

Abstract
The purpose of the study was to explore the feasibility of a protocol for the isolation and molecular characterization of single circulating tumor cells (CTCs) from cancer patients using a single-cell next generation sequencing (NGS) approach.
To reach this goal we used as a model an artificial sample obtained by spiking a breast cancer cell line (MDA-MB-231) into the blood of a healthy donor.
Tumor cells were enriched and enumerated by CellSearch® and subsequently isolated by DEPArray™ to obtain single or pooled pure samples to be submitted to the analysis of the mutational status of multiple genes involved in cancer.
Upon whole genome amplification, samples were analysed by NGS on the Ion Torrent PGM™ system (Life Technologies) using the Ion AmpliSeq™ Cancer Hotspot Panel v2 (Life Technologies), designed to investigate genomic “hot spot” regions of 50 oncogenes and tumor suppressor genes.
We successfully sequenced five single cells, a pool of 5 cells and DNA from a cellular pellet of the same cell line with a mean depth of the sequencing reaction ranging from 1581 to 3479 reads.
We found 27 sequence variants in 18 genes, 15 of which already reported in the COSMIC or dbSNP databases. We confirmed the presence of two somatic mutations, in the BRAF and TP53 gene, which had been already reported for this cells line, but also found new mutations and single nucleotide polymorphisms. Three variants were common to all the analysed samples, while 18 were present only in a single cell suggesting a high heterogeneity within the same cell line.
This paper presents an optimized workflow for the molecular characterization of multiple genes in single cells by NGS. The described pipeline can be easily transferred to the study of single CTCs from oncologic patients.
Keywords: Single cell, Circulating tumor cells, NGS, Somatic mutations, Microgenomics
1. Introduction
The molecular analysis of solid tumors is of great impact in the field of oncology research notwithstanding some unsolved challenging aspects in both sample selection and technical approaches. Solid tumors are in fact a heterogeneous mixture of tumor and normal cells differentially contributing to the final results of molecular biology tests on nucleic acids. As a result molecular signatures represent an average value masking the individual signals deriving from each single cancer cell [1]. The analysis of somatic mutations at the single-cell level can evidence whether one or more variants detected in the bulk tissue are simultaneously present in the same cell or if, on the contrary, derive from different cell clones.
Next-generation sequencing (NGS) has become a major tool for nucleic acid analysis and promises to disclose the basis of diseases, opening perspectives for increased personalized therapies and screening [2]. NGS has been so far applied to nucleic acids deriving from a high number of cells providing global information on the average state of the entire population of cells [1]. By combining whole genome amplification (WGA) with NGS, it is now possible to achieve individual cancer cell sequencing [3].
Single-cell sequencing has the potential to address some important questions in oncology such as understanding tumor heterogeneity, reconstructing cell lineages and defining the genome of rare tumor cell populations [3], [4], [5].
In addition, single-cell technologies will contribute to elucidate rare cell events such as scarce cancer cells in clinical samples and the dissemination of circulating tumor cells (CTCs) in the blood of cancer patients [1], [6]. Since CTCs reflect the heterogeneity of primary or metastatic tumors, their analysis at the single-cell level represent a real time liquid biopsy of the disease giving an insight in cancer progression, helping in the choice of target therapies or in the prediction of the efficacy of chemotherapeutic agents [1] and thus representing a key tool in the development of precision medicine.
Another aspect that can be considered as an application of single-cell sequencing is the analysis of cell lines derived from tumors that are commonly used as models in cancer research. This approach can be successfully adopted to identify those cell lines that more closely resemble the characteristics of the tumor under study [7]. Even if at a much lower extent than tissues, the cell line population usually considered as composed of identical cellular elements can hide a heterogeneity that could be disclosed only by single-cell analysis.
The MDA-MB-231 is an adherent human breast cancer cell line of epithelial origin, which is highly aggressive, invasive and poorly differentiated. MDA-MB-231 cells display a mesenchymal-like phenotype thus representing a good model for metastatic breast cancer [8].
In the present study we investigated the mutational status of multiple genes on an artificial sample obtained by spiking the MDA-MB-231 cell line into the blood of a healthy donor. The tumor cells were immunomagnetically isolated by CellSearch® (Veridex, USA), sorted by a dielectrophoretic system (DEPArray™, Silicon Biosystems, Italy) and then submitted to WGA and targeted deep sequencing. We used this model with the aim to demonstrate the feasibility of a workflow for the isolation and molecular characterization of single circulating tumor cells from cancer patients using a single-cell NGS approach.
2. Methods
2.1. Cell culture and spiking experiments
Experiments were conducted on the MDA-MB-231 human breast cancer cell line. MDA-MB-231 cells were obtained from Di.V.A.L. Toscana (Laboratory for Drug Validation and Antibody production, University of Florence) and maintained in DMEM (Lonza, Basel, Switzerland) + 10% FBS (PAA Laboratories, Austria) at 37 °C in 5% CO2. Cells were detached from plastic surface by Accutase (PAA Laboratories) to better preserve membrane antigens. MDA-MB-231 cells (n = 500) were spiked in blood samples from a healthy volunteer collected in CellSave® tubes (Veridex LLC, Raritan, NJ) and processed as described below. The remaining cells from the same culture were centrifuged and the resulting cell pellet was submitted to DNA extraction by QIAamp DNA Mini Kit (Qiagen, Germany).
2.2. Cell enrichment and enumeration
Cell enrichment and enumeration was performed by the CellSearch® System. 7.5 mL of whole blood were processed using the CellSearch™ Epithelial Cell kit (Veridex LLC), which selects EpCAM positive cells using ferrofluids particles coated with EpCAM antibody. Cells were stained with the nuclear dye 4′,6′-diamino-2-phenylindole (DAPI), anti-cytokeratin 8, 18 and 19-phycoerythrin (PE) labelled antibodies, and CD45 antibody labelled with allophycocyanin (APC). After enrichment, isolated and stained cells were resuspended in the MagNest Device (Veridex LLC), labelled cells were analyzed in the CellTracks® Analyzer II (Veridex LLC) and cells identified and enumerated according to the criteria specified by the manufacturer's instructions.
2.3. Single cell sorting
Following CellSearch® enrichment the sample was recovered from the Veridex cartridge and loaded into the DEPArray™ A300K chip (Silicon Biosystems) according to the manufacturer's instructions. The chip is a single-use, microfluidic cartridge containing an array of individually controllable electrodes, each one with embedded sensors. Chip scanning was performed by an automated fluorescence microscope to generate an image gallery, with cells selected according to their morphology (round shape, round nucleus within the cytoplasm) and staining pattern deriving from that of the CellSearch® system: DAPI positive, PE positive (CK8, CK18, CK19 positive cells), APC negative (CD45 negative cells).
After tumor cell identification, single cells or groups of few cells were recovered into 200 μl tubes.
2.4. Whole genome amplification
Single cells or pools of cells were submitted to WGA using the Ampli1™ WGA kit (Silicon Biosystems) according to the manufacturer's instructions, in order to obtain a sample suitable for sequencing analysis. The procedure is based on a ligation-mediated PCR following a site specific DNA digestion by MseI enzyme. The kit has no needs for precipitation steps avoiding DNA loss and provides a library of fragments of about 0.2-2 kb representing the entire genome. The kit uses a mixture of Taq polymerase with a proofreading enzyme, Pwo polymerase, that has been reported to have error rates more than 10 times lower than the error rate observed with Taq polymerase [9].
2.5. WGA quality control
The quality of the output product of the WGA reaction was assessed by the Ampli1™ QC kit (Silicon Biosystems) according to the manufacturer's instructions. The kit is a PCR-based assay which implies the amplification of two distinct regions of the human genome to produce two amplicons (A and B) of 373 and 167 bp respectively. PCR products A and B were analyzed by capillary electrophoresis on the Agilent 2100 Bionalyzer. The presence of both amplification products indicates a successful WGA and consequently the suitability of the sample for downstream analysis.
2.6. Next generation sequencing
Sequencing analysis was performed on the Ion Torrent PGM™ system (Life Technologies). We amplified the samples using the Ion AmpliSeq™ Cancer Hotspot Panel v2 (Life Technologies) designed to target 207 amplicons covering mutations from 50 oncogenes and tumor suppressor genes. DNA quantification was assessed using the Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). Ten nanograms of DNA were used to prepare barcoded libraries using the Ion AmpliSeq™ Library kit 2.0 and Ion Xpress™ barcode adapters (Life Technologies). The libraries were purified with Agentcourt AMPure XP (Beckman Coulter) and quantified with the Ion Library Quantitation Kit (Life Technologies) on the StepOne Plus system (Applied Biosystem).
Template preparation was performed with the Ion OneTouch™ 2 System and Ion One Touch ES. Finally sequencing was performed on PGM using Ion PGM™ Sequencing 200 kit v2 (Life Technologies) on the Ion 316 chip V1. The run was set in order to achieve a 1000X coverage for each sample.
Due to the WGA method, involving an enzymatic cleavage of DNA by the MseI restriction enzyme, we could not sequence 49/207 amplicons of the panel. Table 1 lists the genes of the AmpliSeq™ Cancer Hotspot Panel v2 which are respectively totally, partially or non-affected by the enzymatic digestion. 31 genes had no amplicon cleaved by the enzyme; 17 genes had some amplicons cut by MseI, and two genes (NPM1 and JAK2) were impossible to analyse because of the enzymatic cleavage.
Table 1.
Classification of genes of the AmpliSeq™ Cancer Hotspot Panel v2 on the basis of the presence of the MseI restriction site in one or more amplicons.
| 31 Unaffected genes | 17 Partially affected genes | 2 Totally affected genes |
|---|---|---|
| ABL1 | APC | NPM1 |
| AKT1 | ATM | JAK2 |
| ALK | CDH1 | |
| BRAF | EGFR | |
| CDKN2A | ERBB4 | |
| CSF1R | FBXW7 | |
| CTNNB1 | FGFR2 | |
| ERBB2 | KDR | |
| EZH2 | KIT | |
| FGFR1 | KRAS | |
| FGFR3 | MET | |
| FLT3 | PIK3CA | |
| GNA11 | PTEN | |
| GNAQ | RB1 | |
| GNAS | RET | |
| HNF1A | SMAD4 | |
| HRAS | VHL | |
| IDH1 | ||
| IDH2 | ||
| JAK3 | ||
| MLH1 | ||
| MPL | ||
| NOTCH1 | ||
| NRAS | ||
| PDGFRA | ||
| PTPN11 | ||
| SMARCB1 | ||
| SMO | ||
| SRC | ||
| STK11 | ||
| TP53 |
2.7. Data analysis
All samples were processed using the Torrent Suite Software 3.6 and variant calling was performed running the Torrent Variant Caller plugin version 3.6.56708. Moreover, samples were analysed using NextGENe® software 2.3.1 (SoftGenetics, LLC, State College, PA). We considered only variants with balanced numbers of forward and reverse reads.
Each variant was investigated about its potential pathogenetic role using available gene mutations and SNPs databases and prediction algorithms (COSMIC, dbSNP, 1000GENOME, SIFT, Polyphen).
3. Results
3.1. Design of the workflow for single cell recovery
We defined a workflow for targeted NGS sequencing composed of the following steps covering the procedure from the blood draw to the deep sequencing data analysis. Fig. 1 reports the entire pipeline.
Fig. 1.

Flow chart representing the single steps of the proposed procedure for single cell analysis by NGS.
3.1.1. Tumor cell enrichment and recovery
The preanalytical variables are taken into account by the use of a stabilizing blood collection tube which also provides cell fixation. Cells spiked into the blood of a healthy donor are enriched, fluorescently labelled and counted by the CellSearch® system. The platform provides the immunomagnetic capture of cells and their selection on the basis of positive staining for cytokeratins and negative signal for CD45. The left-over cartridge is handled in order to achieve the right volume for single cell sorting by the DEPArray™, which combines imaging technologies with the ability to manipulate and recover individual cells from a heterogeneous sample. The system performs a scanning of the sample by an automated fluorescence microscope generating an image gallery. Cells are selected according to their morphology (round shape, round nucleus within the cytoplasm) and fluorescence staining pattern deriving from that of the CellSearch® system. Selected cells are trapped in dielectrophoretic “cages” and moved singularly by manipulating the applied electric field [10].
The workflow has several critical points related to the complete CTC recovery from the Cellsearch® cartridge, to the subsequent volume reduction to fit the input volume of the DEPArray™ chip (14 μl) and to the volume reduction of single cell samples after DEPArray™ recovery.
In particular, after enrichment by the CellSearch® system we identified 130 cells (percentage of recovery = 26%). The cell suspension recovered from the CellSearch® was injected in the DEPArray chip: we identified 86 cells with a recovery ratio (defined as the ratio between the number of single cells identified by the DEParray™ after CellSearch® enrichment and the total number of cells counted by the CellSearch®) of 66% as already reported in a previous study [11].
We sorted 5 single MDA-MB-231 cells and a pool of 5 cells to be submitted to deep sequencing. Fig. 2A shows the images of 2 single cells.
Fig. 2.

(A) Cell staining pattern visualized by the DEPArray™: the cell show positive fluorescent signals for nuclear DAPI and cytokeratins (PE) and no signal for CD45 (APC). (B) Analysis of Ampli1™ QC PCR products for MDA-MB-231 single cell 1 and 2 by capillary electrophoresis. “A = PCR product A; “B” = PCR product B.
3.1.2. WGA and quality control
We amplified the whole genome of our samples in a 3-day procedure required for the Ampli1 WGA™ kit. Handling time was reduced, but two overnight incubations were required. After the WGA reaction we verified the quality of the output product before proceeding to sequencing by a PCR based assay using the Ampli1™ QC kit. The five single cells and the pool of 5 cells showed both the expected PCR products A and B by capillary electrophoresis. Fig. 2B shows the capillary electrophoresis of the two products of the Ampli1™ QC kit in two single cells.
3.1.3. Next generation sequencing on single and pooled cells
We successfully sequenced 5 single cells, a pool of 5 cells and DNA from a pellet of MDA-MB-231 cells with a mean depth of the sequencing reaction of 2966, 2862, 1581, 1884, 1854, 3479 and 2100 reads respectively.
On the whole 27 sequence variants were detected in 18 genes (Table 2). Fifteen variants had been already reported in the COSMIC or dbSNP databases, while 12 were described for the first time.
Table 2.
List of the sequence variants detected in single and pooled MDA-MB-231 cells. White boxes represent wild type sequences, while grey boxes indicate the presence of the mutation. In single cell samples, “homo” and “het” indicate the presence of the mutation in homozygosis or heterozygosis respectively. For all samples, the number of forward and reverse reads of the mutated base has been reported for each sequence variant. Sequence variants with possible deleterious effect on the protein phenotype are written in bold, while benign variants are reported in plain text. Somatic mutations specifically investigated by the Ion AmpliSeq™ Cancer Hotspot Panel are highlighted in light grey.
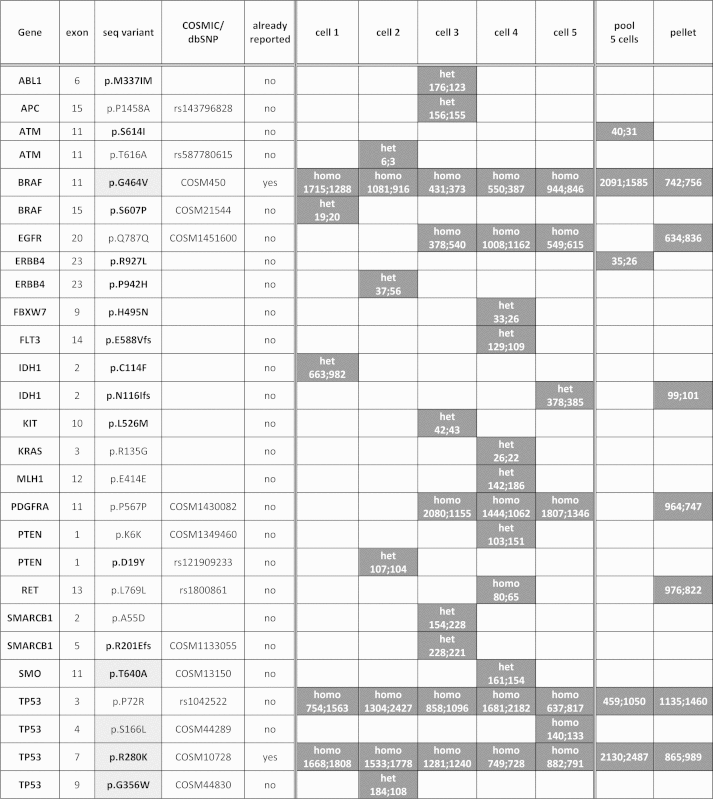 |
Only two somatic mutations, p.G464 V in exon 11 of the BRAF gene and p.R280K in exon 7 of the TP53 gene, had been already described in this cells line, while the other mutations and single nucleotide polymorphisms (SNPs) have never been reported before.
Two somatic mutations (p.G464 V in exon 11 of the BRAF gene and p.R280K in exon 7 of the TP53 gene) and a SNP (p.P72R in exon 3 of the TP53 gene), were detected in single and pooled cells as well as in the reference cellular pellet; three synonymous variants (p Q787Q in EGFR, p.P567P in PDGFRA and p.L769L in RET) and the deleterious mutation p.N116Ifs in IDH1 were detected in at least two samples (single or pooled cells or in the reference pellet), while 18 variants were present only in a single cell sample (Table 2). Two variants (p.S614I in ATM and p.R927L in ERBB4) were found only in the pool (Table 2). All the variants detected in the cellular pellet were present in at least one single cell sample (Table 2).
In addition to the identified variants, Table 2 reports for all samples the number of forward and reverse reads of the mutated base and the homozygous or heterozygous status of the variant detected in each single cell.
Sequence variants common to the 6 samples were detected with a high number of reads, while a few variants found just in a single sample had a low number of reads of the mutated nucleotide.
Table 3 reports the mean coverage of each amplicon per sample. In addition, for samples with a detected sequence variant, we reported the coverage of the specific position.
Table 3.
List of the sequence variants detected in single and pooled MDA-MB-231 cells with details about the coverage of the next generation sequencing reaction. For single wild type cells we reported the mean coverage of the sequenced amplicon; while for mutated single or pooled cells we reported the mean coverage of the amplicon as well as the coverage of the specific mutated nucleotide position. When more than a cell is considered for calculation, the number of cells is reported in brackets.
| Gene | Sequence variant | Single wild type cells | Single mutated cells |
Pool 5 cells |
||
|---|---|---|---|---|---|---|
| Mean coverage | Mean coverage | Single base coverage | Mean coverage | Single base coverage | ||
| ABL1 | p.M337IM | 9965 (n = 4) | 5493 | 4718 | 8377 | |
| APC | p.P1458A | 7483 (n = 4) | 5721 | 4784 | 11545 | |
| ATM | p.S614I | 336 (n = 5) | 1101 | 985 | ||
| ATM | p.T616A | 372 (n = 4) | 191 | 176 | 1101 | |
| BRAF | p.G464V | 1937 (n = 5) | 1712 (n = 5) | 4363 | 3688 | |
| BRAF | p.S607P | 549 (n = 4) | 808 | 739 | ||
| EGFR | p.Q787Q | 6050 (n = 2) | 1664 (n = 3) | 1423 (n = 3) | ||
| ERBB4 | p.R927L | 558 (n = 5) | 1356 | 1200 | ||
| ERBB4 | p.P942H | 538 (n = 4) | 640 | 559 | 1356 | |
| FBXW7 | p.H495N | 2788 (n = 4) | 600 | 545 | 4475 | |
| FLT3 | p.E588Vfs | 1932 (n = 4) | 1095 | 975 | 3502 | |
| IDH1 | p.C114F | 9337 (n = 4) | 4856 | 3306 | 15573 | |
| IDH1 | p.N116Ifs | 8012 (n = 4) | 10155 | 9003 | 15573 | |
| KIT | p.L526M | 1227 (n = 4) | 929 | 818 | 2405 | |
| KRAS | p.R135G | 597 (n = 4) | 416 | 350 | 1980 | |
| MLH1 | p.E414E | 7435 (n = 4) | 4831 | 4222 | 14523 | |
| PDGFRA | p.P567P | 2586 (n = 2) | 3513 (n = 3) | 2973(n = 3) | 3933 | |
| PTEN | p.K6K | 3823 (n = 4) | 3066 | 2837 | 7575 | |
| PTEN | p.D19Y | 3562 (n = 4) | 4109 | 3380 | 7575 | |
| RET | p.L769L | 52 (n = 4) | 167 | 150 | 45 | |
| SMARCB1 | p.A55DA | 9550 (n = 4) | 6130 | 5279 | 10136 | |
| SMARCB1 | p.R201Efs | 7089 (n = 4) | 4686 | 4055 | 6902 | |
| SMO | p.T640A | 3114 (n = 4) | 9737 | 8450 | 4748 | |
| TP53 | p.P72R | 3321 (n = 5) | 2668 (n = 5) | 1989 | 1514 | |
| TP53 | p.S166L | 14711 (n = 4) | 6567 | 5564 | 17535 | |
| TP53 | p.R280K | 2886 (n = 5) | 2508 (n = 5) | 5417 | 4670 | |
| TP53 | p.G356W | 3243 (n = 4) | 3998 | 3408 | 2897 | |
4. Discussion
We described a workflow for the molecular characterization of multiple genes in single cells by NGS. The study represents a proof-of-principle demonstrating the possibility to investigate the molecular status of about 50 genes related to cancer development in a single tumor cell. The described pipeline can be easily transferred to the study of single circulating tumor cells from oncologic patients. In fact, we demonstrated the feasibility of deep sequencing on single and pooled pure MDA-MB-231 cells which represent a model of metastatic breast cancer cells.
One of the purposes of the study was to bypass some of the limitations of the CellSearch®, one of the most widely used systems for CTC enrichment, in the molecular analysis of patients samples. In fact, while the instrument was extensively evaluated for its ability to accurately count CTCs in blood and for the consequent prognostic performances [12], [13], [14], it is unable to provide pure samples to be submitted to molecular characterization: contaminating leucocytes are still present in the enriched samples and the low number of isolated CTCs substantially limits subsequent molecular analyses [15], [16].
We already demonstrated the possibility to sequence by Sanger method pure CTC samples enriched by CellSearch® and sorted by DEPArray™ [11] and now we would like to extend the procedure to a higher throughput sequencing system like targeted NGS.
In all our samples we verified the presence of two expected somatic mutations detectable by the Ion AmpliSeq Cancer Hotspot panel on the basis of what is reported for MDA-MB-231 in the COSMIC database, demonstrating that NGS fulfils the analytical features of specificity and accuracy required for single cell analysis.
With the exception of the above mentioned mutations and the p.P72R SNP in the TP53 gene, we evidenced a high heterogeneity among different cells of the same cancer cell line: nineteen variants were in fact present in only one of the five analyzed single cells. This finding modifies the idea of cell lines as a pool of isogenic and homogeneous cells, in fact cells in a culture may be subjected to divergent clonal evolution resulting in a heterogeneous mixture of different genomes. Moreover, all the variants detected in the cellular pellet were confirmed in at least one single cell indicating that single cells represent the different clones of the original population.
Previous studies showed that amplification and sequencing errors are a concern for single cell mutation analysis [17], but the punctual and mean high coverage reached for our samples make us confident on the reliability of even minor variants found only in single samples. In fact achieving high physical coverage of the targeted sequences is crucial for calling mutations at the same regions across multiple single cells Navine, [3].
As a matter of fact, we cannot exclude technical errors deriving from the WGA procedure. As already pointed out by other authors, WGA could affect subsequent sequencing results by introducing a number of technical variables such as allelic dropout, inadequate coverage, false positive and negative results [3]. For this reason we chose to adopt a WGA method which has been shown to reliably amplify the entire cellular genome homogeneously [18].
It is generally accepted that, due to high error rates in NGS, mutations must be detected in multiple single cells (at least 3 cells) to make a mutant call [3]. Nonetheless with the aim to report the entire results of the proposed workflow and to highlight cell heterogeneity, we decided to report all the variants detected in each single cell provided that the number of forward and reverse reads for the mutated base was balanced.
The feasibility of high-resolution single cell analysis is very important when applied to the study of CTCs which are extremely rare in the bloodstream and, due to the challenge in the development of isolation procedures, can be often recovered in low numbers from a single blood draw.
Single cell analysis may assume even higher relevance when dealing with samples from cancer patients in whom such methods could help elucidating tumor heterogeneity in a variety of specimens, such as primary tissues, metastases and circulating tumor cells. CTCs in particular are rare in the circulation and require highly sensitive and specific methods for their isolation and high throughput procedures for nucleic acid characterization in order to highlight their metastatic potential and their usefulness as dynamic markers of tumor development.
Our study is an example of optimization of single-cell analysis procedures which are still in development and hold promise to establish robust single-cell diagnostics which will be crucial for the realization of precision medicine implying novel therapeutic approaches [6].
Conflict of interest
All the authors declare to have no conflict of interest.
Acknowledgement
This work has received financial support from Ente Cassa di Risparmio di Firenze (Firenze, Italy) (2011.1035) and Regione Toscana (project “CYTOPEM” POR CRO FSE 2007-2013).
References
- 1.Navin N., Hicks J. Future medical applications of single-cell sequencing in cancer. Genome Med. 2011;3:31. doi: 10.1186/gm247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richards C. How next-generation sequencing came to be. Drug Target Rev. 2015;2:36–37. [Google Scholar]
- 3.Navin N.E. Cancer genomics: one cell at a time. Genome Biol. 2014;15:452. doi: 10.1186/s13059-014-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shapiro E., Biezuner T., Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y., Waters J., Leung M.L., Unruh A., Roh W., Shi X., Chen K., Scheet P., Vattathil S., Liang H., Multani A., Zhang H., Zhao R., Michor F., Meric-Bernstam F., Navin N.E. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speicher M.R. Single-cell analysis: toward the clinic. Genome Med. 2013;5:74. doi: 10.1186/gm478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domcke S., Sinha R., Levine D.A., Sander C., Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin K.B. The Mesenchymal-Like Phenotype of the MDA-MB-231Cell Line. In: Gunduz M. Prof., editor. Breast Cancer—Focusing Tumor Microenvironment, Stem cells and Metastasis. InTech.; 2011. pp. 385–402. [Google Scholar]
- 9.McInerney P., Adams P., Hadi M.Z. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol. Biol. Int. 2014;2014:287430. doi: 10.1155/2014/287430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs A.B., Romani A., Freida D., Medoro G., Abonnenc M., Altomare L., Chartier I., Guergour D., Villiers C., Marche P.N., Tartagni M., Guerrieri R., Chatelain F., Manaresi N. Electronic sorting and recovery of single live cells from microlitre sized samples. Lab. Chip. 2006;6:121–126. doi: 10.1039/b505884h. [DOI] [PubMed] [Google Scholar]
- 11.Pestrin M., Salvianti F., Galardi F., De Luca F., Turner N., Malorni L., Pazzagli M., Di Leo A., Pinzani P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015;9:749–757. doi: 10.1016/j.molonc.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristofanilli M., Hayes D.F., Budd G.T., Ellis M.J., Stopeck A., Reuben J.M., Doyle G.V., Matera J., Allard W.J., Miller M.C., Fritsche H.A., Hortobagyi G.N., Terstappen L.W. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J. Clin. Oncol. 2005;23:1420–1430. doi: 10.1200/JCO.2005.08.140. [DOI] [PubMed] [Google Scholar]
- 13.Cohen S.J., Punt C.J., Iannotti N., Saidman B.H., Sabbath K.D., Gabrail N.Y., Picus J., Morse M., Mitchell E., Miller M.C., Doyle G.V., Tissing H., Terstappen L.W., Meropol N.J. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008;26:3213–3221. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]
- 14.de Bono J.S., Scher H.I., Montgomery R.B., Parker C., Miller M.C., Tissing H., Doyle G.V., Terstappen L.W., Pienta K.J., Raghavan D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008;14:6302–6309. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 15.Sieuwerts A.M., Kraan J., Bolt-de Vries J., van der Spoel P., Mostert B., Martens J.W., Gratama J.W., Sleijfer S., Foekens J.A. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real-time PCR Breast. Cancer Res. Treat. 2009;118:455–468. doi: 10.1007/s10549-008-0290-0. [DOI] [PubMed] [Google Scholar]
- 16.Sieuwerts A.M., Mostert B., Bolt-de Vries J., Peeters D., de Jongh F.E., Stouthard J.M., Dirix L.Y., van Dam P.A., Van Galen A., de Weerd V., Kraan J., van der Spoel P., Ramírez-Moreno R., van Deurzen C.H., Smid M., Yu J.X., Jiang J., Wang Y., Gratama J.W., Sleijfer S., Foekens J.A., Martens J.W. mRNA and microRNA expression profiles in circulating tumor cells and primary tumors of metastatic breast cancer patients. Clin. Cancer Res. 2011;17:3600–3618. doi: 10.1158/1078-0432.CCR-11-0255. [DOI] [PubMed] [Google Scholar]
- 17.Heitzer E., Auer M., Gasch C., Pichler M., Ulz P., Hoffmann E.M., Lax S., Waldispuehl-Geigl J., Mauermann O., Lackner C., Höfler G., Eisner F., Sill H., Samonigg H., Pantel K., Riethdorf S., Bauernhofer T., Geigl J.B., Speicher M.R. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73:2965–2975. doi: 10.1158/0008-5472.CAN-12-4140. [DOI] [PubMed] [Google Scholar]
- 18.Klein C.A., Schmidt-Kittler O., Schardt J.A., Pantel K., Speicher M.R., Riethmüller G. Comparative genomic hybridization, loss of heterozygosity and DNA sequence analysis of single cells. Proc. Natl. Acad. Sci. U. S. A. 1999;96(8):4494–4499. doi: 10.1073/pnas.96.8.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]