

Abstract
Background
Glaucoma is a complex, multifactorial disease characterised by the loss of retinal ganglion cells and their axons leading to a decrease in visual function. The earliest events that damage retinal ganglion cells in glaucoma are currently unknown. Retinal ganglion cell death appears to be compartmentalised, with soma, dendrite and axon changes potentially occurring through different mechanisms. There is mounting evidence from other neurodegenerative diseases suggesting that neuronal dendrites undergo a prolonged period of atrophy, including the pruning of synapses, prior to cell loss. In addition, recent evidence has shown the role of the complement cascade in synaptic pruning in glaucoma and other diseases.
Results
Using a genetic (DBA/2J mouse) and an inducible (rat microbead) model of glaucoma we first demonstrate that there is loss of retinal ganglion cell synapses and dendrites at time points that precede axon or soma loss. We next determine the role of complement component 1 (C1) in early synaptic loss and dendritic atrophy during glaucoma. Using a genetic knockout of C1qa (D2.C1qa-/- mouse) or pharmacological inhibition of C1 (in the rat bead model) we show that inhibition of C1 is sufficient to preserve dendritic and synaptic architecture.
Conclusions
This study further supports assessing the potential for complement-modulating therapeutics for the prevention of retinal ganglion cell degeneration in glaucoma.
Keywords: Glaucoma, Dendrite, Synapse, Complement, C1, C1qa, Retinal ganglion cell
Background
Glaucoma is a complex multifactorial disease characterized by the progressive dysfunction and loss of retinal ganglion cells and associated visual field deficits. It remains a leading cause of vision loss affecting 70 million people worldwide [1]. In spite of notable technical advances, the earliest neurodegenerative events that injure retinal ganglion cells during glaucoma [1–5] are unclear. Degeneration of the retinal ganglion cell during glaucoma appears to be partially compartmentalized, with different mechanisms influencing the soma, axon and dendritic tree. In human glaucoma, early changes to retinal ganglion cells have been observed both in the optic nerve head, including axon transport deficits [6–9], as well as in the retina with dendritic atrophy [10–12]. The precise relationship of axonal damage to events at the cell soma and retinal ganglion cell synaptic connectivity has been the focus of a number of recent studies in animal models of glaucoma [13–16] in which dendritic remodelling has been shown to precede gross cell loss. Recently, we demonstrated retinal ganglion cell dendritic atrophy during DBA/2J glaucoma and suggested that dendritic remodelling may be an early feature of glaucoma [16]. However, the mechanisms by which these early retinal ganglion cell changes occur are not known.
It has been proposed that activation of immune responses may be key events that damage retinal ganglion cells during early stages of glaucoma. Using a rat model of ocular hypertension (based on the sclerosis of episcleral vessels), Johnson et al. showed gene expression changes with significant enrichment of immune response pathways and cell proliferation pathways, among others [17]. The exploration of gene expression at the level of the optic nerve head confirmed immune cell responses and microglial activation, in the absence of significant astrocyte activation [18]. Guo and colleagues extended this work by showing an early immune cell response, coupled with gene expression patterns, that suggested retinal ganglion cell dysfunction and dendritic remodelling [19, 20]. Immune cell and/or microglial activation may therefore be a pertinent, early disease feature in animal models of glaucoma. Early changes to microglia [17–29] and peripherally derived immune cells [4, 30, 31] have been well documented in animal glaucoma models and in human glaucoma [32–34], consistent with this scenario of early immune activation.
The DBA/2 J mouse is a widely used model of chronic glaucoma showing hallmark features of the human disease. In DBA/2 J mice, mutant alleles of two genes (GpnmbR150X and Tyrp1b) cause iris pigment dispersion that results in ocular hypertension in the majority of eyes by 8 to 9 months of age. Glaucomatous damage is usually present by 12 months, signified by the loss of retinal ganglion cells and axonal degeneration within the optic nerve [35], which appears to initiate at the optic nerve head [2]. The interaction of multiple cell-types residing within the optic nerve head and retina are thought to be critical in glaucoma although the roles of specific cell-types are not well understood [3, 36–39].
We have previously used microarray gene expression of optic nerve head and retina tissue from pre-glaucomatous (8.5 months old) and glaucomatous (10.5 months old) DBA/2 J mice [3] in order to demonstrate that changes to inflammatory pathways, including the endothelin pathway, cell-adhesion pathways and the complement system occur early in the disease. In particular, induction of genes encoding proteins of the complement cascade was the earliest identifiable molecular change in glaucomatous DBA/2 J retinas [3]. DBA/2 J mice deficient for C1qa show robust protection from glaucomatous retinal ganglion cell loss and optic nerve degeneration [3]. The complement cascade has been heavily implicated in human and animal models of glaucoma, has increased expression in the eyes of patients with end stage glaucoma, and in primate and murine glaucomatous eyes [30, 40–49].
The role of the complement cascade in glaucoma is complex. In addition to its role in inflammatory signalling, complement pathways play a critical role in synaptic development and pruning [46, 50–53]. During central nervous system development neurons make many immature synaptic connections, followed by the selective elimination of those that are redundant. In the retinas of C1qa–deficient mice, retinal ganglion cells show defective synaptic elimination with limited refinement of the retinogeniculate pathway. TGF-β is thought to be important in the refinement process through activation/regulation of the complement cascade in neurons, microglia [51] or astrocytes [50]. The colocalization of C1q, part of the initiating complex of the classical pathway of the complement cascade, with synaptic markers such as PSD95 early in glaucomatous pathogenesis [50] supports the concept that complement activation underpins synapse elimination in glaucoma.
Here we determine the role of C1 in early synaptic loss and dendritic atrophy during glaucoma. We quantified synaptic and dendritic atrophy during early stages of glaucoma using two models of ocular hypertension. We demonstrate that synaptic and dendritic atrophy occur prior to any detectable damage to the axon or soma in both DBA/2 J glaucoma and in a rat model where ocular hypertension (OHT) was induced using an injection of magnetic microspheres [54]. Importantly, inhibiting C1 functions using either genetic ablation (DBA/2 J mutant for C1qa) or pharmacological inhibition (C1 esterase inhibitor in the rat OHT model) was sufficient to preserve dendritic and synaptic architecture. Taken together with the strong protective effect of C1qa knockout in retinal ganglion cell death and optic nerve damage, this suggests that inhibition of C1 should be considered as a therapeutic strategy for glaucoma.
Methods
Mouse strain, breeding and husbandry
Mice were housed and fed, as published [3], in a 14 h light/10 h dark cycle with food and water available ad libitum. All breeding and experimental procedures were undertaken in accordance with the Association for Research for Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic Research. The Institutional Animal Care and Use Committee (IACUC) at The Jackson Laboratory approved this study. The DBA/2 J (D2, n (mice) = 24), DBA/2 J-Gpnmb+ (D2-Gpnmb+, n = 20), and D2.C1qa (n = 16) strains were utilized and have been described in detail elsewhere [3]. We used D2-Gpnmb+ mice as a control, non-glaucomatous substrain of DBA/2 J [55].
Rat husbandry and IOP monitoring
Rat work was conducted in accordance with the Animals (Scientific Procedures) Act 1986. Brown Norway rats (Charles River, UK) were housed under a 12 h light/dark cycle with IOP measured at the same time of day in order to reduce variation in IOP. Breeding diet (SDS, RM3) and water were provided ad libitum.
Glaucoma induction and IOP monitoring in the rat
Baseline IOP for both eyes was established prior to glaucoma induction and IOP measured 24 h after induction and every 3 days thereafter. IOP was measured using a rebound tonometer (TonoLab, Finland) calibrated for use on the rat eye. Animals were awake and unrestrained during measurement, requiring only topical 0.4 % oxybuprocaine hydrochloride eye drops (Midoptic, UK). IOP was taken as the average of 5 repeat readings. Glaucoma was induced as described for the magnetic microbead model [54]. Briefly, the left anterior chamber was injected with 10 μl of bead solution (sterile ~4.5 μm diameter paramagnetic microspheres, Invitrogen UK, in balanced salt solution) using a 32-gauge needle and Hamilton syringe (n = 19). Injections were conducted under isofluorane-induced anaesthesia with topical 0.5 % chloramphenicol (Mid Optic, UK) applied to the cornea. Beads were drawn into the iridocorneal angle using a small handheld magnet in order to block trabecular meshwork outflow and induce OHT (n = 20). The right eye remained an un-operated, normotensive (NT) control.
C1 inhibitor administration
C1 inhibitor (C1 esterase inhibitor; CINRYZE; Shire (previously ViroPharma)) or vehicle only (PBS) was administered to rats 24 h prior to induced ocular hypertension (OHT) and then once every 4 days for 16 days. Intravitreal injections of 5 μl of C1 inhibitor (100 Units/ml (n = 17)) or PBS (n = 10) were performed on the left eye under isofluorane-induced anaesthesia.
DiOlistic labelling of flat mount retinas
The protocol for retinal dissection and DiOlistic labelling was identical for both mouse and rat eyes except where stated otherwise. DiI and DiO coated tungsten bullets were prepared as described previously using 8 mg DiI and 16 mg DiO (both Life technologies, US) dissolved in 100 μl dichloromethane to 100 mg of tungsten particles (1.7 μm; Bio-Rad). Mice were killed by cervical dislocation, their eyes enucleated and retinas immediately dissected in chilled HBSS. For rats, a rising concentration of CO2 was used, with cervical dislocation confirming death. Retinas were dissected as described above. Retinas were then flat mounted ganglion cell layer (GCL) up on Millicell cell culture inserts (0.4 μm pore diameter, Life technologies, US). Retinas were then ballistically labelled with DiO/DiI bullets at a pressure of 120 psi using a Helios gene gun system (Biorad, US). 3.0 μm pore size cell culture inserts were placed between the gun and retina to prevent aggregated particles over labelling the retina. The culture inserts were then placed in Neurobasal-A media (Life technologies, US) containing 2 mM L-glutamate, 2 % B27 supplement and 1 % N2 supplement and incubated for 30 min at 37 °C, 4 % CO2 to facilitate dye diffusion. Retinas were then fixed in 4 % PFA for 30 min before mounting on glass slides with Fluoromount and coverslips.
Immunofluorescent staining of retinal sections
The protocol for immunohistochemical staining of retinal sections was identical for both mice and rats except where stated otherwise. Mice were sacrificed by cervical dislocation, their eyes enucleated and placed in 4 % PFA ON. For rats, a rising concentration of CO2 was used, with cervical dislocation confirming death. Following this, eyes were cryoprotected in 30 % sucrose, frozen in OCT and cryosectioned at 12 μm. Eyes from the following number of animals (and central retina sections) were used: (in mice [animals (sections)]; 4mo D2 n = 6[34], 9mo D2 n = 3[25], 4mo D2-Gpnmb+n = 9[36], 9mo D2-Gpnmb+n = 5[28], 4mo D2.C1qa n = 5[29], 9mo D2.C1qa n = 3[30]. Rats: normotensive n = 9[43], normotensive plus C1 inhibitor n = 3[49], OHT n = 3[38], OHT plus C1 inhibitor n = 3[36]). Central retina sections were brought to RT for 1 h, permeabilized with 0.1 % Triton-X for 30mins, blocked with 5 % chick serum in PBS and stained ON at RT in primary antibody (for mice; rabbit anti-PSD-95; 51-6900, Life Technologies, USA, or goat anti-IBA1; ab5076, Abcam, UK; for rats; rabbit anti-PSD-95; ab18258, Abcam, UK). After primary antibody incubation, sections were washed 5 times in PBS, stained for 4 h at RT with secondary antibody (for mice; goat anti-rabbit AF488; Life Technologies, USA, for rats; goat anti-rabbit AF555, Abcam, UK). Slides were then washed a further 5 times with PBS, stained with DAPI (mice) or Hoechst 33342 (rats) for 15mins, mounted with fluoromount, coverslipped and sealed with nail-polish. For PSD95 quantification in DBA/2 J mice and rats, images of central retina showing the IPL and ONL were collected under identical conditions on a Leica SP8 confocal microscope and Leica DM6000B respectively (image area = 553.57 μm2). Fluorescent intensity of antibody staining was quantified using ImageJ. Three-dimensional reconstruction of IBA1+ cells was performed using IMARIS.
Retinal ganglion cell morphological analysis
Retinas were imaged using either a Leica SP8 or Zeiss LSM 510 confocal microscope with ×20 objective (mice: 4mo D2 n = 63, 9mo D2 n = 58, 4mo D2-Gpnmb+n = 27, 9mo D2-Gpnmb+n = 37, 4mo D2.C1qa n = 30, 9mo D2.C1qa n = 52. Rats: normotensive n = 43, normotensive plus C1 inhibitor n = 49, OHT n = 38, OHT plus C1 inhibitor n = 36). Z-stack imaged (slice thickness 1 μm) were captured for mouse retinal ganglion cells and rat retinal ganglion cells ensuring that the entire dendritic tree was in frame. A number of RGC morphological features were measured using FIJI. Dendritic field area was measured by connecting the outermost dendritic tips using the convex polygon tool. The dendritic tree was traced and the mean dendritic length calculated using the Neuron J plugin. Sholl analysis was conducted using the bitmap Sholl analysis plugin for FIJI [56]. Statistical analysis was conducted using SPSS 20 (IBM, US).
Classifying retinal ganglion cell subgroups
Retinal ganglion cells can be classified on the basis of their dendritic tree and soma morphology. To account for any bias that may arise from selective labelling of cells, we classified retinal ganglion cells from our control groups (in mice D2-Gpnmb+ and in rats, the normotensive group) based on classifications set out by Sun et al. [57, 58]. There was no obvious bias in the cell types expected (in mouse; our study (expected), RGCA 9 % (10 %), RGCB 38 % (30 %), RGCC 41 % (37 %), RGCD 13 % (23 %), in rat; RGCA 20.9 % (17.5 %), RGCB 32.6 % (29.2 %), RGCC 34.9 % (35.2.3 %), RGCD 11.6 % [18]). Once cells start to degenerate classifying based on morphology may lead to misrepresentation of cells at different stages of disease. Given that our control groups showed equal, unbiased sampling, we did not classify disease susceptible DBA/2 J or D2.C1qa-/- retinal ganglion cells. As lamination is unlikely to drastically change during disease, we confirmed that an even proportion of ON/OFF/ON-OFF cells were present across our samples (in mouse; ON/OFF/ON-OFF, D2-Gpnmb+ 87 %/4 %/9 %, D2 86 %/7 %/7 %, D2.C1qa 86 %/6 %/8 %, in rat; normotensive 71 %/17 %/12 %, normotensive plus C1 inhibitor 74 %/12 %/14 %, OHT 82 %/7 %/10 %, OHT plus C1 inhibitor 84 %/8 %/8 %). This is further expanded on in the Discussion.
Axon labelling with PPD and grading of glaucomatous damage
The processing of optic nerves and staining with paraphenylenediamine (PPD) which darkly stains the axoplasm and myelin sheath of damaged axons has been reported previously [59]. In brief, intracranial portions of optic nerves were fixed in 4 % PFA at RT for 48 h, processed and embedded in plastic. A segment of optic nerve from within a region up to 1 mm from the posterior surface of the sclera was sectioned (1 μm thick sections) and stained with PPD. Typically 30-50 sections are taken from each nerve. Homology between sections is considered during grading. Optic nerves were analysed and only eyes that had a corresponding nerve grade of ‘no or early damage’ (i.e. less than 5 % axons damaged) were selected in order to evaluate early disease events prior to axonal damage.
Results
Synapse reduction precedes optic nerve damage and is C1qa dependent
Dendrites degenerate prior to significant axon degeneration in DBA/2 J mice [16]. However, the factors that drive this dendritic atrophy in glaucoma are not known. Given the role of the complement cascade in synapse loss during development and neurodegenerative diseases [50], and the early induction of the complement components in the inner plexiform layer of glaucomatous retinas [3], we hypothesized that complement may mediate both synapse loss and dendritic atrophy in glaucomatous retinas. To test this, we first assessed the synaptic density of the inner plexiform and ganglion cell layer of 9 month-old DBA/2 J mice (an age at which IOP elevation is established) using a synaptic marker, PSD-95. To focus on very early stages of glaucoma, eyes were selected that had no detectable signs of glaucomatous axon damage (seeMethods). Previous studies have shown that at 9 months, axonal transport is intact in these eyes [4, 16]. DBA/2 J eyes were compared to age matched control eyes (D2.Gpnmb+) and pre-glaucomatous DBA/2 J eyes (4 months prior to IOP increase). In both these control strains, at the ages selected, there was no detectable axon damage.
To test the role of complement activation during synapse loss in the IPL, we measured synaptic density using PSD-95. Twelve-micron sections were labelled with anti-PSD-95 antibody and DAPI (to demarcate the boundaries of the inner plexiform layer). At a young, pre-glaucomatous age (4 months old) there was no difference in labelling intensity in the IPL between D2.Gpnmb+ and DBA/2 J mice consistent with similar synaptic development in these genotypes. However, in 9 months old, DBA/2 J eyes with no discernible optic nerve disease or RGC loss there was a significant reduction in synaptic integrity (20 % compared to 4 months old DBA/2 J, P < 0.001 and 18 % compared to aged matched D2.Gpnmb+, P < 0.001) (Fig. 1). Given that C1QA expression increases in the IPL early in glaucoma [3, 50] and that DBA/2 J mice deficient in C1qa are protected from optic nerve degeneration, we tested the role of C1qa in synapse degeneration in DBA/2 J glaucoma. In D2.C1qa-/- eyes, PSD-95 labelling intensity did not reduce significantly with age (6 % 4-9 months comparison, P = 0.26) (Fig. 1a) consistent with a role for C1qa in glaucomatous synapse elimination. There was no significant difference in outer plexiform layer (OPL) PSD-95 intensity (data not shown). Supporting a possible involvement of retinal microglia in early retinal ganglion cell synapse loss, high-resolution imaging combined with 3D reconstruction revealed high-resolution rendered images of IBA1+ microglia containing PSD-95 (Fig. 1c) supporting the concept that activated microglia phagocytose C1q tagged synapses in the early stages of DBA/2 J glaucoma.
Fig. 1.
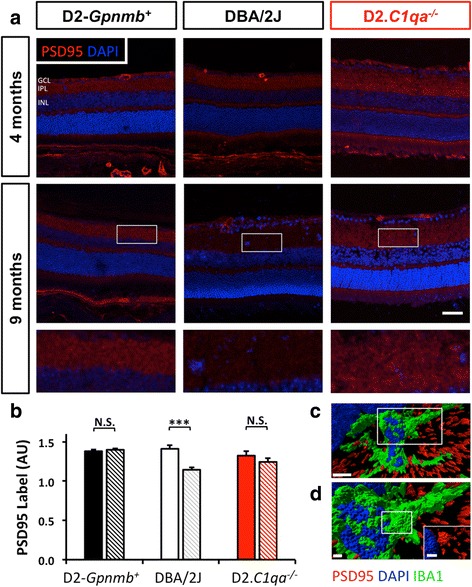
DBA/2 J mice exhibit synapse loss early during glaucoma pathogenesis which is absent in D2.C1qa -/- retinas. a. Immunohistochemical labelling (PSD-95, red) to demonstrate synaptic integrity in 9 months old DBA/2 J retinas. All eyes shown had no significant retinal ganglion cell or axon loss. Using C1qa mutant DBA/2 J mice (D2.C1qa -/-) we explored whether synaptic pruning was present in these retinas. At 4 months D2.Gpnmb +, DBA/2 J and D2.C1qa -/- retinas show comparable synapse density (a, top row). By 9 months, there is significant synapse loss in retinas of DBA/2 J mice that is absent in retinas of either D2.Gpnmb + or D2.C1qa -/- (a, bottom two rows). b. The degree of PSD-95 labelling is shown. The region of interest is marked (white boxes in upper row). c. Rendered confocal images show PSD-95 localisation within IBA1+ microglia in the IPL of DBA/2 J mice. Inset shows a higher resolution image of a region of interest (white rectangle, d). d. Inset from C showing PSD95 engulfed by IBA1+ microglia, inset within d (white rectangle) shows PSD95 and DAPI only. Scale bar = 50 μm (a), 5 μm (c) and 1 μm (d and inset), error bars = SEM, *** = P < 0.001, GCL = ganglion cell layer, IPL = inner plexiform layer, INL = inner nuclear layer
Early RGC dendrite loss is preserved in D2.C1qa-/- retinas
We next explored whether complement C1q deficiency prevents dendritic atrophy in DBA/2 J glaucoma. Dendritic integrity was quantified in DiOlistically labelled retinas from 9 month-old DBA/2 J, D2.Gpnmb+, and D2.C1qa-/- eyes with no detectable optic nerve damage. Two hundred and sixty seven cells were identified as retinal ganglion cells (confirmed by the presence of an axon running in the nerve fibre layer towards the optic disc) imaged at 40× and then compressed to give a 2D image for quantification. Dendritic architecture was assessed by total dendritic field area, total dendritic length, and dendritic complexity [60]. At 4 months of age, there were no significant differences in any of these phenotypes between mice of each genotype. By contrast, 9 month-old DBA/2 J mice had a significant reduction in retinal ganglion cell dendritic integrity compared to control D2.Gpnmb+ mice (significant reduction in dendritic field area (P < 0.01), and trending decreases in AUC and total dendritic length which do not reach significance), consistent with earlier work on non-glaucomatous retinas at 12 months of age [16].
Dendritic atrophy was not present in D2.C1qa-/- retinal ganglion cells, the dendritic trees of which were unchanged compared to the RGCs from D2.Gpnmb+ control mice (Fig. 2), confirming the critical importance of C1qa and its protein product C1q for early dendritic atrophy in DBA/2 J glaucoma. In addition there was no significant change in soma sizes across groups (data not shown). A panel of representative, DiOlistically labelled retinal ganglion cells is shown in Fig. 3.
Fig. 2.
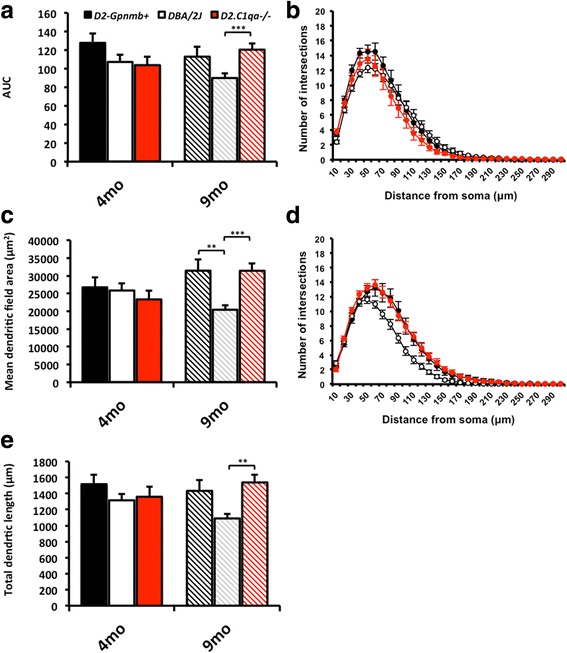
C1qa plays a role in dendritic pruning early during glaucoma pathogenesis. We tested whether C1qa has a role in dendrite remodelling using mice deficient in C1qa (D2.C1qa -/-). a Area under the curve (AUC) of the Sholl analysis (at 4 months of age (b), and at 9 months of age (d)) shows a decrease in dendritic complexity in DBA/2 J retinal ganglion cells that is absent in D2.Gpnmb + and D2.C1qa -/- retinas. This is paralleled in both the retinal ganglion cell dendritic field area (c) and total dendritic length (e). Error bars = SEM, ** = P < 0.01, *** = P < 0.001
Fig. 3.
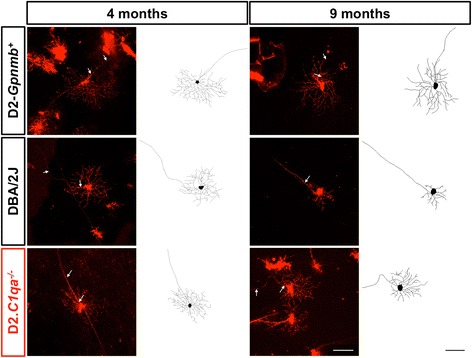
Representative DiOlistic and tracings of mouse retinal ganglion cells. Retinal ganglion cells from D2.Gpnmb +, DBA/2 J and D2.C1qa -/- retinas were DiOlistically labelled to analyse dendritic morphology. Top row: representative images and tracings from 4 month (left) and 9 month (right) D2.Gpnmb + retinal ganglion cells. Centre row: representative images and tracings from 4 month (left) and 9 month (right) DBA/2 J retinal ganglion cells. Bottom row: representative images and tracings from 4 month (left) and 9 month (right) D2.C1qa -/- retinal ganglion cells. Scale bar = 100 μm, white arrows = axon
C1 inhibitor protects retinal ganglion cells from dendritic and synaptic atrophy
To further evaluate the role of C1q in dendrite and synapse integrity in glaucoma, we used an experimentally induced bead-model of ocular hypertension in the rat. In this model paramagnetic beads (~4.5 μm in diameter) are injected into the anterior chamber and pulled into the trabecular meshwork under the influence of an external magnetic field to generate sustainable increases in IOP [54] (Fig. 4). As with the DBA2J mouse, a reduction in PSD95 labelling indicated significant synapse loss in the IPL with retinal ganglion cell dendritic atrophy in glaucomatous eyes compared to the control, (un-injected), contralateral eyes (Figs. 5, 6 and 7). There was no significant difference in OPL PSD-95 intensity (data not shown).
Fig. 4.
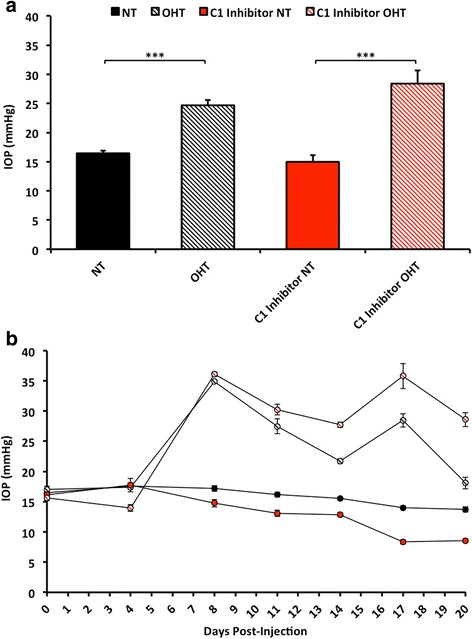
Ocular hypertension occurs in rat eyes treated with C1 inhibitor. a Average IOP over a 20 day time course. b Individual IOP recordings at each day of measurement (0 = day 0, base line reading). Points represent an average of 5 readings per eye. Note that C1 inhibitor administration does not significantly affect intraocular pressure. Error bars = StDev. *** = P < 0.001
Fig. 5.
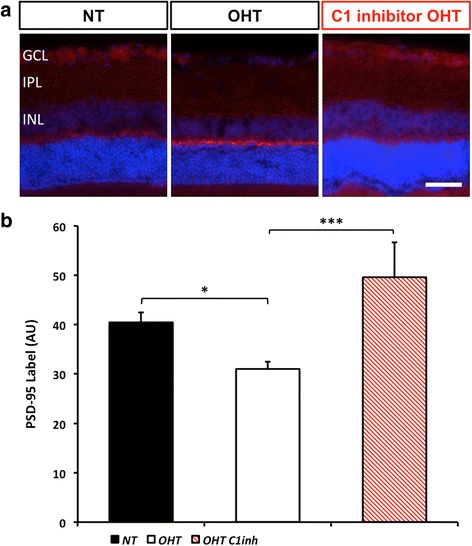
Ocular hypertensive rat eyes exhibit early inner plexiform layer synapse loss which is absent in C1 inhibitor treated eyes. Administration of a commercial FDA approved C1 inhibitor reduced synapse loss. a Representative images of rat retinas labelled with PSD-95 to show synapses. b Analysis of synaptic density. NT = normotensive eyes, sham control, OHT = ocular hypertensive eyes. Scale bar = 50 μm, * = P < 0.05, *** = P < 0.001, GCL = ganglion cell layer, IPL = inner plexiform layer, INL = inner nuclear layer
Fig. 6.
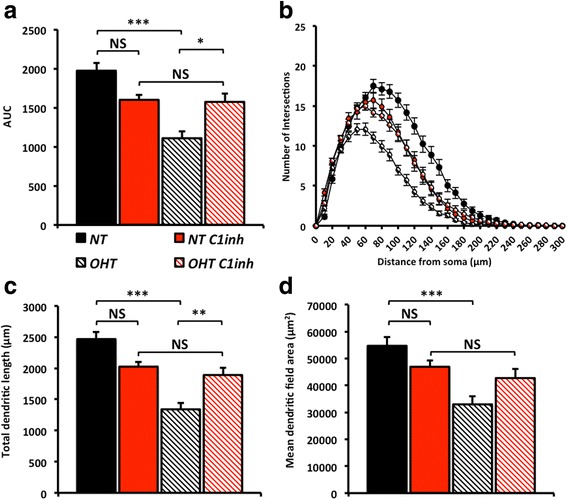
Inhibition of C1 prevents retinal ganglion cell dendrite pruning in ocular hypertensive rat eyes. To test dendrite loss in the rat bead model of glaucoma we DiOlistically labelled retinal ganglion cells following an ocular hypertensive insult. There was a dramatic decrease in dendritic integrity of retinal ganglion cells in ocular hypertensive eyes compared with normotensive eyes as assessed by Sholl analysis (a, area under the curve (AUC), b, Sholl analysis). This is paralleled in both the retinal ganglion cell dendritic field area (c) and total dendritic length (d). NT = normotensive eyes, sham control, OHT = ocular hypertensive eyes. Error bars = SEM, * = P < 0.05, ** = P < 0.01, *** = P < 0.001
Fig. 7.
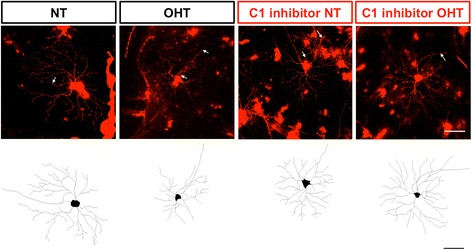
Representative DiOlistic and tracings of rat retinal ganglion cells. Retinal ganglion cells from normotensive eyes and ocular hypertensive eyes were DiOlistically labelled to analyse dendritic morphology. Top row: representative images and Bottom row: tracings from normotensive (far left), ocular hypertensive (centre left) normotensive (centre right), and ocular hypertensive (far right) retinal ganglion cells treated with C1 inhibitor. Scale bar = 100 μm, white arrows = axon
To evaluate the role of C1q in synaptic loss and dendritic atrophy in the rat model, and to assess therapeutic benefit of pharmacologic inhibition of the C1 complex, we administered human C1 inhibitor intraviterally 1 day prior to the induction of ocular hypertension and then at 4 day intervals for a period of 28 days for those animals with sustained elevation in IOP. C1 inhibitor-treated eyes were significantly protected from RGC dendritic and synaptic atrophy compared to normotensive (NT) eyes. No significant dendritic or synaptic atrophy was observed in treated normotensive eyes (that received C1 inhibitor without OHT) (Figs. 5, 6 and 7). In addition there was no significant change in soma sizes across groups (data not shown). Importantly, the C1 inhibitor had no significant effect on the IOP profile (Fig. 4). Collectively, our data support a major role for C1 and the classical pathway of the complement cascade in dendritic and synaptic pruning during early glaucoma.
Discussion
Multiple compartments of the retinal ganglion cell are suggested to be impacted in glaucoma [61]. While the optic nerve head is considered a primary and critical site of damage in glaucoma [2, 6, 8, 9, 62] early damage may also manifest at other locations including the retinal ganglion cell soma, dendrites and synapses [3, 10–12, 16, 63–67]. In this study, we use both a genetic (DBA/2 J mice) and an inducible (rat bead) model of glaucoma to show that significant changes occur to retinal ganglion cell synapses and dendrites prior to optic nerve degeneration. This dendritic and synaptic atrophy is disease- but not age-related as there is no detectable atrophy in age and strain matched, non-glaucomatous controls (D2.Gpnmb+). At this stage of the disease there is no damage as detectable by PPD staining of the optic nerve or retinal ganglion cell loss, predicting that observed effects [68] might be subtle. It cannot be ruled out that there was regional bias in the DiOlistic labelling in areas of the retina that are resistant to damage or contain retinal ganglion cells of varying morphologies. This seems unlikely given that DiOlistic labelling is diffuse (typically 5-20 cells per retina labelled evenly across the retina) and random. The preferential damage or loss of selective types of retinal ganglion cells has been investigated previously in the DBA/2 J mouse model, with neither cell type specific loss of dendritic morphology or somal shrinkage apparent [13, 69]. In addition, we have previous shown that genetic labelling of retinal ganglion cells introduces an inherent bias that might misrepresent cells at different stages of the disease in DBA/2 J glaucoma [16]. However, new, unbiased genetic tools that allow the visualization of specific retinal ganglion cell subtypes are being developed and provide scope for further investigation once available across mouse strains and in the rat [68]. These early changes to retinal ganglion cell structures in the retina are consistent with the early loss of pattern electroretinogram (PERG) that occurs in DBA/2 J mice [2, 70]. Since dendritic integrity is essential for the efficient generation of action potentials in the retinal ganglion cells, we suggest that dendritic degeneration may be an important contributor to the reduced PERG signal in early glaucoma [71, 72].
Determining the timing of changes to retinal ganglion cell compartments in the retina in relation to axonal changes in the optic nerve head in humans and animal models will be important to fully understand the earliest drivers of glaucoma. Although early soma and axon loss have been reported in 9 months old mice in other colonies [73], this is not the general case in our DBA/2 J colony [35]. However, in eyes with no significant optic nerve damage (based on optic nerve damage assessment just behind the orbit) or axon transport disruption, discrete and focal axon damage is observed in the glial lamina region of the optic nerve head at this stage [2]. The extent of the changes to retinal ganglion cell synapses and dendrites in the retina are greater than the number of focally damaged axons in the optic nerve head suggesting synaptic and dendritic atrophy are amongst the earliest pathological events in DBA/2 J glaucoma. To explore whether this is exclusive to the DBA/2 J mouse we also studied these changes in an inducible model of rat ocular hypertension, showing dendritic atrophy in eyes with consistent elevations in IOP but not in sham treated, normotensive or contralateral eyes.
Since early synaptic and dendritic atrophy were associated with an increase in complement components in the IPL [50] the complement pathway was a promising candidate for mediating early retinal ganglion cell degeneration. The key finding in the present study is that early synaptic and dendritic atrophy were prevented through inhibition of the C1 complex, the initiating complex of the classical complement pathway. D2.C1qa-/- mice developed the anterior segment changes that occur in normal DBA/2 J mice with subsequent elevation in IOP [3]. Only a small percentage of D2.C1qa-/- mice go on to develop optic nerve degeneration and retinal ganglion cell loss [3]. At 9 months of age (prior to any retinal ganglion cell loss and optic nerve degeneration) we could not discern any significant reduction in retinal ganglion cell dendritic integrity or synapse number compared to age matched, control D2.Gpnmb+ retinas, i.e. DBA/2 J dendritic and synaptic atrophy is prevented by C1qa deficiency. The functional implications of this protective effect (e.g. if the PERG is restored in DBA/2 J mice deficient in C1qa) have yet to be elucidated.
Given our findings in DBA/2 J glaucoma we sought to test whether pharmacological inhibition of C1 could be of therapeutic value in glaucoma. We demonstrate that intravitreal administration of human C1 inhibitor prior to and during the onset of OHT preserved dendritic and synaptic integrity in eyes with sustained elevations in IOP. Global genetic inhibition of C1 in the mouse protects all compartments of the retinal ganglion cell. It is important to note that, C1 inhibitor treatment in the rat did not protect the optic nerve, most likely reflecting the reduced accessibility of the drug outside of the retina (data not shown). This shows inhibiting complement in the retina is sufficient to protect dendrites and synapses even whilst axon degeneration is occurring. Our results further support compartmentalised changes in the retinal ganglion cell possibly due to independent, but not necessarily mutually exclusive, intraocular pressure-related events in the retina and optic nerve head. Studies employing the systemic administration of C1 inhibitors would clarify the protective effects of C1 inhibition in neural compartments outside the retina.
Complement activation has been shown in human and animal models of glaucoma [3, 49, 74], as well as in other chronic neurodegenerative diseases such as Alzheimer’s disease [75–82]. The classical pathway of the complement cascade is heavily implicated in glaucoma, and in DBA/2 J mice deficient in C1qa there is a significant decrease in retinal ganglion cell loss and optic nerve degeneration compared to regular DBA/2 J mice [3]. The complement cascade is activated by the classical, alternative or lectin pathways, converging at the cleaving of C3 into effector fragments that can modulate immune response such as chemo-attraction, phagocytosis or cell lysis. The precise mechanisms by which C1qa or the C1 complex contribute to glaucomatous retinal ganglion cell damage is not known but the opsonisation of synapses for removal by microglia [46, 50, 51, 53, 83], seems one likely route. C3-independent roles for the C1 complex have been also identified. For instance, C1q can bind apoptotic cells to promote their phagocytic uptake by myeloid-derived macrophages. C1q can also bind the frizzled receptor and modulate WNT signalling [84]. Components of the C1 complex are up-regulated in multiple cell types in the retina and optic nerve head during early stages of glaucoma, particularly myeloid derived cells and retinal ganglion cells. However, the extrinsic or intrinsic factors that induce or modulate complement induction are also not known. In the brain, C1qa expression increase in IBA1+ cells and neurons with age, and C1qa deficiency can mitigate age-related cognitive decline [53, 85]. Therefore, the complement cascade is playing multiple roles during different stages of glaucoma and further work is required to elucidate them.
Conclusion
This study demonstrates in two different animal models of glaucoma that early synaptic and dendritic atrophy is dependent on C1QA, a component of the C1 complex of the complement cascade. It provides further evidence for the importance of complement activation in glaucoma, raising the possibility that complement-modulating therapeutics could play a role in the prevention of retinal ganglion cell damage.
Acknowledgements
The Authors would like to thank Mimi de Vries, Catherine Braine and Harriet Jackson for their help with this investigation.
Funding
EY021525 (GRH), EY011721 (SWMJ), BB/F016352/1 (JRT), Glaucoma Research Foundation (GRH). Simon John is an Investigator of HHMI.
Abbreviations
- ACUC
Animal care and use committee
- AUC
Area under the curve
- C1
Complement component 1
- DiI - 1
1'-Dioctadecyl-3,3,3',3'-Tetramethylindocarbocyanine Perchlorate
- DiO - 3
3′-dioctadecyloxacarbocyanine perchlorate
- GCL
Ganglion cell layer
- IBC
Institutional biosafety committee
- OHT
Ocular HyperTension
- IOP
IntraOcular pressure
- IPL
Inner plexiform layer
- NT
NormoTensive/NormoTension
- OPL
Outer plexiform layer
- RGC
Retinal ganglion cell
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PAW–designed, conceived and performed mouse experiments (husbandry, DiOlistics, immunofluorescence, optic nerve assessment), wrote the manuscript; JRT–performed mouse and rat experiments (clinical examination, husbandry, DiOlistics, immunofluorescence, microscopy), wrote the manuscript; KWP–performed mouse experiments (immunofluorescence, microscopy); SDC–designed and performed rat experiments (clinical examination, husbandry, DiOlistics, immunofluorescence, microscopy); BPM–designed and conceived experiments, wrote the manuscript; JEM–designed and conceived experiments, wrote the manuscript; SWMJ–designed and conceived experiments, wrote the manuscript; GRH–designed and conceived experiments, wrote the manuscript. All Authors read and approved the final manuscript.
Contributor Information
Simon W. M. John, Email: simon.john@jax.org
Gareth R. Howell, Email: gareth.howell@Jax.org
References
- 1.Ha Q. Number of people with glaucoma worldwide. Br J Ophthalmol. 1996;80(5):389–93. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2 J glaucoma. J Cell Biol. 2007;179(7):1523–37. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Investig. 2011;121(4):1429–44. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell GR, Soto I, Zhu X, Ryan M, Macalinao DG, Sousa GL, et al. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J Clin Invest. 2012;122(4):1246–61. doi: 10.1172/JCI61135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–79. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- 6.Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980;19(2):137–52. [PubMed] [Google Scholar]
- 7.Gaasterland D, Tanishima T, Kuwabara T. Axoplasmic flow during chronic experimental glaucoma. 1. Light and electron microscopic studies of the monkey optic nervehead during development of glaucomatous cupping. Invest Ophthalmol Vis Sci. 1978;17(9):838–46. [PubMed] [Google Scholar]
- 8.Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974;13(10):771–83. [PubMed] [Google Scholar]
- 9.Anderson DR, Hendrickson AE. Failure of increased intracranial pressure to affect rapid axonal transport at the optic nerve head. Invest Ophthalmol Vis Sci. 1977;16(5):423–6. [PubMed] [Google Scholar]
- 10.Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci. 1998;39(12):2304–20. [PubMed] [Google Scholar]
- 11.Weber AJ, Harman CD. Structure–function relations of parasol cells in the normal and glaucomatous primate retina. Invest Ophthalmol Vis Sci. 2005;46(9):3197–207. doi: 10.1167/iovs.04-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber AJ, Harman CD. BDNF preserves the dendritic morphology of alpha and beta ganglion cells in the cat retina after optic nerve injury. Invest Ophthalmol Vis Sci. 2008;49(6):2456–63. doi: 10.1167/iovs.07-1325. [DOI] [PubMed] [Google Scholar]
- 13.Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2 J mice. J Cell Biol. 2005;171(2):313–25. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan JE. Retina ganglion cell degeneration in glaucoma: an opportunity missed? A review. Clin Experiment Ophthalmol. 2012;40(4):364–8. doi: 10.1111/j.1442-9071.2012.02789.x. [DOI] [PubMed] [Google Scholar]
- 15.Morquette B, Morquette P, Agostinone J, Feinstein E, McKinney RA, Kolta A, et al. REDD2-mediated inhibition of mTOR promotes dendrite retraction induced by axonal injury. Cell Death Differ. 2015;22(4):612–25. doi: 10.1038/cdd.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams PA, Howell GR, Barbay JM, Braine CE, Sousa GL, John SW, et al. Retinal ganglion cell dendritic atrophy in DBA/2 J glaucoma. PLoS One. 2013;8(8) doi: 10.1371/journal.pone.0072282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson EC, Jia L, Cepurna WO, Doser TA, Morrison JC. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2007;48(7):3161–77. doi: 10.1167/iovs.06-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson EC, Doser TA, Cepurna WO, Dyck JA, Jia L, Guo Y, et al. Cell proliferation and interleukin-6-type cytokine signaling are implicated by gene expression responses in early optic nerve head injury in rat glaucoma. Invest Ophthalmol Vis Sci. 2011;52(1):504–18. doi: 10.1167/iovs.10-5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, Cepurna WO, Dyck JA, Doser TA, Johnson EC, Morrison JC. Retinal cell responses to elevated intraocular pressure: a gene array comparison between the whole retina and retinal ganglion cell layer. Invest Ophthalmol Vis Sci. 2010;51(6):3003–18. doi: 10.1167/iovs.09-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y, Johnson EC, Cepurna WO, Dyck JA, Doser T, Morrison JC. Early gene expression changes in the retinal ganglion cell layer of a rat glaucoma model. Invest Ophthalmol Vis Sci. 2011;52(3):1460–73. doi: 10.1167/iovs.10-5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2 J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008;49(4):1437–46. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- 22.Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. 2011;519(4):599–620. doi: 10.1002/cne.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosco A, Crish SD, Steele MR, Romero CO, Inman DM, Horner PJ, et al. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. PLoS One. 2012;7:e43602. doi: 10.1371/journal.pone.0043602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosco A, Romero CO, Ambati BK, Vetter ML. In vivo dynamics of retinal microglial activation during neurodegeneration: confocal ophthalmoscopic imaging and cell morphometry in mouse glaucoma. J Vis Exp. 2015;99 doi: 10.3791/52731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yue YK, Mo B, Zhao J, Yu YJ, Liu L, Yue CL, et al. Neuroprotective Effect of Curcumin Against Oxidative Damage in BV-2 Microglia and High Intraocular Pressure Animal Model. J Ocul Pharmacol Ther. 2014;30(8):657–64. doi: 10.1089/jop.2014.0022. [DOI] [PubMed] [Google Scholar]
- 26.Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I. Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefes Arch Clin Exp Ophthalmol. 2014;252(5):761–72. doi: 10.1007/s00417-014-2588-4. [DOI] [PubMed] [Google Scholar]
- 27.Qu J, Jakobs TC. The Time Course of Gene Expression during Reactive Gliosis in the Optic Nerve. PLoS One. 2013;8(6) doi: 10.1371/journal.pone.0067094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor S, Calder CJ, Albon J, Erichsen JT, Boulton ME, Morgan JE. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp Eye Res. 2011;92(5):338–43. doi: 10.1016/j.exer.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naskar R, Wissing M, Thanos S. Detection of early neuron degeneration and accompanying microglial responses in. Invest Ophthalmol Vis Sci. 2002;43(9):2962–8. [PubMed] [Google Scholar]
- 30.Howell GR, MacNicoll KH, Braine CE, Soto I, Macalinao DG, Sousa GL, et al. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol Dis. 2014;71:44–52. doi: 10.1016/j.nbd.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gramlich OW, Ding QJ, Zhu W, Cook A, Anderson MG, Kuehn MH. Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathol Commun. 2015;3:56. doi: 10.1186/s40478-015-0234-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neufeld AH. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch Ophthalmol. 1999;117(8):1050–6. doi: 10.1001/archopht.117.8.1050. [DOI] [PubMed] [Google Scholar]
- 33.Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. 2000;32(1):42–50. doi: 10.1002/1098-1136(200010)32:1<42::AID-GLIA40>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 34.Wong M, Huang P, Li W, Li Y, Zhang SS, Zhang C. T-helper1/T-helper2 cytokine imbalance in the iris of patients with glaucoma. PLoS One. 2015;10(3) doi: 10.1371/journal.pone.0122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, et al. Inherited glaucoma in DBA/2 J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005;22(5):637–48. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- 36.Quigley HA, Addicks EM, Green WR, Maumenee AE. Optic-nerve damage in human glaucoma. 2. The site of injury and susceptibility to damage. Arch Ophthalmol. 1981;99(4):635–49. doi: 10.1001/archopht.1981.03930010635009. [DOI] [PubMed] [Google Scholar]
- 37.Quigley HA, Addicks EM. REGIONAL DIFFERENCES IN THE STRUCTURE OF THE LAMINA CRIBROSA AND THEIR RELATION TO GLAUCOMATOUS OPTIC-NERVE DAMAGE. Arch Ophthalmol. 1981;99(1):137–43. [DOI] [PubMed]
- 38.Quigley HA, Hohman RM, Addicks EM, Massof RW, Green WR. MORPHOLOGIC CHANGES IN THE LAMINA CRIBROSA CORRELATED WITH NEURAL LOSS IN OPEN-ANGLE GLAUCOMA. Am J Ophthalmol. 1983;95(5):673–91. [DOI] [PubMed]
- 39.Soto I, Pease ME, Son JL, Shi X, Quigley HA, Marsh-Armstrong N. Retinal Ganglion Cell Loss in a Rat Ocular Hypertension Model Is Sectorial and Involves Early Optic Nerve Axon Loss. Invest Ophthalmol Vis Sci. 2011;52(1):434–41. doi: 10.1167/iovs.10-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doudevski I, Rostagno A, Cowman M, Liebmann J, Ritch R, Ghiso J. Clusterin and complement activation in exfoliation glaucoma. Invest Ophthalmol Vis Sci. 2014;55(4):2491–9. doi: 10.1167/iovs.13-12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Križaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, et al. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. 2014;39(2):105–19. doi: 10.3109/02713683.2013.836541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue T, Kawaji T, Tanihara H. Elevated levels of multiple biomarkers of Alzheimer’s disease in the aqueous humor of eyes with open-angle glaucoma. Invest Ophthalmol Vis Sci. 2013;54(8):5353–8. doi: 10.1167/iovs.13-12245. [DOI] [PubMed] [Google Scholar]
- 43.Howell GR, Soto I, Ryan M, Graham LC, Smith RS, John SW. Deficiency of complement component 5 ameliorates glaucoma in DBA/2 J mice. J Neuroinflammation. 2013;10(1):76. doi: 10.1186/1742-2094-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheetz TE, Fingert JH, Wang K, Kuehn MH, Knudtson KL, Alward WL, et al. A genome-wide association study for primary open angle glaucoma and macular degeneration reveals novel Loci. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0058657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren L, Danias J. A role for complement in glaucoma? Adv Exp Med Biol. 2010;703:95–104. doi: 10.1007/978-1-4419-5635-4_7. [DOI] [PubMed] [Google Scholar]
- 46.Rosen AM, Stevens B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv Exp Med Biol. 2010;703:75–93. doi: 10.1007/978-1-4419-5635-4_6. [DOI] [PubMed] [Google Scholar]
- 47.Tezel G, Yang X, Luo C, Kain AD, Powell DW, Kuehn MH, et al. Oxidative stress and the regulation of complement activation in human glaucoma. Invest Ophthalmol Vis Sci. 2010;51(10):5071–82. doi: 10.1167/iovs.10-5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nikolskaya T, Nikolsky Y, Serebryiskaya T, Zvereva S, Sviridov E, Dezso Z, et al. Network analysis of human glaucomatous optic nerve head astrocytes. BMC Med Genomics. 2009;2:24. doi: 10.1186/1755-8794-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stasi K, Nagel D, Yang X, Wang RF, Ren L, Podos SM, et al. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci. 2006;47(3):1024–9. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- 50.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 51.Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16(12):1773–82. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107(17):7975–80. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 54.Samsel PA, Kisiswa L, Erichsen JT, Cross SD, Morgan JE. A novel method for the induction of experimental glaucoma using magnetic microspheres. Invest Ophthalmol Vis Sci. 2011;52(3):1671–5. doi: 10.1167/iovs.09-3921. [DOI] [PubMed] [Google Scholar]
- 55.Howell GR, Libby RT, Marchant JK, Wilson LA, Cosma IM, Smith RS, et al. Absence of glaucoma in DBA/2J mice homozygous for wild-type versions of Gpnmb and Tyrp1. BMC Genet. 2007;8:45. doi: 10.1186/1471-2156-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreira T, Ou Y, Li S, Giniger E, van Meyel DJ. Dendrite architecture organized by transcriptional control of the F-actin nucleator Spire. Development. 2014;141(3):650–60. doi: 10.1242/dev.099655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun W, Li N, He S. Large-scale morophological survey of rat retinal ganglion cells. Vis Neurosci. 2002;19(4):483–93. doi: 10.1017/S0952523802194107. [DOI] [PubMed] [Google Scholar]
- 58.Sun WZ, Li N, He SG. Large-scale morphological survey of mouse retinal ganglion cells. J Comp Neurol. 2002;451(2):115–26. doi: 10.1002/cne.10323. [DOI] [PubMed] [Google Scholar]
- 59.Smith R, John S, Nishina P, Sundberg J. Systematic evaluation of the mouse eye. Anatomy, pathology and biomethods. Boca Raton: CRC Press; 2002. [Google Scholar]
- 60.Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87(4):387. [PMC free article] [PubMed] [Google Scholar]
- 61.Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways-a rôle for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24(6):639–62. doi: 10.1016/j.preteyeres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Nickells RW. From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Can J Ophthalmol. 2007;42(2):278–87. doi: 10.3129/canjophthalmol.i07-036. [DOI] [PubMed] [Google Scholar]
- 63.Feng L, Zhao Y, Yoshida M, Chen H, Yang JF, Kim TS, et al. Sustained ocular hypertension induces dendritic degeneration of mouse retinal ganglion cells that depends on cell type and location. Invest Ophthalmol Vis Sci. 2013;54(2):1106–17. doi: 10.1167/iovs.12-10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morgan JE. Retinal ganglion cell death in experimental glaucoma. Br J Ophthalmol. 2000;84(3):303–10. doi: 10.1136/bjo.84.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010;107(11):5196–201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crish SD, Calkins DJ. Central Visual Pathways in Glaucoma: Evidence for Distal Mechanisms of Neuronal Self-Repair. J Neuroophthalmol. 2015;35(Suppl 1):S29–37. doi: 10.1097/WNO.0000000000000291. [DOI] [PubMed] [Google Scholar]
- 67.Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong RO. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci. 2013;33(44):17444–57. doi: 10.1523/JNEUROSCI.5461-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El-Danaf RN, Huberman AD. Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J Neurosci. 2015;35(6):2329–43. doi: 10.1523/JNEUROSCI.1419-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, et al. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28(11):2735–44. doi: 10.1523/JNEUROSCI.4443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saleh M, Nagaraju M, Porciatti V. Longitudinal evaluation of retinal ganglion cell function and IOP in the DBA/2 J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2007;48(10):4564–72. doi: 10.1167/iovs.07-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dowling JE, Werblin FS. Synaptic organization of the vertebrate retina. Vision Res. 1971; Suppl 3:1-15 [DOI] [PubMed]
- 72.Porciatti V. The mouse pattern electroretinogram. Doc Ophthalmol. 2007;115(3):145–53. doi: 10.1007/s10633-007-9059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2 J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fuchs M, Scholz M, Sendelbeck A, Atorf J, Schlegel C, Enz R, et al. Rod photoreceptor ribbon synapses in DBA/2 J mice show progressive age-related structural changes. PLoS One. 2012;7(9) doi: 10.1371/journal.pone.0044645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tenner AJ, Fonseca MI. The double-edged flower: roles of complement protein C1q in neurodegenerative diseases. Adv Exp Med Biol. 2006;586:153–76. doi: 10.1007/0-387-34134-X_11. [DOI] [PubMed] [Google Scholar]
- 76.Gasque P, Neal JW, Singhrao SK, McGreal EP, Dean YD, Van BJ, et al. Roles of the complement system in human neurodegenerative disorders: pro-inflammatory and tissue remodeling activities. Mol Neurobiol. 2002;25(1):1–17. doi: 10.1385/MN:25:1:001. [DOI] [PubMed] [Google Scholar]
- 77.Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 2001;35(1):72–9. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 78.Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R, et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett. 2001;305(3):165–8. doi: 10.1016/S0304-3940(01)01842-0. [DOI] [PubMed] [Google Scholar]
- 79.Shen Y, Meri S. Yin and Yang: complement activation and regulation in Alzheimer’s disease. Prog Neurobiol. 2003;70(6):463–72. doi: 10.1016/j.pneurobio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 80.Shen X, Ying H, Qiu Y, Park JS, Shyam R, Chi ZL, et al. Processing of optineurin in neuronal cells. J Biol Chem. 2011;286(5):3618–29. doi: 10.1074/jbc.M110.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Strohmeyer R, Shen Y, Rogers J. Detection of complement alternative pathway mRNA and proteins in the Alzheimer’s disease brain. Brain Res Mol Brain Res. 2000;81(1-2):7–18. doi: 10.1016/S0169-328X(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 82.Tooyama I, Sato H, Yasuhara O, Kimura H, Konishi Y, Shen Y, et al. Correlation of the expression level of C1q mRNA and the number of C1q-positive plaques in the Alzheimer Disease temporal cortex. analysis of C1q mrna and its protein using adjacent or nearby sections. Dement Geriatr Cogn Disord. 2001;12(4):237–42. doi: 10.1159/000051265. [DOI] [PubMed] [Google Scholar]
- 83.Schafer DP, Stevens B. Synapse elimination during development and disease: immune molecules take centre stage. Biochem Soc Trans. 2010;38(2):476–81. doi: 10.1042/BST0380476. [DOI] [PubMed] [Google Scholar]
- 84.Naito AT, Sumida T, Nomura S, Liu ML, Higo T, Nakagawa A, et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell. 2012;149(6):1298–313. doi: 10.1016/j.cell.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]