

Summary
Prior studies suggest that the transcription factor ATF4 negatively regulates synaptic plastic and memory. By contrast, we provide evidence from direct in vitro and in vivo knockdown of ATF4 in rodent hippocampal neurons and from ATF4 null mice that implicate ATF4 as essential for normal synaptic plasticity and memory. In particular, hippocampal ATF4 down-regulation produces deficits in long-term spatial memory and behavioral flexibility without affecting associative memory or anxiety-like behavior. ATF4 knockdown or loss also causes profound impairment of both long-term potentiation (LTP) and long-term depression (LTD) as well as decreased glutamatergic function. We conclude that ATF4 is a key regulator of the physiological state necessary for neuronal plasticity and memory.
Introduction
Activating transcription factor 4 (ATF4) belongs to the ATF/cAMP response element binding protein (CREB) family (Hai and Hartman, 2001). Though originally described as a repressor of CRE-dependent transcription (Karpinski et al., 1992), ATF4 can also act as a transcriptional activator (Bouman et al., 2011).
Transcriptional regulation by ATF4 occurs through formation of homo- and heterodimers with a variety of partners via its basic leucine zipper (bZIP) domain (Ameri and Harris, 2008). ATF4 has roles in a variety of tissues. ATF4-null mice are blind due to lens dysgenesis and have severely impaired bone development (Elefteriou et al., 2006; Tanaka et al., 1998). In the mammalian nervous system, ATF4 has been implicated in synaptic plasticity and memory formation (Chen et al., 2003; Costa-Mattioli et al., 2007). Studies on Aplysia identified ApCREB2, a homolog of ATF4, as an inhibitor of CREB-dependent long-term facilitation (Bartsch et al., 1995; Lee et al., 2003b). Consistent with this, several studies in rodents either manipulating eIF2α phosphorylation (which regulates translation of ATF4 among other proteins) (Costa-Mattioli et al., 2005; Costa-Mattioli et al., 2007) or using a broad dominant-negative inhibitor of C/EBP proteins (Chen et al., 2003) led to suggestions that ATF4 negatively regulates long-term memory and synaptic plasticity. In contrast, recent studies have supported the view that ATF4 is required for object recognition memory (ILL-Raga et al., 2013), fear extinction memory (Wei et al., 2012), and memory flexibility (Trinh et al., 2012). In all these studies, interpretation is hampered by use of indirect ATF4 modulation with the potential to affect additional proteins. Thus, the precise role of ATF4 in synaptic plasticity and memory formation has yet to be determined.
To directly define ATF4’s role in neuronal plasticity, we used lentivirally-delivered shRNAs to specifically interfere with its expression in rodent hippocampal neurons, in long-term cultures and in adult animals. Where possible, we also used ATF4 null mice. We recently reported that direct ATF4 down-regulation reduces the density of dendritic mushroom spines in vitro and in vivo accompanied by a decrease in post-synaptic markers for excitatory glutamatergic synapses (Liu et al., 2014). These effects were partially mediated by reduction in levels of total and active Cdc42, a small Rho family GTPase involved in regulation of the actin cytoskeleton. Here, we extend our findings to investigate the role of ATF4 in synaptic plasticity and memory formation.
Results
ATF4 down-regulation impairs long-term spatial memory and memory flexibility without affecting associative memory and anxiety-like behavior
To assess ATF4’s role in hippocampal-dependent behavior and synaptic plasticity, we used lentivirally-delivered shRNAs to specifically down-regulate ATF4 expression in rodent neurons in vitro and in vivo. These shRNAs efficiently reduce ATF4 protein levels in cultured neurons and their actions on dendritic spines and post-synaptic markers are rescued by shRNA-resistant ATF4 expression constructs (Liu et al., 2014). We confirmed effective ATF4 knockdown by shATF4 in cultured hippocampal neurons at 2 weeks after infection (Figure 1A). The infection efficiency was ~90%. We also observed that an shRNA targeted to another sequence of rat ATF4 had no effect on ATF4 expression (Figure 1A) and used this as a control (shCTRL2) along with another control (shCTRL1) in which 5 bases of shATF4 were changed (Liu et al., 2014). To verify infection efficiency and spreading of the lentiviral particles in vivo, we injected adult mouse hippocampi with an empty lenti-GFP vector, sacrificed them 1 month later and assessed GFP expression. This revealed positive neurons in all regions of the hippocampus, particularly in the hilus of the dentate gyrus (DG) and in CA3 (Figure 1B). Immunoreactivity was also observed throughout CA1 and CA2, while no positive cells were detected in surrounding areas. The infection efficiency was approximately 60% of total cells and the vast majority of labeled cells had a neuronal morphology.
Figure 1. Hippocampal-specific ATF4 down-regulation.
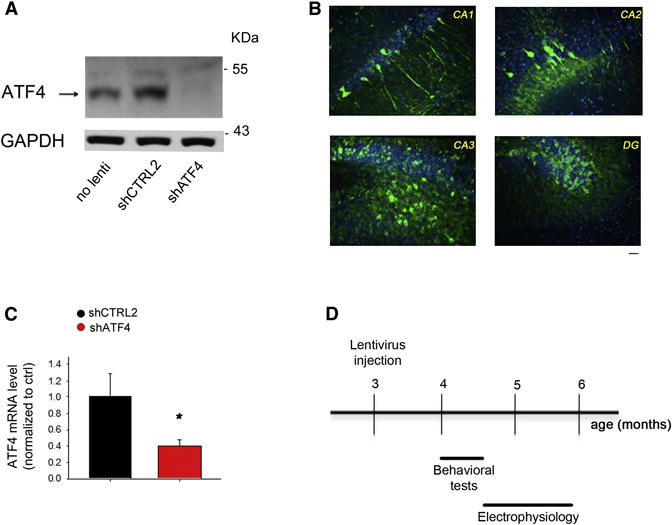
(A) Representative western blot showing ATF4 protein knockdown in primary hippocampal neuronal cultures infected at 5 DIV for 2 weeks with shATF4-lentivirus compared to shCTRL2-infected and non-infected cultures.
(B) Distribution and expression of GFP in mouse hippocampus 1 month after infection with GFP-expressing lentivirus. Sections of lentivirus-injected animals were immunolabeled with antibody against GFP (green) and counterstained with Topro3 (nuclei, blue). Scale bar 20μm.
(C) RT-qPCR analysis of ATF4 mRNA levels in mouse hippocampus 4 weeks after lenti-shATF4 injection. Data are expressed as mean ± SEM (shCTRL2 n=7, shATF4 n=7) (*p<0.05).
(D) Scheme of experimental design: 3-month-old hippocampi of C57BL/6 wt male mice were stereotaxically inoculated either with lenti-shCTRL2, -shCTRL1, or -shATF4. 4 weeks after injection, cognitive functions of the animals were assessed with a battery of behavioral tests, at the end of which they were sacrificed for electrophysiological analyses.
We next prepared high-titre lentiviruses expressing shATF4 or shCTRL2 and bilaterally injected them into hippocampi of adult mice. ATF4 mRNA levels were assessed in the hippocampi by RT-qPCR after four weeks. Compared to shCTRL2-injected hippocampi, those receiving shATF4 showed a 60% reduction of ATF4 mRNA levels (Figure 1C). Given these results, we next evaluated the consequences of hippocampal ATF4 down-regulation on learning and memory and on the electrophysiological correlates of synaptic plasticity (Figure 1D). One month after injection of shCTRL1, shCTRL2, or shATF4 lentiviruses we tested hippocampus-dependent cognition with a battery of behavioral tests. In the standard Morris water maze task in which mice were trained to locate a hidden platform using extra-maze visual cues, we found that all three groups learned the task equally well (Figure 2A). After the last training trial, retention of long-term spatial memory was analyzed during a probe trial consisting of a 60s free swim without the platform. Analysis of the paths revealed that shCTRL1- and shCTRL2-injected mice spent much more time exploring the target quadrant (TQ) than the other three quadrants (Figure 2B). In contrast, shATF4-injected mice spent almost the same amount of time searching in each quadrant, suggesting a strong impairment of reference memory (Figure 2B).
Figure 2. ShATF4-injected mice display spatial long-term memory deficits and memory inflexibility.
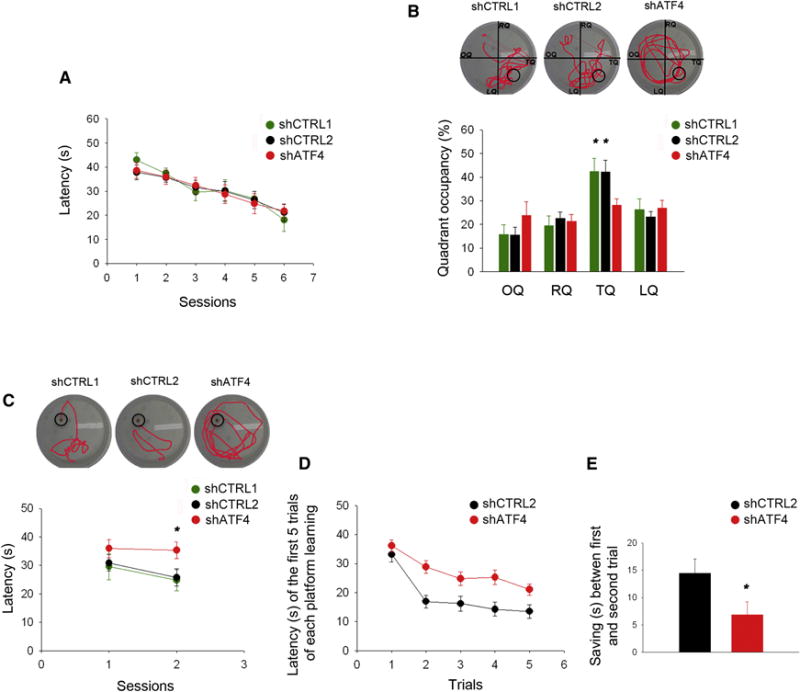
(A) Latency in the training phase of the standard Morris water maze test plotted against number of sessions. No difference in the learning capacity is detected between shCTRL1-, shCTRL2- and shATF4-injected animals (shCTRL1 n=10, shCTRL2 n=12, shATF4 n=9).
(B) Percentage of time spent in each quadrant of the pool during the probe trial (OQ, opposite quadrant; RQ, right quadrant; TQ, target quadrant; LQ, left quadrant). ShATF4-injected mice spend significantly less time exploring the target quadrant compared to the shCTRL1-, and shCTRL2-injected animals (shCTRL1 n=10, shCTRL2 n=12, shATF4 n=9).
(C) Latency in the re-training phase of the reversal-learning test is plotted against session number. ShATF4-injected mice take significantly more time to reach the new platform position compared to shCTRL1- and shCTRL2-injected animals (shCTRL1 n=10, shCTRL2 n=12, shATF4 n=9).
(D) Latency of the first 5 trials of new platform training in the Delayed Matching to Place task. Values are average latencies obtained from the last two training sessions (the third and the fourth platform locations). ShATF4-injected mice take significantly more time to learn new platform positions compared to shCTRL1- and shCTRL2-injected animals (shCTRL2 n=8, shATF4 n=13).
(E) The decrease in latency (saving time) between the first and second trial of each session in the Delayed Matching to Place task. Values are average saving times from the last two training sessions, i.e., the third and the fourth platform locations (shCTRL2 n=8, shATF4 n=13).
All data are expressed as mean ± SEM (* p<0.05).
To determine whether other forms of hippocampal-dependent spatial memory were compromised, we tested the mice for memory flexibility in a reversal-learning task. We moved the hidden platform to the opposite quadrant and trained the mice to reach it in the same manner as in the standard Morris water maze. Unlike the initial training period (Figure 2A), on the second day of reversal training, we saw a significant decrease in the learning capability of shATF4-injected animals compared to controls (Figure 2C). While control animals showed a decreased latency in finding the new platform position, ATF4 knockdown mice did not, pointing to ATF4 involvement in hippocampus-dependent memory flexibility.
The behavioral flexibility deficits of shATF4-injected animals, were confirmed using the Delayed Matching-to-Place (DMP) task, which measures the ability to learn a new platform location in the water maze based on a small number of trials (Zeng et al., 2001). shCTRL2-injected animals reduced their latency of finding the new platform position appreciably faster than the shATF4-injected mice (Figure 2D). This difference was mainly due to the difference in the latency between the first and second trials of the last two platform positions (Figure 2E).
To rule out the possibilities that spatial memory deficits in ATF4 down-regulated mice were due to differences in vision, motivation or swimming ability, animals were tested in a visible platform task. No differences in latency or speed were observed between the three groups in tests performed at the end of the reversal learning tasks (Figures S1A,B), and at the beginning of the DMP task (Figures S1C,D).
ATF4 down-regulated animals did not differ from controls in a fear conditioning paradigm in which mice learn to predict an aversive event (foot shock) by associating it with a conditioning stimulus (context or sound) 24 h after training (Figure S1E), suggesting that ATF4 is not involved in formation of associative memory (Contarino et al., 2002). As an internal control, we tested the animals in the 48 h cued fear conditioning paradigm, an amygdala-dependent task. Again, no differences were found (Figure S1F).
To determine whether alterations in cognition in ATF4 down-regulated mice are due to or accompanied by alterations in anxiety-like behavior, we tested them in the open field task (Campos et al., 2014). This revealed that all three groups (controls and ATF4 knockdown) spent similar times in the central area of the enclosure (Figure S1G) with the same number of entries into this zone (Figure S1H). Anxiety was further assessed using the elevated plus maze test (Figures S1I,J,K,L). No differences were seen in the time spent in the open and closed arms (Figure S1I), in number of entries into the open and closed arms (Figure S1J), or in the speed and transit time in the maze (Figures S1K,L). Taken together, our behavioral studies indicate that ATF4 is required for formation of spatial long-term memory and behavioral flexibility, but not associative memory or anxiety-like behavior.
ATF4 is required for LTP and LTD at CA3-CA1 synapses
LTP and LTD are considered critical components of hippocampal synaptic plasticity involved in learning and memory (Shors and Matzel, 1997). We compared these phenomena in acute hippocampal slices from 4–5-month-old mice that had received either shATF4 or shCTRL2. Similar studies were carried out with hippocampal slices from 2-month-old ATF4 null mice and aged-matched controls. We first assessed basal synaptic transmission by measuring the input/output (I-O) relationship and PPF at different inter-stimulus intervals. We found no differences in the I-O and PPF curves of ATF4 down-regulated (Figures S2A,B,C) and ATF4 null mice (Figures S2D,E,F) compared with their respective controls.
The magnitude of LTP in the Schaffer collateral-CA1 pathway elicited by either a strong stimulation protocol (3 theta-bursts, 15s intervals; Figure 3A) or a weaker one (100 Hz for 1s, Figure 3C) was significantly reduced in ATF4 down-regulated mice compared to controls. Similar differences were observed when we compared LTP in 2-month-old ATF4 null and WT mice (Figures 3B,D).
Figure 3. ATF4 is required for LTP and LTD induction at CA3-CA1 synapses.
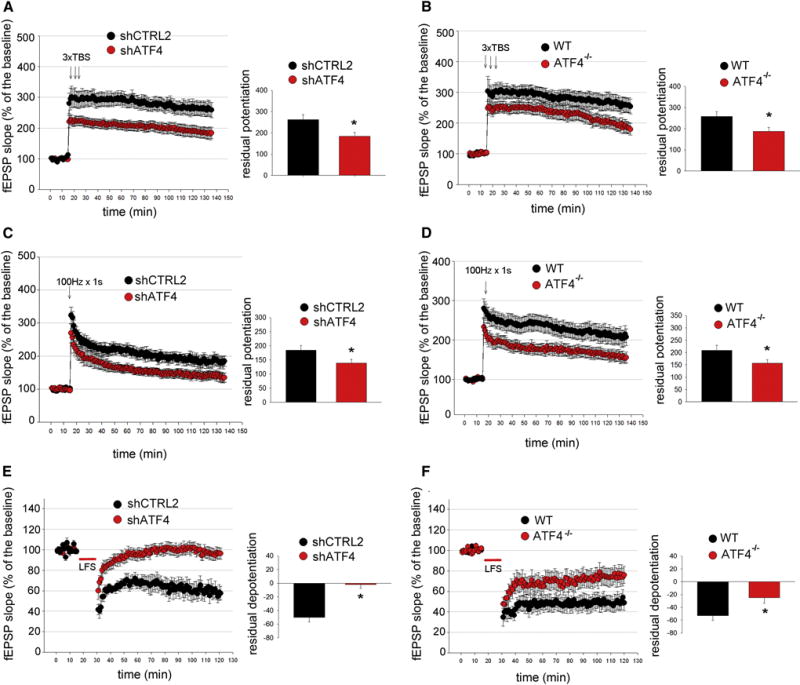
(A and B) LTP induction elicited by 3 TBS is significantly reduced in hippocampal slices obtained from animals receiving shATF4 (A, shCTRL2 n=12, shATF4 n=16) and from ATF4 KO mice (B, wt n=9, KO n=9). Bar graphs indicate the average of the last 15 min of recordings.
(C and D) LTP induction elicited by 100 Hz for 1 s is significantly decreased in hippocampal slices from animals injected with shATF4 (C, shCTRL2 n=8, shATF4 n=9) and from ATF4 KO mice (D, wt n=12, KO n=12).
***(E and F) LTD induction elicited by a 15 min, 1 Hz stimulus train is decreased in hippocampal slices obtained from animals injected with shATF4 (E, shCTRL2 n=6, shATF4 n=8) and from ATF4 KO mice (F, wt n=10, KO n=10).
All fEPSPs are expressed as mean ± SEM (* p<0.05).
Because behavioral flexibility deficits have been associated with LTD deregulation (Nicholls et al., 2008), we examined ATF4’s role in LTD in the same hippocampal pathway. In slices from animals treated with control shRNA, low-frequency stimulation (1Hz/15 min) of CA3 neurons induced sustained depression of synaptic strength in CA1 neurons (Figure 3E). In contrast, this response was almost fully blocked in slices from ATF4-silenced animals (Figure 3E). LTD in acute hippocampal slices from ATF4 null mice was also decreased compared to age-matched WT animals (Figure 3F). Together these data strongly support the idea that ATF4 is required for normal LTP and LTD induction at CA3-CA1 synapses.
ATF4 knockdown reduces AMPAR-mediated mEPSCs
Glutamatergic neurotransmission is a key component of synaptic plasticity primarily mediated by AMPA and NMDA receptors (Malenka and Bear, 2004). ATF4 knockdown in cultured hippocampal neurons did not alter total protein levels of AMPA receptor subunits GluR1 and GluR2 (Figures S3A,B) or of NMDA receptors subunits NMDAR1, R2A, and R2B (Figures S3C,D,E). We have reported that shATF4 reduces the density of post-synaptic markers (GluR1 and PSD95 puncta) and of dendritic mushroom spines in cultured neurons and mushroom spine density in mouse hippocampus (Liu et al., 2014). These changes could result in decreased glutamatergic synapse function. To test this, we performed whole-cell patch-clamp to record AMPA receptor (AMPAR)-mediated miniature EPSCs (mEPSCs) independently of action potentials. Both the amplitude and frequency of mEPSCs were significantly reduced (20% and 40%, respectively) in ATF4 down-regulated cultured hippocampal neurons compared to controls (Figures 4A,B,C). To confirm that these results were not due to off-target effects, we performed a rescue experiment in which the neurons were co-infected with lentiviruses expressing shATF4 and an ATF4 transcript (ATF4add) conservatively mutated to render it unresponsive to shATF4 (Liu et al., 2014). This results in knockdown of endogenous and over-expression of exogenous ATF4 (Figure 4D). Adding back ATF4 restored both the frequency and amplitude of mEPSCs to control values (Figures 4A,B,C). While ATF4add over-expression rescued the effects of shATF4 on densities of mushroom spines and PSD-95 puncta, ATF4add or ATF4 over-expression itself did not affect these properties (Liu et al., 2014). The present data also indicate that ATF4add over-expression doesn’t increase mEPSC frequency or amplitude beyond that in control cultures (Figures 4A,B,C).
Figure 4. ATF4 knockdown reduces AMPAR-mediated mEPSCs and this is rescued by transcriptionally active, but not by transcriptionally inactive, shRNA-resistant ATF4.
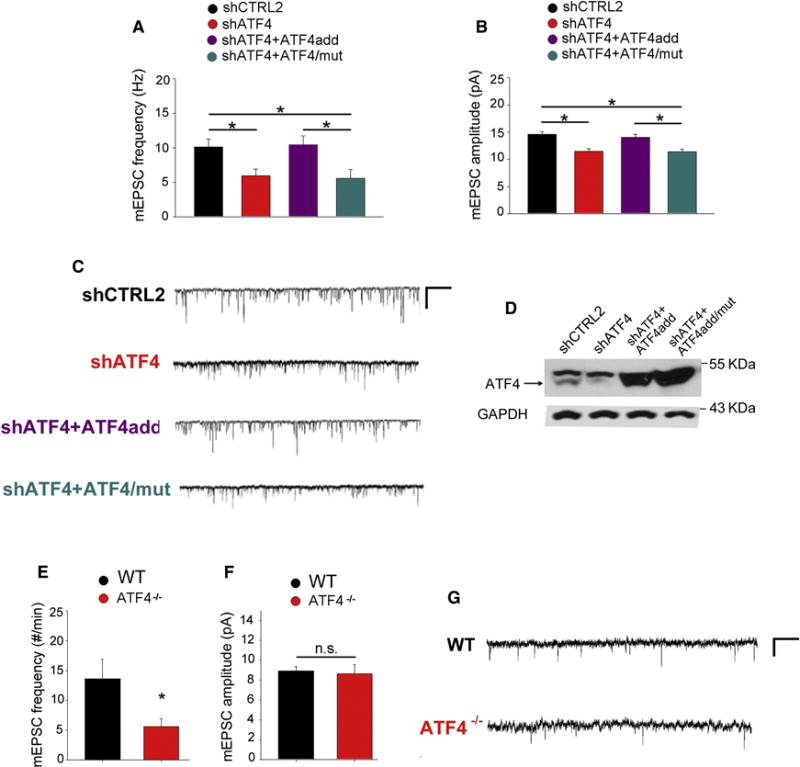
(A and B) Summary bar graphs of the frequency (A) and the amplitude (B) of mEPSCs recorded from cultured rat hippocampal neurons infected with lentivirus expressing either shCTRL2 (n=56, black bars), shATF4 (n=54, red bars), shATF4+ATF4add (n=38, cyan bars), or shATF4+ATF4/mut (n=30, purple bars).
(C) Sample traces of recordings shown in A and B.
(D) Representative western blot of primary hippocampal neurons infected at 5 DIV for 2 weeks with indicated lentiviral constructs.
(E and F) Summary bar graphs of the frequency (E) and the amplitude (F) of mEPSCs recorded from pyramidal neurons in hippocampal slices from 1-month-old wt (n=15, black bars) and ATF4 null mice (n=11, red bars).
(G) Sample traces of recordings shown in E and F.
Bars represent mean ± SEM (* p<0.05).
To test whether the effects of ATF4 on mEPSCs requires its transcriptional activity we co-infected cultured hippocampal neurons with shATF4 and a mutant ATF4 transcript (ATF4add/mut) that is not recognized by shATF4 and that encodes a mutant ATF4 that does not bind DNA (Liu et al., 2014). This results in knockdown of endogenous ATF4 and over-expression of transcriptionally inactive exogenous ATF4 (Figure 4D). Whole-cell patch-clamp recordings revealed that ATF4add/mut, unlike ATF4add, did not reverse the effect of ATF4 knockdown on frequency and amplitude of mEPSCs (Figures 4A,B,C), suggesting that the transcriptional activity of ATF4 is required.
When we recorded mEPSCs in CA1 pyramidal neurons in acute hippocampal slices from 1-month-old ATF4 null mice (Figures 4E,F,G) there was a robust decrease in mEPSC frequency but no significant change in mEPSC mean amplitude. The reasons for the difference in amplitude effect between the knockdown and knockout animals are unclear, but could reflect compensatory changes in the null animals or differences in the preparations (i.e. dissociated cultured rat hippocampal neurons vs acute hippocampal slices from ATF4 KO mice). Irrespective of these differences, our results strongly suggest that maintenance of glutamatergic synapse functionality requires ATF4 and its transcriptional activity.
Discussion
Our data show that ATF4 down-regulation in hippocampal neurons leads to deficits in long-term spatial memory and memory flexibility. ATF4 knockdown or deletion also leads to profound impairment in induction of both forms of synaptic plasticity, LTP and LTD. In addition, whole-cell patch-clamp recording revealed that ATF4 silencing or knockout significantly reduces function of glutamatergic synapses. ATF4 null mice were also used to the extent possible to confirm the knockdown results. However, because ATF4 KO causes visual and skeletal problems (Tanaka et al., 1998) ATF4 null mice could not be tested in hippocampal-dependent behavioral tasks.
LTP and LTD at CA3-CA1 synapses are critical components of synaptic plasticity hypothesized to underlie memory formation (Disterhoft and De Jonge, 1987; Maren and Baudry, 1995; Shors and Matzel, 1997). Our data show that long-term hippocampal ATF4 down-regulation leads to impairments in LTP and LTD and to spatial memory and memory flexibility deficits. The mechanisms underlying these effects may relate to the significant decrease in mushroom spine density seen after ATF4 knockdown (Liu et al., 2014). Mushroom spines are considered the most active spines and the substrate upon which memory is based. (Arellano et al., 2007; Schikorski and Stevens, 1999; von Bohlen Und Halbach, 2009). At the cellular level, modification of spine number and shape leads to functional changes at synapses (Bourne and Harris, 2007; Matus, 1999; Star et al., 2002). Our prior study showed that shATF4-mediated reduction in mushroom spine density correlates with a parallel reduction in post-synaptic markers for excitatory glutamatergic synapses (PSD95 and GluR1 puncta) (Liu et al., 2014).
In agreement with the hypothesis that shATF4-mediated reduction in mushroom spine density and excitatory synapses is reflected in impairment of glutamatergic neurotransmission, we found significant reduction in the frequency and amplitude of AMPAR-mediated mEPSCs in ATF4-downregulated cultured hippocampal neurons and in mEPSC frequency in hippocampal neurons from ATF4 null mice. We also observed that maintenance of spontaneous glutamatergic synapse activity requires transcriptionally competent ATF4 and that ATF4 over-expression does not elevate these parameters beyond baseline values.
Glutamatergic transmission is crucial for both hippocampal LTP and LTD (Lee et al., 2003a). Our findings suggest that reduction/loss of ATF4 in hippocampal neurons causes decreased mushroom spine density, diminished excitatory synapse numbers and reduced glutamatergic transmission which in turn leads to impaired synaptic plasticity. By what mechanism(s) does ATF4 knockdown cause these changes? Our prior data show that ATF4 knockdown leads to increased turnover of the actin-regulatory protein Cdc42 and to a decrease in neuronal levels of both total and activated Cdc42 (Liu et al., 2014). Knockdown of Cdc42 qualitatively mimicked the effects on ATF4 knockdown on densities of mushroom spines and PSD95 puncta. It thus appears that the effects of ATF4 down-regulation/loss on synaptic plasticity and memory are at least in part mediated by reduction in total and activated Cdc42 levels (see Graphical Abstract). This is consistent with the report that conditional reduction of Cdc42 in mouse forebrain excitatory neurons led to reduced spine density, impaired LTP induction, and defective remote memory recall with no effect on anxiety-like behavior or contextual memory (Kim et al., 2014).
In addition to affecting spatial memory, we found that ATF4 down-regulation leads to significant impairment of behavioral flexibility and a complete block of LTD. The association between LTD and behavioral flexibility is supported by findings that mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility (Nicholls et al., 2008; Zeng et al., 2001), while mice with enhanced LTD show improvement in spatial reversal learning (Duffy et al., 2008). We found that LTD in ATF4 null mice was also significantly impaired, although less than in ATF4 knockdown animals. This difference, most likely due to compensatory mechanisms, raises the question of whether ATF4 null mice would show behavioral inflexibility, a question that cannot be directly addressed due to their blindness.
We did not see effects of ATF4 knockdown on 24 hour contextual fear conditioning, a hippocampal-dependent paradigm (Phillips and LeDoux, 1992). This suggests either that ATF4 is not involved in formation of associative memory or, since this task involves a fear component, that other brain regions not targeted by the lentivirus, such as the amygdala, compensate for the hippocampal deficit in ATF4 expression.
ApCREB2, the Aplysia homolog of ATF4, was described as an inhibitor of CREB-dependent long-term facilitation (Bartsch et al., 1995; Lee et al., 2003b) and transgenic mice expressing a broad-spectrum dominant-negative inhibitor of C/EBP proteins in excitatory forebrain neurons show reduced ATF4 expression, increased induction of synaptic potentiation, enhanced memory and impaired LTD (Chen et al., 2003). Interpretation of these findings is confounded by possible interaction of the inhibitor with other members of the C/EBP family.
Phosphorylation of the translation factor eIF2α decreases overall protein translation and selectively promotes ATF4 translation (Lu et al., 2004). Several studies examined memory and synaptic plasticity after modifying eIF2α phosphorylation in rodent brain (Costa-Mattioli et al., 2005; Costa-Mattioli et al., 2007). Knockout of eIF2α kinase GCN2 or knock-in of phosphorylation-resistant eIF2α decreased hippocampal ATF4 by 40–50% and it was suggested that the observed changes in synaptic plasticity and memory were due to altered ATF4 levels. Our results, based on direct regulation of ATF4 differ from the eIF2α results in the consistent observation in both weak and strong protocols that ATF4 knockdown/deletion diminished initial LTP induction and had no evident effect on late phase LTP. In contrast, there were effects on late phase LTP when eIF2α phosphorylation was modulated and GCN2 null mice showed increased LTP induction in the weak protocol. There was also no effect of GCN2 deletion on LTD. Furthermore, while ATF4 knockdown impairs spatial long-term memory, but not learning, GCN2 knockout or treatment with eIF2α phosphatase inhibitor Sal003 (which elevates ATF4) diminished both learning and memory whereas knock-in of phosphorylation-resistant eIF2α increased both learning and memory. These differences suggest that the effects of regulating eIF2α phosphorylation on synaptic plasticity and memory may be due to factors other than or in addition to modulation of ATF4 expression. Elevation of eIF2α phosphorylation diminishes overall cap-dependent protein translation (Holcik and Sonenberg, 2005) and increases translation of transcripts in addition to those encoding ATF4 (Scheuner et al., 2001).
There is a considerable body of work suggesting a positive role for ATF4 in memory formation, specifically in consolidation of object recognition memory (ILL-Raga et al., 2013), formation of fear extinction memory (Wei et al., 2012), and memory flexibility (Trinh et al., 2012). These studies target pathways, such as PERK and HRI kinases. Additionally, a recent study on Aplysia sensorimotor long-term facilitation (LTF) provided evidence that LTF in this system requires increased post-synaptic ApCREB2 expression/activity (Hu et al., 2015). Interestingly, elevating ApCREB2 post-synaptically increased synaptic strength while doing this pre-synaptically decreased synaptic strength.
Implicit in our findings is that the effects we describe are due to pre-existing changes caused by long-term reduction/loss of ATF4 expression. That is, ATF4 permissively regulates the basal machinery required for synaptic plasticity rather than directly mediating plasticity events. Consistent with this idea, both ATF4 down-regulated and ATF4 KO mice exhibited a significant drop in the induction of both hippocampal LTP and LTD without evident changes in the late phases of these events. It remains to be seen whether dynamic changes in ATF4 expression occur during vertebrate learning and memory formation and their possible role therein.
In conclusion, we find that ATF4 plays an essential role in hippocampal-dependent long-term spatial memory and behavioral flexibility as well as in LTP, LTD and glutamatergic synapse function. Combining these data with our previously published ones (Liu et al., 2014), we present a model in which dysregulation of Cdc42 levels in the absence of ATF4 causes reductions in the densities of dendritic mushroom spines and post-synaptic GluR1 puncta, which in turn consequently produce a drop in glutamatergic synaptic functionality, eventually leading to deficits in synaptic plasticity and memory (see Graphic Abstract).
Experimental Procedures
Mice
ATF4 KO mice were purchased from Jackson Laboratory and the colony was expanded in the Columbia University Animal Facility. Wild-type (wt) C57BL/6 male mice were purchased from Jackson Laboratory. All the animals were maintained on a 12h light/dark schedule and allowed ad libitum access to food and water. All the experiments were conducted during the light phase and performed blind to the group of subjects.
Surgical procedures
3-m.o C57BL/6 male mice were deeply anesthetized with ketamine-xylazine, placed in a stereotactic apparatus and injected bilaterally in the hippocampus with 2μl of viral preparation (titers between 107 and 109 IU/μl) (− 2.45 mm, +/− 1.8 mm from Bregma, and − 2 mm from the outer surface of the skull) with a 31G needle attached to a 50 μl Hamilton syringe, at a rate of 0.5 μl/min over 4 mins.
Behavioral tests
Detailed behavioral procedures are described in the Supplemental Experimental Procedures.
DNA constructs
Lentiviral constructs were produced as previously described (Liu et al., 2014). The shRNAs were cloned in pLVTHM vectors while ATF4add and ATF4add/mut constructs were cloned in the pWPI vector (Addgene). Lenti-ATF4add was generated by introducing point mutations into the recognized site of ATF4 (CCTGACTCTGCTGCTTAT to CCAGAGTCAGCTGCTTAC) using the QuickChange Site-directed Mutagenesis kit (Stratagene). These point mutations do not change amino acid coding in the sequence. Lenti-ATF4add/mut was derived from lenti-ATF4add by introducing point mutations into the DNA binding site (292RYRQKKR298 to 292GYLEAAA298).
Primary hippocampal neuronal cultures and Western blotting
Detailed procedures are described in the Supplemental Experimental Procedures.
RT-qPCR
Total RNA was extracted from mouse hippocampi 1 month after lentiviral infection according to the RNeasy Mini Protocol (Quiagen). mRNA was then reverse-transcribed into cDNA using the 1st Strand cDNA Synthesis System for quantitative RT-qPCR (Origene) following the protocol instructions.
Electrophysiology
Detailed electrophysiological procedures are described in the Supplemental Experimental Procedures.
Statistical analysis
Data are shown as means ± SEMs. For in vivo experiments comparison between two groups was performed with a two-tailed unpaired Student’s t-test, and for in vitro experiments with a two-tailed paired Student’s t-test. Comparison between multiple groups and comparison of curves was performed using two-way ANOVA, followed by Bonferroni post hoc test when applicable. Statistical significance was set for p<0.05.
Supplementary Material
Acknowledgments
We thank Drs. O Arancio and M Fa for discussions of behavioral and electrophysiological aspects of this work, Dr. F Saeed and A Staniszewski for help with rodent surgery and behavior, respectively, C Shu for her help in mouse maintenance and M. Grasso for Graphical abstract. This work was partially supported by AG08702 (MLS), the Henry and Marilyn Taub Foundation (MLS), The Parkinson’s Disease Foundation (LAG), and Udall Center P50 NS38370 (LAG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contribution
S.P. and C.C. contributed equally in designing and conducting the experiments and performing data analysis. S.P. conducted stereotactic injections, histology and behavioral experiments and C.C. conducted the electrophysiological experiments. J.L. provided DNA constructs and part of neuron cultures. L.A.G. and M.L.S conceived the project and directed its overall execution. All authors participated in writing and editing of the manuscript.
References
- Arellano JI, Benavides-Piccione R, Defelipe J, Yuste R. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Frontiers in neuroscience. 2007;1:131–143. doi: 10.3389/neuro.01.1.1.010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell death and differentiation. 2011;18:769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Current opinion in neurobiology. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Campos AC, Vaz GN, Saito VM, Teixeira AL. Further evidence for the role of interferon-gamma on anxiety- and depressive-like behaviors: Involvement of hippocampal neurogenesis and NGF production. Neuroscience letters. 2014;578:100–105. doi: 10.1016/j.neulet.2014.06.039. [DOI] [PubMed] [Google Scholar]
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I, et al. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron. 2003;39:655–669. doi: 10.1016/s0896-6273(03)00501-4. [DOI] [PubMed] [Google Scholar]
- Contarino A, Baca L, Kennelly A, Gold LH. Automated assessment of conditioning parameters for context and cued fear in mice. Learn Mem. 2002;9:89–96. doi: 10.1101/lm.43002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, De Jonge M. Associative learning and long-term potentiation: cellular mechanisms compared. International journal of neurology. 1987:21–22. [PubMed] [Google Scholar]
- Duffy S, Labrie V, Roder JC. D-serine augments NMDA-NR2B receptor-dependent hippocampal long-term depression and spatial reversal learning. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2008;33:1004–1018. doi: 10.1038/sj.npp.1301486. [DOI] [PubMed] [Google Scholar]
- Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, Karsenty G. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell metabolism. 2006;4:441–451. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- G IR, Kohler C, Radiske A, Lima RH, Rosen MD, Munoz FJ, Cammarota M. Consolidation of object recognition memory requires HRI kinase-dependent phosphorylation of eIF2alpha in the hippocampus. Hippocampus. 2013;23:431–436. doi: 10.1002/hipo.22113. [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nature reviews Molecular cell biology. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Hu JY, Levine A, Sung YJ, Schacher S. cJun and CREB2 in the Postsynaptic Neuron Contribute to Persistent Long-Term Facilitation at a Behaviorally Relevant Synapse. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:386–395. doi: 10.1523/JNEUROSCI.3284-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinski BA, Morle GD, Huggenvik J, Uhler MD, Leiden JM. Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4820–4824. doi: 10.1073/pnas.89.11.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IH, Wang H, Soderling SH, Yasuda R. Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. eLife. 2014:e02839. doi: 10.7554/eLife.02839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003a;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee JA, Kim H, Lee YS, Kaang BK. Overexpression and RNA interference of Ap-cyclic AMP-response element binding protein-2, a repressor of long-term facilitation, in Aplysia kurodai sensory-to-motor synapses. Neuroscience letters. 2003b;337:9–12. doi: 10.1016/s0304-3940(02)01285-5. [DOI] [PubMed] [Google Scholar]
- Liu J, Pasini S, Shelanski ML, Greene LA. Activating transcription factor 4 (ATF4) modulates post-synaptic development and dendritic spine morphology. Frontiers in cellular neuroscience. 2014;8:177. doi: 10.3389/fncel.2014.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. The Journal of cell biology. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Maren S, Baudry M. Properties and mechanisms of long-term synaptic plasticity in the mammalian brain: relationships to learning and memory. Neurobiology of learning and memory. 1995;63:1–18. doi: 10.1006/nlme.1995.1001. [DOI] [PubMed] [Google Scholar]
- Matus A. Postsynaptic actin and neuronal plasticity. Current opinion in neurobiology. 1999;9:561–565. doi: 10.1016/S0959-4388(99)00018-5. [DOI] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, Kandel ER. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58:104–117. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behavioral neuroscience. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative fine-structural analysis of olfactory cortical synapses. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:4107–4112. doi: 10.1073/pnas.96.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Matzel LD. Long-term potentiation: what’s learning got to do with it? The Behavioral and brain sciences. 1997;20:597–614. doi: 10.1017/s0140525x97001593. discussion 614–555. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nature neuroscience. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes to cells : devoted to molecular & cellular mechanisms. 1998;3:801–810. doi: 10.1046/j.1365-2443.1998.00230.x. [DOI] [PubMed] [Google Scholar]
- Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E. Brain-specific disruption of the eIF2alpha kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell reports. 2012;1:676–688. doi: 10.1016/j.celrep.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bohlen Und Halbach O. Structure and function of dendritic spines within the hippocampus. Annals of anatomy = Anatomischer Anzeiger : official organ of the Anatomische Gesellschaft. 2009;191:518–531. doi: 10.1016/j.aanat.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, Meyers D, Mukherjee C, Sbardella G, Castellano S, et al. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:11930–11941. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.