

Abstract
Worldwide growth and performance-enhancing substances are used in cattle husbandry to increase productivity. In certain countries however e.g., in the EU, these practices are forbidden to prevent the consumers from potential health risks of substance residues in food. To maximize economic profit, ‘black sheep‘ among farmers might circumvent the detection methods used in routine controls, which highlights the need for an innovative and reliable detection method. Transcriptomics is a promising new approach in the discovery of veterinary medicine biomarkers and also a missing puzzle piece, as up to date, metabolomics and proteomics are paramount. Due to increased stability and easy sampling, circulating extracellular small RNAs (smexRNAs) in bovine plasma were small RNA-sequenced and their potential to serve as biomarker candidates was evaluated using multivariate data analysis tools.
After running the data evaluation pipeline, the proportion of miRNAs (microRNAs) and piRNAs (PIWI-interacting small non-coding RNAs) on the total sequenced reads was calculated. Additionally, top 10 signatures were compared which revealed that the readcount data sets were highly affected by the most abundant miRNA and piRNA profiles. To evaluate the discriminative power of multivariate data analyses to identify animals after veterinary drug application on the basis of smexRNAs, OPLS-DA was performed. In summary, the quality of miRNA models using all mapped reads for both treatment groups (animals treated with steroid hormones or the β-agonist clenbuterol) is predominant to those generated with combined data sets or piRNAs alone. Using multivariate projection methodologies like OPLS-DA have proven the best potential to generate discriminative miRNA models, supported by small RNA-Seq data. Based on the presented comparative OPLS-DA, miRNAs are the favorable smexRNA biomarker candidates in the research field of veterinary drug abuse.
Abbreviations: CLEN, treated group with clenbuterol-hydrochloride; CON, control group; DA, discriminant analysis; EU, European Union; exRNA, extracellular RNA; miRNA, microRNA; OPLS, orthogonal partial least-squares; PCA, principal component analysis; P + EB, treated group with steroid hormone implant: progesterone plus estradiol benzoate; piRNA, PIWI-interacting small non-coding RNA; PLS, partial least-squares projection; rpm, reads per million; small RNA-Seq, small RNA-Sequencing; smexRNA, circulating extracellular small RNA
Keywords: Biomarker signatures, Circulating small RNAs, Multivariate data analysis, Small RNA-Sequencing, Transcriptomics, Veterinary diagnostics
1. Introduction
Monitoring of chemical contaminations, species fraud and product mislabelings in food is a complex task for control laboratories. Recent pan-European food safety affairs, for example the horsemeat scandal in 2013, underline the need for sophisticated and reliable analytical methods as well as sufficiently frequent routine investigations in food producing animals [1]. For official laboratories, the conventional methods for screening for forbidden veterinary drug compounds are RIA (radio immuno assay) and ELISA (enzyme-linked immunosorbent assay), and for the confirmation, it is mass spectrometry (MS) combined with gas (GC–MS) or liquid chromatography (LC–MS) [2]. These verifying approaches persue the direct tracking of targeted chemical compounds and/or their metabolites in various food, feed or biological samples. As corresponding analytical protocols are based on the direct detection of the target substance in a sample matrix, the chemical and physical properties of this substance must be known in advance. For example, to test the compliance with regulations in antibiotics surveillance, a maximum threshold of antibiotic residues may not be exceeded in the detection window. However, in the case of an illegal abuse, where unknown substances or undefined drug cocktails with low-dose single compounds were administered, chromatographic systems are limited. This is especially the case when the substance itself has already been metabolized (but the physiological effect is still existent), or due to signal to noise ratio in MS and the unknown mass of the applied drug(s). Next to chromatographical methods or immunoassays, new and innovative techniques have emerged in veterinary medicine in the last years. Since recently, veterinary drug abuse can be detected by finding endogenous molecular biomarkers on the transcriptomic, proteomic or metabolomic level that indirectly indicate exogenous physiological modifications [3]. With the objective of controlling veterinary drug abuse, metabolomics approaches have so far shown to be effective in detecting growth-promotor abuse in bovines [4], [5], [6], and in racehorses [7], [8]. Rapid technological advancements in these “-omics” sciences allow now a comprehensive high-throughput screening for differentially expressed biomarkers. Thus, according to a physiological condition, disease status, or drug application, the biomarker signature is capable of revealing specific biological traits or a measurable change in the organism [9]. Seen from the genetic point of view, the transcription of genes is a fast and highly dynamic process that adapts to environmental stimuli, such as medication, making the transcriptome ideally suitable for the discovery of new biomarkers. The transcriptome covers inter alia a RNA class called microRNAs (miRNA). These small, non-protein coding molecules with a length of typically 18 to 25 nucleotides act as modulators of mRNA targets on the post-transcriptional level. By suppressing the mRNA translation or promoting mRNA destabilization, miRNAs play key roles in regulating gene expression in a multitude of healthy and pathologic biological processes [10]. The successful identification of miRNA biomarkers is already evident in clinical diagnostics, such as early disease detection, progression monitoring and prognosis [11]. In veterinary drug analysis, it was also possible to establish miRNA supported biomarkers in bovine liver to detect anabolic steroid treatment [12]. In the year 2008, miRNAs were also detected as free, extracellular RNA (exRNA) in the bloodstream [13] and the potential usability of circulating nucleic acids as biomarkers was promptly recognized and investigated. Since then, circulating extracellular small RNAs (smexRNA) have been detected in other human body fluids, e.g., milk, saliva, tears, cerebrospinal fluid, urine etc. [14]. Among these smexRNAs are also a recently very emerging class of transcriptional molecules, the PIWI-interacting small non-coding RNAs (piRNAs). They are slightly longer than miRNAs (25 to 32 nucleotides), but also show post-transcriptional regulatory functions. Initially detected in the germ line of drosophila, piRNAs are involved in RNA silencing and therefore in gene expression regulation (as reviewed in [15]). In biomarker development, it was already verified that circulating piRNAs own the potential to serve as human biomarkers for several cancer types, for example gastric cancer [16]. Focusing animal sciences, proteomics and metabolomics are now gradually finding their way into veterinary medicine and food safety analyses, but transcriptomics and especially the analysis of small RNAs and/or smexRNAs have not yet fully arrived.
Worldwide growth and performance-enhancing substances are used in cattle husbandry to increase productivity. Livestock farming strives to promote faster weight gains, increased feed conversion efficiencies and heavier carcasses to maximize economic profit. However, the use of anabolic agents is prohibited in certain countries, including the European Union (EU) since the EU Council Directive 88/146/EC188 entered into force in 1988. From that year on, all growth promoting agents including steroid hormones and β-adrenergic agonists have been prohibited from animal breeding practices across European markets. This ban was mainly due to precautionary food safety reasons to prevent consumers from possible health risks caused by residue carryover [17]. Also the import of products derived from hormone-treated cattle is legally forbidden in the EU. Due to financial benefits, an abuse by application of illicit substances is still frequently suspected in meat production [18]. To circumvent supervisory authorities and positive test results, alternative compounds as well as application scenarios have emerged. Applying transcriptomics in the field of food safety constitutes a new innovative screening strategy for a reliable and effective control method to maintain legislation. First studies demonstrated that the monitoring of mRNA expression ratios has already proven to be a useful tool for biomarker development to trace growth-promotor abuse [19], [20], [21] however, far less is known about the applicability of smexRNAs in this context.
If the aim is to measure small non-coding RNAs in a high-throughput approach, small RNA-Sequencing (small RNA-Seq) is the strategy of choice. This allows the holistic and parallel sequencing-by-synthesis analysis of the whole transcriptome of multiplexed samples. To study the influence of anabolic substances on the gene expression profiles at the small RNA level in meat-producing livestock, an animal trial was conducted to simulate the real environment during a potential drug abuse situation. In general, ultrahigh-throughput studies result in immensely huge data output that is highly multivariate (k variables ≫ n observations). To get the most value out of complex small RNA-Seq data and reveal knowledge that is hiding behind, we implemented multivariate projection methodologies to circumvent this bottleneck in biomarker development. The aim is to find a valid and stable biomarker signature, which explicitly leads back to the treatment. Thereby, treated or diseased subjects will be compared with untreated control samples. To select the most significant single biomarkers and combine this pattern to a biomarker signature, the applicability of multivariate projection methodologies in omics studies is beneficial and productive [22]. Most applied multivariate projection methodologies are principal-component analysis (PCA), hierarchical clustering (HCA), and partial least-squares (PLS) projections to latent structures [22]. Recently, orthogonal partial least squares (OPLS) demonstrated to be a useful discriminant analysis (DA) tool for complex data structures [23], [24]. The goal of OPLS-DA is to establish a model that is able to distinguish the classes of observations (non-treated from treated), to visualize large-volume data sets and to highlight meaningful interpretation possibilities.
The OPLS algorithm [25] is an improved and complexity-reduced interpretation of PLS regression models with an integrated orthogonal correction filter [26], allowing easier interpretation and augmenting classification performance [27]. Therefore, systemic variation from the input data set X, which is not correlating with the response set Y, is eliminated [25]. High-quality OPLS-DA models have the ability to separate the modelled variation in X into two parts, one that is correlated to Y and therefore predictive, and another that is orthogonal to Y. Thus, the correlated and therefore predictive variation in X is displayed by the predictive components and represents the variation between classes (non-treated animals and treatment groups). The variation in X that is orthogonal to Y is modeled by the orthogonal components and reflects the variation within classes [28].
Not only miRNAs but also piRNAs were investigated in this study to evaluate the potential of both smexRNA biomarker candidates. The decisive advantages of smexRNAs in bio-fluids compared to RNAs sampled from tissue are easy accessibility and an increased stability in the body and after sample collection [29]. We examine and discuss the potential of smexRNAs as novel source of biomarkers in veterinary diagnostics, to battle against illegal application of growth and performance enhancing substances to bovines.
2. Material and methods
2.1. Design of the animal study
In this study, 21 male Friesian Holstein veal calves (bos taurus) were randomly divided into three groups of 7 animals each (n = 7). All animals had a similar age (161 ± 15 days) and an average body weight of 151.4 ± 19.2 kg at the beginning of the trial. One group remained completely untreated and served as control group (CON). The second group was treated with Component E–C (IVY Animal Health, USA), a hormonal implant that consisted of a combination of 100 mg of progesterone plus 10 mg of estradiol benzoate (steroid hormone group, P + EB). One implant per animal was deposited between the skin and the cartilage on the backside of the middle third of the pinna of the ear. The third group received an oral dose of clenbuterol-hydrochloride (clenbuterol group, CLEN) (10 μg/kg body weight) (Boehringer Ingelheim, Germany) in daily intervals for 36 days. This animal study was approved by the ethical committee of the German Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (permit number 84-02.04.2012.A040). The animals were housed and fed according to good animal attendance practice and all efforts were made to prevent suffering.
2.2. Plasma sampling
To generate plasma, peripheral whole blood was collected from the jugular vein using 9 ml K3E K3EDTA-Vacuette tubes (Greiner bio-one, Germany) and single use needles (Greiner bio-one, Germany) with a subsequent separation of cellular components by centrifugation for 15 min at 3500 × rcf at room temperature with transportable centrifuges (EBA20, Hettich, Germany). Plasma was stored at −80 °C until RNA extraction. Samples were taken at d + 17 after the initial treatments.
2.3. Total RNA isolation
ExRNAs from plasma were isolated by an optimized method that enabled small RNA-Seq as previously implemented by our group [30]. RNA eluates were stored at −80 °C until further usage.
2.4. Small RNA Sequencing, Data Evaluation, Mapping and Annotation
The sample pre-processing pipeline, analytical small RNA-Seq steps on a HiSeq sequencing platform (Illumina, USA) and the bioinformatics steps to generate annotated readcount tables of 21 bovine plasma samples were realized as described and discussed by our group [30]. As inter-sample normalization strategy, total readcounts were adjusted to library sizes in reads per million (rpm) to correct differences in library sizes [31].
2.5. Univariate and multivariate data analysis
SigmaPlot 11.0 (Systat Software Inc., USA) was used for statistical data analysis and SIMCA 13.0.3.0 software (Umetrics AB, Sweden) for running the multivariate data analysis. For model generation, library size-normalized data sets were first logarithmically transformed and then pareto-scaled [24]. Different miRNA and piRNA models, depending on the input data quantities, were built. These were either readcount tables with all annotated reads (all reads) or with more than 50 rpm at an average (>50 readcounts). 50 rpm was set as a noise cut-off that is commonly used in small RNA-Seq data analysis. Discriminative model results were shown in scores scatter plots, displaying the CON group in blue, the P + EB group in red and the CLEN group in green. The quality of OPLS-DA models was controlled by evaluating R2 and Q2 values. The R2(cum) value represents the cumulative percentage of the modelled variation in Y, using the X model. Therefore, the R2(cum) value is the measure of fit and describes how well the model fits the X data. A large value close to 1 is a requisite condition for good models. The Q2(cum) value is the cumulative percentage of the variation in Y that can be predicted by the model according to cross validation using the X model. Q2(cum) is the measure of predictability and explains how well the generated model predicts new data. A large value (>0.5) indicates good predictability [28].
3. Results and discussion
3.1. Abundance of smexRNAs
After passing the sequencing quality checkpoints and successful alignment, miRNA and piRNA data from all sequenced 21 animals were library-size normalized and compiled in readcount tables. Analysis of the proportion of miRNAs on the total sequenced reads resulted in a content of 5.7% ± 2.4 (SD) (median = 6.6) in the CON group, 3.5% ± 1.7 (SD) (median = 3.6) in the P ± EB group and 6.6% ± 2.7 (SD) (median = 7.9) in the CLEN group (Fig. 1). There is a statistically significant difference between the CON group and the steroid hormone treated group (p = 0.047) and also between the two treatment groups (p = 0.042). Concerning piRNAs, CON contained 0.7% ± 0.4 (SD) (median = 0.6), P ± EB 1.0% ± 0.7 (SD) (median = 0.8) and CLEN 1.1% ± 1.0 (SD) (median = 0.6) without statistically significant differences (Fig. 1).
Fig. 1.

Abundance of circulating miRNAs and piRNAs. Box plots illustrate the circulating miRNA and piRNA proportions in plasma of untreated control animals (CON), steroid hormone- (P + EB) and clenbuterol (CLEN)-treated animals (n = 7 each). Steroid hormones decreased the miRNA quantity (p = 0.047) and clenbuterol application resulted in increased miRNA concentrations (p = 0.042).
As the magnitude of piRNAs was comparable to previously published data of nine healthy bovines [32], the proportion of piRNAs seemed not to alter even under the influence of anabolic stimulants. It was a recognizable effect that the steroid treatment led to a significant decrease of miRNA quantity compared to the CON group. In the CLEN-treated animals, gene expression changes towards an up-regulation of miRNAs were noticed compared to the P + EB treated individuals. Therefore, the different kinds of treatment substances seemed to have an opposite impact on miRNA translation.
3.2. Top 10 abundance lists
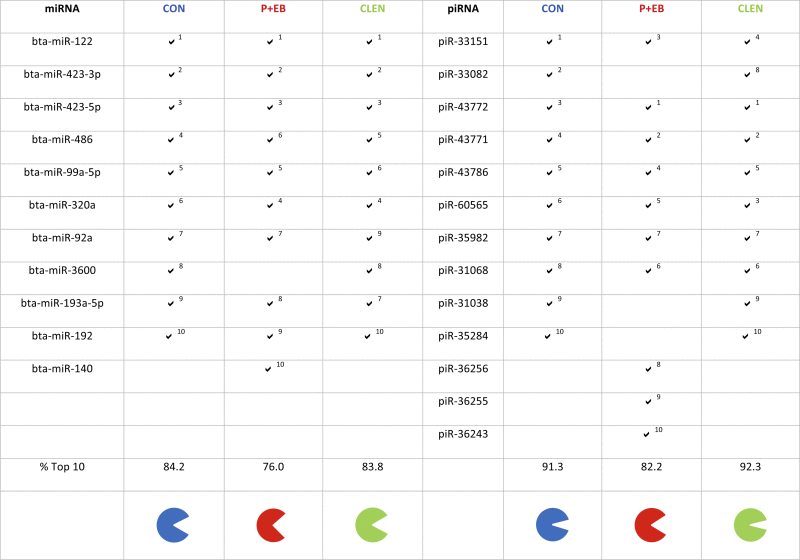
Rpm-normalized miRNA reads were sorted according to their decreasing readcount numbers to generate top 10 abundance lists. This revealed that the largest proportion of the data sets was reflected by the top 10 ranks: 84.2% in the CON group, 76.0% in the P + EB group and 83.8% in the CLEN group (Table 1). By comparing the most abundant CON miRNAs with the P + EB and CLEN treatment group, it could be stated that the composition is nearly the same (CON vs. P + EB) or exactly matching (CON vs. CLEN). The top 10 signature of the CON group confirmed miR-3600, which was substituted with miR-140 in the P + EB group. To evaluate piRNAs that were high ranking in terms of abundance, the same data organization was conducted. The top 10 piRNA list accounted for 91.3% of the total reads data set in the CON group, for 82.2% in the P + EB group and for 92.3% in the CLEN group. The CON and CLEN piRNA list exhibited the same pattern and the P + EB group varied in three piRNAs: piR-31038, piR-35284 and piR-33082. A statistically significant difference between the groups could not be detected. Therefore, in summary for both treatment groups, the readcount data sets were highly affected by the most abundant miRNA and piRNA profiles. Moreover, top 10 expressed data did not show significant deviations from the CON group, indicating that the smexRNA profiles of treated animals were not subject of fluctuations as great as assumed and the major components were constantly expressed.
Table 1.
Comparison of the top 10 expressed miRNAs and piRNAs in the three analyzed groups: (CON) control group, (P + EB) steroid hormone-treated group, (CLEN) clenbuterol-treated group. Checkmarks signify presence of matching small RNAs and superscript numbers give ranking information. Pie charts depict the percentage of the top 10 on the total annotated miRNAs and piRNAs, respectively.
 |
3.3. Differential expression
To evaluate the discriminative power of multivariate data analyses to identify animals after veterinary drug application on the basis of smexRNAs, OPLS-DA was performed after data pre-processing. miRNA and piRNA scores scatter plots were analyzed regarding between class variation (horizontal direction) and within class variation (vertical direction) depending on the read input of either all aligned reads (all reads) or size-filtered data sets with reads that had averagely more than 50 rpm (>50 readcounts) (Fig. 2, Fig. 3, Fig. 4), , ). Fig. 5 gives an overview over model quality parameters of all examined discriminative analyses.
Fig. 2.

Combined miRNA and piRNA data set. OPLS-DA of sequenced plasma samples using full [C and D] and readcount-filtered datasets [A and B]. [A and C] represent scores scatter plots discriminating control animals (CON, blue) from steroid hormone-treated animals (P + EB, red). [B] and [D] display samples from the CON and the clenbuterol-treated population (CLEN, green). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.

MiRNA data set. OPLS-DA of sequenced plasma samples using full [C and D] and readcount-filtered datasets [A and B]. [A and C] represent scores scatter plots discriminating control animals (CON, blue) from steroid hormone-treated animals (P + EB, red). [B] and [D] display samples from the CON and the clenbuterol-treated population (CLEN, green).
Fig. 4.

PiRNA data set. OPLS-DA of sequenced plasma samples using full [C and D] and readcount-filtered datasets [A and B]. [A and C] represent scores scatter plots discriminating control animals (CON, blue) from steroid hormone-treated animals (P + EB, red). [B] and [D] display samples from the CON and the clenbuterol-treated population (CLEN, green).
Fig. 5.

Model quality overview. The R2(cum) value (dark colored bars) reflects the goodness of fit and the Q2(cum) value (light colored bars) the goodness of prediction. Quality parameters were evaluated for the data set with all reads and with reads over averagely more than 50 readcounts (>50 readcounts). Red colored bars display the values from the P + EB study and green colored bars the values from the CLEN study.
First, the discriminative power of combined data sets including miRNAs and piRNAs was examined. As shown in Fig. 2[A] and [B], the separation between the CON animals and the treated groups, based on miRNA observations >50 readcounts, is imperfect. Although moderate goodness of fit and prediction could be attested for the CLEN study model (CLEN: R2(cum) = 0.752, Q2(cum) = 0.458), it was not feasible for the P + EB study (P + EB: R2(cum) = 0.319, Q2(cum) = 0.001). Better discriminative and quality results were accomplished with models that included all reads (Fig. 2[C] and [D]). DA could not manage to perfectly separate the CLEN-treated animals from the CON group (CLEN: R2(cum) = 0.794; Q2(cum) = 0.355), while acceptance of all reads in the discriminative analysis led to a model that allowed separation of P + EB classes with high quality (P + EB: R2(cum) = 924, Q2(cum) = 0.657).
In a second step, to examine a potentially improved discriminative power of data sets with uniquely miRNAs or piRNAs respectively, equivalent OPLS-DA models were generated. As shown in Fig. 3[A] and [B], the separation between the CON animals and the treated groups, based on miRNA observations >50 readcounts, is not more specific. The model quality parameters attest better fit and prediction for the CLEN study (CLEN: R2(cum) = 0.607, Q2(cum) = 0.377; P + EB: R2(cum) = 0.210, Q2(cum) = 0.035) but still, best discriminative and quality results were accomplished with models that included all available reads. For both multivariate data analyses studies, the miRNA models were able to distinguish between classes (Fig. 3[C] and [D]), which was reflected by good fit (CLEN: R2(cum) = 0.927; P + EB: R2(cum) = 0.893) as well as good predictability (CLEN: Q2(cum) = 0.782; P + EB: Q2(cum) = 0.706). Generally, large R2 and Q2 values at the level of 0.5 or above are necessary for high quality OPLS-DA models. Therefore, the quality parameters of the miRNA OPLS-DA models for the P + EB and the CLEN-treated animals indicated that the models fitted the data very well and that new variables could be predicted. Using all reads, the scores scatter plots illustrated a grouping of the CON and the treated animals, highlighting that multivariate data analysis tools were clearly capable to reveal treatment-dependent differences at the miRNA level. Moreover, fusion of data sets did not deliver better fitting and predicting results compared to miRNAs only, neither for the different treatment groups nor for the two compared data inputs.
Besides miRNAs, OPLS-DA models were generated and evaluated for piRNA data only (Fig. 4). Here again, a separation of the treatment groups was not feasible for the data set >50 readcounts. According to that effect, R2 and Q2 could not meet quality standards (CLEN: R2(cum) = 0.221, Q2(cum) = 0.055; P + EB: R2(cum) = 0.536, Q2(cum) = 0.096). Compared to the miRNA models, the piRNA models with all reads could not cluster the treated animals. For the P + EB group, a better OPLS-DA model could be generated than for the CLEN group, also regarding quality (CLEN: R2(cum) = 0.461, Q2(cum) = −0.346; P + EB: R2(cum) = 0.706, Q2(cum) = 0.47). The DA of piRNAs could not present acceptable models, as they could not explain the variation of the variables nor could they predict. Obviously, the miRNA abundance and thus the utilizable read numbers for statistical analyses exceed that of piRNAs (Fig. 1). Therefore, miRNA OPLS-DA could be based on increased data volumes supporting a better prediction ability and discrimination. Fusion of data sets delivered better fitting and predicting results compared to piRNAs only, when using all reads. For the CLEN study, the combined model also presented better fit and prediction for the >50 readcount model. For the miRNAs from the P + EB study, merging data sets resulted in slightly increased R2 values using all reads. Best fit and prediction in the CLEN study were achieved while using all miRNA reads (Fig. 3[D]). In summary, adding miRNAs to improve a piRNA based model resulted in better discrimination, fit and predictability, whereas miRNAs alone (all reads) provided best results for both treatment modalities. Referring to this trial, it can be concluded that DA is improving the more data (reads) are fed into OPLS-DA. It became apparent that OPLS-DA is best suited for full datasets of circulating miRNAs in the search for veterinary drug abuse biomarkers.
3.4. Comparison of analysis models
Taking all findings together, it can be stated that the main percentage of the miRNA and piRNA signature is composed of the top 10 candidates (Table 1). Furthermore, the miRNA and piRNA top 10 signature is almost the same, if the CON group was compared with the P + EB animals, or even identical (CON vs. CLEN). No statistically significant expression ratio could be determined. Therefore, the investigated smexRNAs were expressed more stable than assumed. It was though already published that the treatments were effective, as animals showed a significant weight gain (d0 to d + 34) and a potential gene expression biomarker signature was identified on the mRNA level in liver samples in the course of the same animal trial [21]. Nevertheless, overall variation in the expressed miRNA profiles is sufficient for the generation of good-quality OPLS-DA models, but this variation was not explained by the main components of the data set, but rather by the multiplicity of low-abundance miRNAs. Increasing the sequencing depth to exemplary 24-fold (one sample per flow cell lane) could thereby help to improve the detection of low-abundant smexRNAs, the sequencing of more reads and finally the manifestation of differences in low-abundance circulating small RNAs. As explained before, there is a discriminative effect lying in the data, however hiding behind the major expressed circulating miRNAs.
The quality of miRNA models (all reads) for both treatment groups is predominant to those generated with combined data sets or piRNAs alone. Therefore, the presented results highlighted that miRNAs were superior biomarker candidates to piRNAs regarding the annotated number of reads, model quality, data fit and predictive ability. Therefore, based on the presented comparative OPLS-DA data analyses, miRNAs are the favorable smexRNA biomarker candidates in the research field of veterinary drug abuse. As treated animals could be separated from untreated controls, this study was a first hint, that circulating miRNAs could be beneficial biomarker candidates for anabolic misuse in the future, if sequencing depth is chosen properly.
Although there were clear differences between the control and the treated animals revealed by small RNA-Seq (scores scatter plots in Fig. 2, Fig. 3), the verification and validation of a confident biomarker signature is technically very difficult for smexRNAs in bovine plasma. To experimentally manifest the quantitative expression of candidates in plasma via RT-qPCR, a detectable readcount number is prerequisite. As described before, those miRNAs that fulfilled this precondition were stably expressed and did not underlie variation that is owed to the anabolic treatments. Therefore, due to a very low concentration of smexRNAs in bovine plasma, a sufficient sample volume, an efficient RNA isolation method and appropriate sequencing strategies need to be united for successful screening and validation experiments.
In summary, smexRNAs could be seen as potential candidates in the identification of biomarkers with the ability to uncover illegal drug application in veterinary diagnostics. Using multivariate projection methodologies like OPLS-DA have proven the best potential to generate discriminative miRNA models, supported by small RNA-Seq data. PiRNAs were expressed with low copy numbers, which is not ensuring statistical robustness and significance. OPLS-DA enabled insights into the complex structure of sequencing data and clarified that value could be gained from the presented experiments, namely information about differentiation of treatment groups. However, the quantitative analysis in plasma is challenging as the content of miRNAs or piRNAs seemed robust in bovine plasma (Fig. 1, [32]) and differences in the abundance of minor expressed smexRNAs could not be revealed. Therefore, the assumed modifications of the smexRNA profile by growth-promoting substances was overestimated. Yet, it must not be forgotten that blood and hence plasma underlie extreme systemic influences. The bloodstream is permanently in direct contact with the complete organ system. Hence, the circulating small RNA profile could be heavily altered only throughout one circulation through the body. As described before, steroidal hormone implants as well as the oral clenbuterol doses were effective, but it might be the case that the potential alterations in the small RNA signatures could not be captured due to rapid turnover of the circulation system. To the present date, it could not be described in literature, that smexRNAs are the direct targets of stimulants like steroid hormones or β-2-adrenergic agonist. Therefore, for transcriptional biomarker development, a long-term and permanent miRNA pattern needs to be detected, which is not influenced by the animal circulatory system.
4. Conclusion
In transcriptional biomarker discovery, easy collectable sample specimen like whole blood, serum or plasma offer advantages in veterinary routine diagnostics but meet methodological difficulties, mainly due to matrix complexity, low RNA concentration and bioinformatical challenges. As miRNAs in plasma were tested to be highly stable and resistant to degradation [29], smexRNAs are seen as very potential candidates in the search for the next generation of transcriptional biomarkers. The presented experimental pipeline offered to analyze circulating miRNAs and piRNAs in bovines under anabolic stimulation. The usability of small RNA-Seq in the search for novel miRNA biomarkers in veterinary medicine was demonstrated here, as OPLS-DA discriminative models could be successfully created that also showed high goodness of fit and predictability. Next steps in the experimental lineup would be to deeper sequence plasma samples in order to provide detailed information about the composition of the smexRNA profile at low-abundance levels. Furthermore, RNA isolation systems from bio fluids still need enhanced performances to extract sufficient concentrations of smexRNAs that can be measured with RT-qPCR.
Acknowledgments
The study was organized and conducted in cooperation with KDK (Kontrollgemeinschaft Deutsches Kalbfleisch e.V., Germany), SGS (Société Générale de Surveillance, Switzerland) and DENKAVIT Ingredients (Germany). Boehringer Ingelheim (Germany) supported the study by supplying clenbuterol-hydrochloride.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bdq.2015.08.001.
Contributor Information
Spornraft Melanie, Email: melanie.spornraft@wzw.tum.de.
Kirchner Benedikt, Email: benedikt.kirchner@wzw.tum.de.
Michael W. Pfaffl, Email: michael.pfaffl@wzw.tum.de.
Riedmaier Irmgard, Email: irmgard.riedmaier@wzw.tum.de.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- 1.Premanandh J. Food Control. 2013;34:568–569. [Google Scholar]
- 2.Le Bizec B., Pinel G., Antignac J.P. J. Chromatogr. A. 2009;1216:8016–8034. doi: 10.1016/j.chroma.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Riedmaier I., Pfaffl M.W. Methods. 2013;59:3–9. doi: 10.1016/j.ymeth.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 4.In: R. Schilt (Ed.), Residues of veterinary drugs in food, Proceedings of the Euroresidue VII Conference, Egmond ann Zee, The Netherlands, 14–16 May, 2012, National Institute of Public Health and the Environment, Egmond aan Zee, 2012.
- 5.Regal P., Blokland M.H., Fente C.A., Sterk S.S., Cepeda A., van Ginkel Leen A. J. Agric. Food Chem. 2015;63(1):370–378. doi: 10.1021/jf503773u. [DOI] [PubMed] [Google Scholar]
- 6.Dervilly-Pinel S., Weigel A., Lommen S., Chereau L., Rambaud M., Antignac J.P., Nielen, Michel W.F., Le Bizec B. Anal. Chim. Acta. 2011;700:144–154. doi: 10.1016/j.aca.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Barrey E., Triba M., Messier F., Le Moyec L. Equine Vet. J. 2014;46:2–3. [Google Scholar]
- 8.Boyard-Kieken F., Dervilly-Pinel G., Garcia P., Paris A.C., Popot M.A., Le Bizec B., Bonnaire Y. J. Sep. Sci. 2011;34:3493–3501. doi: 10.1002/jssc.201100223. [DOI] [PubMed] [Google Scholar]
- 9.Pfaffl M.W. Methods. 2013;59:1–2. doi: 10.1016/j.ymeth.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Price C., Chen J. Genes Dis. 2014;1:53–63. doi: 10.1016/j.gendis.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarzenbach H., Nishida N., Calin G.A., Pantel K. Nat. Rev. Clin. Oncol. 2014;11:145–156. doi: 10.1038/nrclinonc.2014.5. [DOI] [PubMed] [Google Scholar]
- 12.Becker C., Riedmaier I., Reiter M., Tichopad A., Pfaffl M.W., Meyer Heinrich H.D. Analyst. 2011;136:1204–1209. doi: 10.1039/c0an00703j. [DOI] [PubMed] [Google Scholar]
- 13.Chim Stephen S.C., Shing Tristan K.F., Hung Emily C.W., Leung T.-Y., Lau T.K., Chiu Rossa W.K., Dennis Lo Y.M. Clin. Chem. 2008;54:482–490. doi: 10.1373/clinchem.2007.097972. [DOI] [PubMed] [Google Scholar]
- 14.Weber J.A., Baxter D.H., Zhang S., Huang D.Y., How Huang K., Jen Lee M., Galas D.J., Wang K. Clin. Chem. 2010;56:1733–1741. doi: 10.1373/clinchem.2010.147405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gou L.T., Dai P., Liu M.F. Wiley Interdiscip. Rev. RNA. 2014;5:733–745. doi: 10.1002/wrna.1252. [DOI] [PubMed] [Google Scholar]
- 16.Cui L., Lou Y., Zhang X., Zhou H., Deng H., Song H., Yu X., Xiao B., Wang W., Guo J. Clin. Biochem. 2011;44:1050–1057. doi: 10.1016/j.clinbiochem.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Serratosa J., Blass A., Rigau B., Mongrell B., Rigau T., Tortadès M., Tolosa E., Aguilar C., Ribó O., Balagué J. Revue Scientifique Et Technique (Int. Off. Epizoot.) 2006;25:637–653. [PubMed] [Google Scholar]
- 18.R.W. Stephany, Handbook of Experimental Pharmacology, 2010, 355–367. [DOI] [PubMed]
- 19.Riedmaier I., Pfaffl M.W., Meyer Heinrich H.D. Drug Test Anal. 2011;3:676–681. doi: 10.1002/dta.304. [DOI] [PubMed] [Google Scholar]
- 20.Riedmaier I., Benes V., Blake J., Bretschneider N., Zinser C., Becker C., Meyer H.H.D., Pfaffl M.W. Anal. Chem. 2012;84:6863–6868. doi: 10.1021/ac301433d. [DOI] [PubMed] [Google Scholar]
- 21.Riedmaier I., Spornraft M., Pfaffl M.W. Food additives & contaminants. Part A. Chem. Anal. Control Expos. Risk Assessment. 2014;31:641–649. doi: 10.1080/19440049.2014.886341. [DOI] [PubMed] [Google Scholar]
- 22.Eriksson L., Antti H., Gottfries J., Holmes E., Johansson E., Lindgren F., Long I., Lundstedt T., Trygg J., Wold S. Anal. Bioanal. Chem. 2004;380:419–429. doi: 10.1007/s00216-004-2783-y. [DOI] [PubMed] [Google Scholar]
- 23.Boccard J., Rutledge D.N. Anal. Chim. Acta. 2013;769:30–39. doi: 10.1016/j.aca.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 24.Regal P., Anizan S., Antignac J.P., Le Bizec B., Cepeda A., Fente C. Anal. Chim. Acta. 2011;700:16–25. doi: 10.1016/j.aca.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Trygg J., Wold S. J. Chemometr. 2002;16:119–128. [Google Scholar]
- 26.Wold S., Antti H., Lindgren F., Öhman J. Chemometr. Intell. Lab. Sys. 1998;44:175–185. [Google Scholar]
- 27.Bylesjö M., Rantalainen M., Cloarec O., Nicholson J.K., Holmes E., Trygg J. J. Chemometr. 2006;20:341–351. [Google Scholar]
- 28.User Guide to SIMCA By MKS Umetrics Version 13 downloaded from: http://131.130.57.230/clarotest190/claroline/backends/download.php?url=L1N0YXQvU0lNQ0EvVXNlciBHdWlkZSB0byBTSU1DQSAxMy5wZGY%3D&cidReset=true&cidReq=300152WS13 1992-2012 MKS Umetrics AB User guide edition date: April 23, 2012 AN MKS COMPANY MKS Umetrics AB Stortorget 21 SE-211 34 Malmö Sweden Phone: +46 (0)40 664 2580 Email: info@umetrics.com.
- 29.Mitchell P.S., Parkin R.K., Kroh E.M., Fritz B.R., Wyman S.K., Pogosova-Agadjanyan E.L., Peterson A., Noteboom J., O'Briant K.C., Allen A., Lin D.W., Urban N., Drescher C.W., Knudsen B.S., Stirewalt D.L., Gentleman R., Vessella R.L., Nelson P.S., Martin D.B., Tewari M. Proc. Natl. Acad. Sci. U. S. A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spornraft M., Kirchner B., Haase B., Benes V., Pfaffl M.W., Riedmaier I. PLoS One. 2014;9:e107259. doi: 10.1371/journal.pone.0107259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 32.Spornraft M., Kirchner B., Pfaffl M.W., Riedmaier I. Biotechnol. Lett. 2015;37(June (6)):1165–1176. doi: 10.1007/s10529-015-1788-2. Epub 2015 February 21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.