or five rounds of 4- to 12-h incubations with 30 μl of protein G beads. Beads were washed with buffer A, boiled in gel sample buffer, and pooled (beads). Seven-eighths of the beads along with 4 μl of the supernatant were analyzed by SDS-PAGE and immunoblotting using anti-Hsc70 mAb, whereas the remaining one-eighth of the beads along with 50 μl of the supernatant were analyzed by SDS-PAGE and immunoblotting using anti-virion serum (Fig. 6B).
Immunoblotting
For immunoblot analysis, proteins were electrophoretically transferred from the gel to a nitrocellulose membrane in transfer buffer (25 mM Tris, pH 8.3, 192 mM glycine). Antibody incubations were performed in 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 5% milk, 0.05% Tween 20, at dilutions that varied with the type of secondary detection used. For Fig. 5A and Fig. 6, A and B, anti-Hsc70 mAb was used at 1:2000 –1:5000 and detected by anti-rat immunoglobulin G conjugated to horseradish peroxidase and developed with luminol-enhanced chemiluminescence substrate (Western Lightning, PerkinElmer). Anti-virion serum was used at 1:1000 for Figs. 2B, 3B, and 6B and detected in Figs. 2B and 6B by anti-rabbit immunoglobulin G conjugated to horse-radish peroxidase, and in Fig. 3B by anti-rabbit immunoglobulin G conjugated to alkaline phosphatase (Bio-Rad) and the colorimetric reagents p-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt (Bio-Rad). For Fig. 6B, anti-μNS serum was used at 1:10,000 and detected by anti-rabbit immunoglobulin G conjugated to horseradish peroxidase.
Statistics
Statistical analyses consisted of one-tailed paired T tests. For Fig. 5B, repeated measures of one-way analysis of variance with post-tests was also performed and yielded p < 0.001 for both two-column comparisons.
RESULTS
Release of δMolecules into the Cytoplasm Is Not Virus Strain Specific
ISVPs of three reovirus strains, T1L, T2J, and T3c9, were attached to cycloheximide-pretreated Mv1Lu cells on ice for 1 h. Following removal of unattached virus, cells were fixed either immediately (0 h post-infection) or after incubation at 37 °C for 2 h in the continued presence of cycloheximide to allow cell entry by virus in the absence of protein synthesis. Entering components were then visualized by immunofluorescence using a combination of conformation-specific mAb 4A3, which recognizes an epitope in the δ region of μ1, and anti-core serum antibodies, which recognize a mixture of epitopes in the core-surface proteins. The two types of antibodies have both different and shared specificities; mAb 4A3 binds to its epitope in ISVP*s and released δ molecules, but not in ISVPs or cores, whereas anti-core serum antibodies bind to their epitopes in ISVP*s and cores, but not in ISVPs or released δ molecules (10, 11). In the current experiments with T1L, which was examined previously (10, 11), and in each of the two newly analyzed strains, we detected 4A3 staining distributed more diffusely throughout the cell at 2 h post-infection and only scattered colocalization between this 4A3 staining and the more punctate staining by anti-core serum antibodies (Fig. 1B; data not shown for T1L because shown in two previous reports (10, 11)). Consistent with previous findings (10, 11), we conclude that the ISVP* is a transient cell-entry intermediate common to different reovirus strains and from which the δ fragment is released into the cytoplasm during or soon after cell entry.
Intact δMolecules Are Released into the Cytoplasm
Preservation of the 4A3 epitope in the preceding experiments suggests that the released δ molecules may be intact. To address whether intact δ molecules are released from particles, [35S]Met/Cys-labeled T1L ISVPs were attached to Mv1Lu cells at 4 °C for 1 h. After removal of unattached virus, cells were either kept on ice (0 h post-infection) or incubated at 37 °C for 2 h to allow cell entry. Cells were then lysed with nonionic detergent (which does not release δ from ISVP*s in vitro (10)), and postnuclear supernatants were subjected to centrifugation to separate intact particles (pelleted) from free proteins (remaining in the supernatant). Both fractions were analyzed by SDS-PAGE and phosphorimaging (Fig. 1C). Enrichment of intact δ relative to the λ band, which represents core proteins, was evident in the supernatant fraction of the 2-h sample. Enrichment of σ1 was also apparent in this fraction, as expected for this protein that is known to be released from ISVP*s (9). Major proteolytic fragments of δ were absent from the supernatant or pellet fractions. The amount of free δ observed by this method reproducibly accounted for at least 20% of recovered δ content (data not shown). These findings show that at least a portion of δ molecules remain intact after release, and thus must have been released in intact form. They do not, however, exclude the possibility that some of the released δ molecules may have been degraded. Removal of δ from ISVP*s in vitro has been previously shown to require proteolysis (9). In contrast, our new data indicate that δ release during infection is at least partly a nondegradative event, and we therefore hypothesize that host factors other than proteases contribute to δ release within cells.
Reticulocyte Lysate Supports δRelease from ISVP*s
An in vitro system using RRL was developed to study δ release, as illustrated in Fig. 2A. Reovirus virions were converted to ISVPs by chymotrypsin treatment and then attached to beads. Bead-bound particles were then converted to ISVP*s by incubation in CsCl-containing buffer at 32 °C and subsequently incubated at the same temperature in a reaction mixture consisting primarily of RRL. Protein content of particles prior to incubation with RRL (input) was compared with that of particles recovered after incubation (output). Using anti-virion serum antibodies for immunoblot detection of proteins separated by SDS-PAGE, we found that the δ content of ISVP*s of reoviruses T1L, T2J, and T3c9 was greatly diminished after incubation with RRL for 30 min (Fig. 2B). Furthermore, the other δ-encompassing fragments of μ1 (μ1C and μ1δ) were also removed from ISVP*s during this incubation (Fig. 2B).
Although this experiment showed that δ and δ-encompassing molecules are released from ISVP*s during incubation in RRL, it did not address the integrity of the released molecules. We therefore performed similar incubations in the presence or absence of RRL using [35S]Met/Cys-labeled particles. In addition to analyzing protein content of the output particles, we analyzed the reaction supernatants directly by SDS-PAGE and phosphorimaging for the presence of released δ (Fig. 2C, lanes 1–3). In these experiments, we failed to observe any proteolytic fragments of δ and reproducibly accounted for at least 69% of the input δ molecules. The release of δ was largely RRL-dependent (Fig. 2C, compare lanes 2 and 3). The released, intact δ remained chymotrypsin-sensitive (Fig. 2C, lane 4) and susceptible to immunoprecipitation by mAb 4A3 (data not shown), properties shared with the δ in ISVP*s (9). The latter finding also agrees with the immunofluorescence data in cells, where released δ molecules were detected using this same mAb (see Fig. 1B).
Release of δin RRL Is Dependent on ISVP → ISVP* Conversion
To correlate other aspects of the in vitro reaction with infection, we compared the capacity of RRL to support release of μ1 and other δ-encompassing species from different reovirus particle types: virions, ISVPs, and ISVP*s (Fig. 3A). RRL did not support release of any μ1 species from virions or ISVPs, whereas release of all δ-encompassing species from ISVP*s was nearly complete after a 30-min incubation with RRL. In addition, conversion of δ to a protease-sensitive conformer, a hallmark of the ISVP → ISVP* conversion (9), temporally correlated with the capacity of RRL to mediate δ release (Fig. 3B). The onset of δ sensitivity to both protease degradation (Fig. 3B, top) and release in RRL (Fig. 3B, bottom) occurred between 6 and 8 min of incubation in the conversion buffer.
δ Release Activity of RRL Is Protease-sensitive, ATP-dependent, and Cation-dependent
We next tested whether a protein component of RRL is required for δ release from ISVP*s. Indeed, pretreatment of RRL with chymotrypsin abolished its capacity for δ release (Fig. 4A). To test whether δ release is energy-dependent, we performed the reaction in RRL in the presence of either ATPγS (a nonhydrolyzable ATP analog) or apyrase (an ATP diphosphatase). Both of these additions inhibited δ release to a substantial degree (Fig. 4B). Following dialysis of RRL against buffer at physiological pH but lacking both salts and ATP, addition of ATP as well as both Mg2+ and K+ ions was required to restore the activity (Fig. 4C). Addition of ATP and either Mg2+ or K+ alone provided at most partial restoration, whereas addition of ATP and both ions restored activity to near that of undialyzed RRL (compare Fig. 4, C and B). To ensure that Mg2+ and K+ ions activated the bona fide δ-release reaction, rather than some δ-degrading protease, we analyzed both output particles (Fig. 4C, top two panels) and reaction supernatants (Fig. 4C, bottom panel). In this manner, we again failed to observe any proteolytic fragments of δ and reproducibly accounted for at least 70% of the input δ molecules. To determine whether these salt requirements reflect a specific role for Mg2+ and K+, or the need for a particular ionic strength, we tested several other mono- and divalent cations. Neither Zn2+ nor Ca2+ could substitute for Mg2+, consistent with a specific role for Mg2+ ions. Similarly, neither Na+ nor Cs+ could substitute for K+; however, Rb+ and NH4+, whose ionic radii are more similar to that of K+, could substitute for K+. These last results suggest that a monovalent cation with ionic radius near that of K+ also has a specific role in the δ-release activity.
Hsc70 and/or Hsp70 Contribute to δRelease in RRL
Several characteristics of the δ-release activity described in preceding sections led us to suspect that a molecular chaperone such as Hsc70 and/or Hsp70 is involved. Molecular chaperones commonly interact with hydrophobic regions of proteins to facilitate protein complex disassembly in an ATP-dependent fashion (25). The removal of δ from ISVP*s demonstrated above is essentially an ATP-dependent disassembly of a large protein complex, and several previous observations have suggested that hydrophobic regions are exposed within the μ1 fragments in ISVP*s (8–10). Moreover, optimal ATPase activity of Hsp70 family members has specific requirements for Mg2+ and K+ ions (26–28), consistent with the salt requirements for δ-release activity in RRL.
To test whether Hsc70 and/or Hsp70 are required for δ release, we subjected RRL to immunodepletion using a combination of anti-Hsc70 and anti-Hsp70 mAbs. This treatment consistently reduced the levels of each protein in the lysate (Fig. 5A). Although mock treatment resulted in a partial loss of δ-release activity, antibody treatment inhibited this activity almost completely, and the differences in activity levels between the paired mock- and antibody-treated lysates (n = 5) were statistically significant (p =0.00013) (Fig. 5B, top and bottom panels). To confirm that reduction of Hsc70 and/or Hsp70 levels was responsible for the observed differences, we complemented the antibody-depleted lysate with purified Hsc70. Addition of Hsc70 restored the δ-release activity of the lysate to near that of the mock-depleted sample, and the differences in activity levels between the paired antibody-treated lysates to which Hsc70 either was or was not added (n = 5) were also statistically significant (p = 0.00025) (Fig. 5B, top and middle panels). Once again in this experiment, to ensure that addition of purified Hsc70 was providing the bona fide δ-release activity, rather than some contaminating protease, we additionally analyzed the reaction supernatants for protein content (Fig. 5B, bottom panel). In this way, we failed to observe any proteolytic fragments of δ and reproducibly accounted for at least 66% of the input δ molecules. We therefore conclude that Hsc70 and perhaps also Hsp70 contribute to δ release from ISVP* particles in RRL.
Hsc70 Associates with Released δMolecules in RRL
Hsp70 family members, including Hsc70, exhibit slow on- and off-rates for substrate binding when bound to ADP, and their on-and off-rates are increased upon ATP binding (14, 29–31). As a result, reagents that deplete ATP, such as apyrase or glucose/hexokinase, typically lock Hsp70 family members onto their substrates (30, 32). We therefore reasoned that if Hsc70 is involved in δ release from ISVP*s, then it should be possible to trap more Hsc70 in complex with released δ upon ATP depletion from the reaction supernatant. To test this possibility, we performed δ-release reactions with [35S]Met/Cys-labeled ISVP*s, and after removal of particles, treated the reaction supernatants either with apyrase, to hydrolyze any remaining ATP, or with ARS, to maintain the ATP supply. We then immunoprecipitated Hsc70 from each of the reaction supernatants using anti-Hsc70 mAb and analyzed half of the sample by SDS-PAGE and immunoblotting to estimate the relative amount of Hsc70 that was pulled down in each case (Fig. 6A, top panel). The other half was analyzed by SDS-PAGE and phosphorimaging to quantify the amount of δ that had co-immunoprecipitated with Hsc70 under each condition. In this manner, we accounted for 5.3 ± 0.6 times more released δ molecules in association with Hsc70 in apyrase-treated supernatants (Fig. 6A, bottom panel, lane 2, and data not shown) than in ARS-treated supernatants (Fig. 6A, bottom panel, lane 1, and data not shown). Moreover, because less of the total Hsc70 was recovered in the immunoprecipitations of the apyrase-treated supernatants (Fig. 6A, top panel, compare lanes 2 and 6) than in those of the ARS-treated supernatants (Fig. 6A, top panel, compare lanes 1 and 5), the preceding ratio may be an underestimate of the increase in Hsc70-associated δ molecules upon apyrase treatment. Nevertheless, even with the confounding difference in Hsc70 recovery, the observed differences in amounts of Hsc70-associated δ molecules in the paired apyrase- or ARS-treated supernatants (n = 3) were statistically significant (p = 0.00080). Although only a small proportion of the total δ molecules recovered from the reaction supernatants was associated with Hsc70 under either condition (Fig. 6A, bottom panel, compare lanes 1 and 5 or lanes 2 and 6), this is not surprising because the results represent an estimate of the amount of Hsc70-associated δ at the moments in time at which these complexes were trapped by either apyrase addition or harvesting. Furthermore, these results are consistent with a model in which multiple rounds of ATP binding and hydrolysis by Hsc70 are required for removing all δ molecules from the population of ISVP*s. Similar amounts of Hsc70 and δ molecules were co-immunoprecipitated when ATP was depleted from the reaction supernatant by glucose/hexokinase treatment instead of apyrase (data not shown), suggesting that the increased stability of the Hsc70-δ complex was because of ATP depletion and not a nonspecific effect.
Hsc70 Associates with Released δMolecules in Cells
To correlate the RRL findings with events in cells during reovirus infection, we addressed whether Hsc70 associates with cytoplasmically released δ molecules. Mv1Lu cells were pretreated with cycloheximide to inhibit protein synthesis and were then either infected as in Fig. 1C or mock infected. After 2 h in the continued presence of cycloheximide, the cells were harvested, and particles were removed from the postnuclear supernatants by pelleting. The cleared supernatants were then subjected to immunoprecipitation using anti-virion serum antibodies. An additional control consisted of a mock immunoprecipitation of the infected cell sample. A fraction of the immunoprecipitated protein was subjected to SDS-PAGE and immunoblotting using the anti-virion serum antibodies. In addition to showing effective pulldown of δ molecules (Fig. 6B, lane 1), this blot confirmed that the pelleting step had cleared virus particles from the supernatant (data not shown). The remainder of the immunoprecipitated protein was subjected to SDS-PAGE and immunoblotting using anti-Hsc70 mAb. This blot showed that Hsc70 was effectively pulled down in complex with released δ molecules, but not in either of the two control samples (Fig. 6B, lanes 1–3). We confirmed that protein synthesis was blocked in this experiment by comparing the levels of viral nonstructural protein μNS in our samples (not detectable, Fig. 6B, lanes 1–3) to those present in cells that were infected in the absence of cyclo-heximide and otherwise prepared equivalently (Fig. 6B, lane 4). We are thus confident that the immunoprecipitated δ represents molecules released from incoming particles, and not newly synthesized protein. These results provide a further important link between δ-release activity in RRL and that during cell entry and thereby strongly suggest a role for Hsc70 in the final step in reovirus outer capsid disassembly in cells, that is, δ release yielding the cytoplasmic viral core.
DISCUSSION
The data presented above demonstrate a role for Hsc70 in release of the δ fragment of outer capsid protein μ1 from the reovirus ISVP* particle in RRL. The data also provide evidence for a role for Hsc70 in δ release during cell entry, representing a final step in outer capsid disassembly to generate the cytoplasmically localized, transcriptionally active reovirus core particle. Upon expression of the viral factory-matrix protein μNS from one of the viral transcripts, this core particle can be recruited into the first viral factory (33), where genome replication and assembly of new viral particles is thought to occur. Although a cytoplasmically localized core has been thought for many years by some to be the outcome of reovirus membrane penetration and outer capsid disassembly (34, 35), this study is the first to provide evidence for a mechanism of δ removal from the penetration-associated ISVP* particle.
This study has not excluded the possibility that in addition to Hsc70, Hsp70 might have a redundant function in δ release from the ISVP*. This would in fact be expected, because even divergent Hsp70 members exhibit many shared target specificities in vitro (36). However, the constitutively expressed protein, Hsc70, likely plays the more important role during reovirus infection, as δ release proceeds efficiently in the absence of ongoing protein synthesis (11; also this study).
It remains possible that other molecular chaperones may contribute to δ release as well, because these proteins commonly act together in multicomponent complexes (25). We have addressed the possibility that Hsp90 might also be involved in δ release; however, reagents known to inhibit Hsp90 function, such as radicicol and geldanamycin, showed no effect on δ release in RRL or in cells (data not shown). On the other hand, we have not succeeded in reconstituting the δ-release reaction using purified Hsc70 alone or in combination with its cochaperone Hsp40 (data not shown), consistent with the requirement for additional factors and/or an alternative cochaperone.
Based on the results in this study, we propose the following model for reovirus uncoating. First, ATP-bound Hsc70 molecules are targeted to the ISVP* by a putative cochaperone.7 Interaction between Hsc70 and ISVP*-associated δ molecules is then followed by ATP hydrolysis, inducing a conformational change in Hsc70 and subsequent dissociation of δ-Hsc70-ADP complexes from the particle. The next round of ATP binding frees Hsc70 from the released δ molecules and allows for another cycle of Hsc70 association with δ molecules remaining on the partially uncoated ISVP*.
The data do not specifically distinguish between a scenario in which δ molecules are removed from the ISVP* after its translocation into the cytoplasm (Fig. 7A) and one in which δ release accompanies, and may even be required for, particle translocation (Fig. 7B). The observation that δ release is temporally linked to cell entry (10, 11) makes for an intriguing hypothesis that δ release by Hsc70 might in fact accompany particle translocation across the membrane (Fig. 7B). This would be consistent with the known activities of molecular chaperones, which include a capacity for protein translocation across the membrane (13). However, the mechanism for translocation of a reovirus particle, which is 70- to 80-nm in diameter, would clearly need to be different from that for a single polypeptide chain, which involves threading of denatured protein through a narrow channel.
FIGURE 7. Models for roles of Hsc70 in cell entry by reovirus.
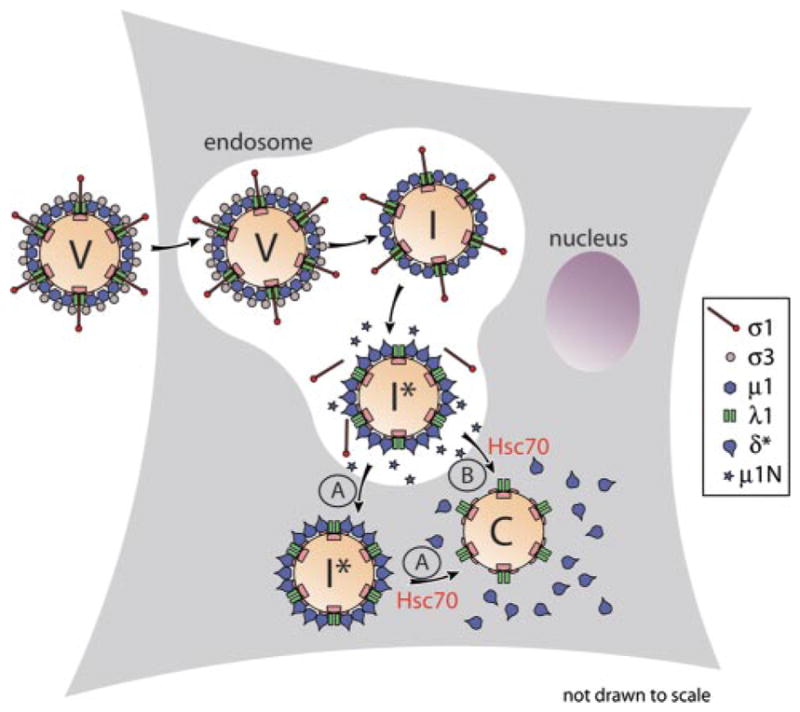
V, virion; I, ISVP; I *, ISVP*; and C, core; A, the ISVP* is translocated into the cytoplasm, and Hsc70 contributes to the subsequent removal of δ molecules from particles; B, Hsc70 contributes to removal of δ molecules from entering particles in concert with particle translocation into the cytoplasm.
Rotaviruses constitute a distinct genus within the same taxonomic family, Reoviridae, as the reoviruses with which this report is concerned. In the past several years, a series of studies by Arias, Lopez, and colleagues has identified a role or roles for Hsc70 in rotavirus entry (19–21). Hsc70 has been shown to interact with a region near the C terminus of VP5, a virion-associated proteolytic fragment of the rotavirus attachment/penetration protein VP4 (20, 21). This interaction appears to be important at a step following attachment (19–21) and perhaps even following membrane penetration (20). Inhibition of Hsc70-VP5 interaction by various means has invariably resulted in a marked decrease in rotavirus infectivity (19–21). Furthermore, premature interaction of Hsc70 with rotavirus particles has resulted in a comparable reduction in infectivity (19–21), which has been shown to be dependent on the ATPase activity of Hsc70 and probably a result of a conformational change in VP5 induced by this interaction (20). Whether Hsc70 directly contributes to rotavirus outer capsid disassembly, as shown for reovirus in this study, remains to be determined or recapitulated in vitro. Further comparisons of the specific role(s) of Hsc70 in the life cycles of these two related, though distinct, viruses should therefore be informative.
As outlined in the Introduction, it is reasonable to think that many different viruses have evolved to utilize molecular chaperones for steps in virus disassembly. Given the varied tasks that different viruses need to accomplish during uncoating and targeting to appropriate intracellular locations for gene expression and replication, it also seems reasonable to expect that different viruses may use different molecular chaperones to accomplish these tasks, or at least different cochaperones of Hsc70. Considering that reovirus is an instructive system for studies of cell entry by nonenveloped animal viruses in general, the insights provided by this study may extend well beyond the reovirus system.
Acknowledgments
We thank Elaine Freimont for technical assistance; Irene Kim and Lan Zhang for helpful comments on the manuscript; and John Parker and Tom Kirchhausen for other helpful discussions; and Branislav Ivanovic for drawings.
Footnotes
This work was supported in part by National Institutes of Health Grants F31 AI064142 (to M. A. A.) and R01 AI46440 (to M. L. N.).
The abbreviations used are: ISVP, infectious subvirion particle; RRL, rabbit reticulocyte lysate; ATPγS, adenosine 5′-O-(thiotriphosphate); mAb, monoclonal antibody; T1L, Type 1 Lang; T2J, Type 2 Jones; T3c9, Type 3 clone 9; ARS, ATP-regenerating system.
M. A. Agosto and M. L. Nibert, unpublished data.
T. Ivanovic and M. L. Nibert, unpublished data.
References
- 1.Bodkin DK, Nibert ML, Fields BN. J Virol. 1989;63:4676–4681. doi: 10.1128/jvi.63.11.4676-4681.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sturzenbecker LJ, Nibert M, Furlong D, Fields BN. J Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nibert ML, Fields BN. J Virol. 1992;66:6408–6418. doi: 10.1128/jvi.66.11.6408-6418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nibert ML, Odegard AL, Agosto MA, Chandran K, Schiff LA. J Mol Biol. 2005;345:461–474. doi: 10.1016/j.jmb.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Nibert ML, Schiff LA, Fields BN. J Virol. 1991;65:1960–1967. doi: 10.1128/jvi.65.4.1960-1967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borsa J, Copps TP, Sargent MD, Long DG, Chapman JD. J Virol. 1973;11:552–564. doi: 10.1128/jvi.11.4.552-564.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joklik WK. Virology. 1972;49:700–715. doi: 10.1016/0042-6822(72)90527-2. [DOI] [PubMed] [Google Scholar]
- 8.Agosto MA, Ivanovic T, Nibert ML. Proc Natl Acad Sci U S A. 2006;103:16496–16501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandran K, Farsetta DL, Nibert ML. J Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odegard AL, Chandran K, Zhang X, Parker JSL, Baker TS, Nibert ML. J Virol. 2004;78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandran K, Parker JSL, Ehrlich M, Kirchhausen T, Nibert ML. J Virol. 2003;77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartl FU. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 13.Young JC, Barral JM, Hartl FU. Trends Biochem Sci. 2003;28:541–547. doi: 10.1016/j.tibs.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Mayer MP, Bukau B. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glotzer JB, Saltik M, Chiocca S, Michou AI, Moseley P, Cotton M. Nature. 2000;407:207–211. doi: 10.1038/35025102. [DOI] [PubMed] [Google Scholar]
- 16.Brodsky JL, Pipas JM. J Virol. 1998;72:5329–5334. doi: 10.1128/jvi.72.7.5329-5334.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan CS, Pipas JM. Virology. 2001;287:1–8. doi: 10.1006/viro.2001.1038. [DOI] [PubMed] [Google Scholar]
- 18.Chromy LR, Oltman A, Estes PA, Garcea RL. J Virol. 2006;80:5086–5091. doi: 10.1128/JVI.80.10.5086-5091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrero CA, Bouyssounade D, Zarate S, Isa P, Lopez T, Espinosa R, Romero P, Mendez E, Lopez S, Arias CF. J Virol. 2002;76:4096–4102. doi: 10.1128/JVI.76.8.4096-4102.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Vargas J, Romero P, Lopez S, Arias CF. J Virol. 2006;80:3322–3331. doi: 10.1128/JVI.80.7.3322-3331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarate S, Cuadras MA, Espinosa R, Romero P, Juarez KO, Camacho-Nuez M, Arias CF, Lopez S. J Virol. 2003;77:7254–7260. doi: 10.1128/JVI.77.13.7254-7260.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurer C, Cimarelli A, Luban J. J Virol. 2002;76:4666–4670. doi: 10.1128/JVI.76.9.4666-4670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sagara J, Kawai A. Virology. 1992;190:845–848. doi: 10.1016/0042-6822(92)90923-d. [DOI] [PubMed] [Google Scholar]
- 24.Furlong DB, Nibert ML, Fields BN. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morimoto RI. Cell. 2002;110:281–284. doi: 10.1016/s0092-8674(02)00860-7. [DOI] [PubMed] [Google Scholar]
- 26.Flaherty KM, Wilbanks SM, DeLuca-Flaherty C, McKay DB. J Biol Chem. 1994;269:12899–12907. [PubMed] [Google Scholar]
- 27.O’Brien MC, McKay DB. J Biol Chem. 1995;270:2247–2250. doi: 10.1074/jbc.270.5.2247. [DOI] [PubMed] [Google Scholar]
- 28.Palleros DR, Reid KL, Shi L, Welch WJ, Fink AL. Nature. 1993;365:664–666. doi: 10.1038/365664a0. [DOI] [PubMed] [Google Scholar]
- 29.Greene LE, Zinner R, Naficy S, Eisenberg E. J Biol Chem. 1995;270:2967–2973. doi: 10.1074/jbc.270.7.2967. [DOI] [PubMed] [Google Scholar]
- 30.Prasad K, Heuser J, Eisenberg E, Greene L. J Biol Chem. 1994;269:6931–6939. [PubMed] [Google Scholar]
- 31.Schmid D, Baici A, Gehring H, Christen P. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 32.Cripe TP, Delos SE, Estes PA, Garcea RL. J Virol. 1995;69:7807–7813. doi: 10.1128/jvi.69.12.7807-7813.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broering TJ, Arnold MM, Miller CL, Hurt JL, Joyce PL, Nibert ML. J Virol. 2004;79:6194–6206. doi: 10.1128/JVI.79.10.6194-6206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borsa J, Sargent MD, Lievaart PA, Copps TP. Virology. 1981;111:191–200. doi: 10.1016/0042-6822(81)90664-4. [DOI] [PubMed] [Google Scholar]
- 35.Chandran K, Nibert ML. Trends Microbiol. 2003;11:374–382. doi: 10.1016/s0966-842x(03)00178-1. [DOI] [PubMed] [Google Scholar]
- 36.Fourie AM, Sambrook JF, Gething MJ. J Biol Chem. 1994;269:30470–30478. [PubMed] [Google Scholar]