

Abstract
Amphibians have been affected globally by the disease chytridiomycosis, caused by the fungus Batrachochytrium dendrobatidis (Bd), and we are just now beginning to understand how immunogenetic variability contributes to disease susceptibility. Lineages of an expressed major histocompatibility complex (MHC) class II locus involved in acquired immunity are associated with chytridiomycosis susceptibility in controlled laboratory challenge assays. Here, we extend these findings to natural populations that vary both in exposure and response to Bd. We find that MHC alleles and supertypes associated with Bd survival in the field show a molecular signal of positive selection, while those associated with susceptibility do not, supporting the hypothesis that heritable Bd tolerance is rapidly evolving. We compare MHC supertypes to neutral loci to demonstrate where selection versus demography is shaping MHC variability. One population with Bd tolerance in nature shows a significant signal of directional selection for the same allele (allele Q) that was significantly associated with survival in an earlier laboratory study. Our findings indicate that selective pressure for Bd survival drives rapid immunogenetic adaptation in some natural populations, despite differences in environment and demography. Our field-based analysis of immunogenetic variation confirms that natural amphibian populations have the evolutionary potential to adapt to chytridiomycosis.
Keywords: major histocompatibility complex, amphibian, chytridiomycosis, Batrachochytrium dendrobatidis, immunogenetics, adaptation
1. Introduction
Emerging infectious diseases are key threats to wildlife populations [1] that often have complex, varied, or uncertain causes [2]. Fungal diseases in particular are on the rise [3], thus understanding the mechanisms of immune system response to fungal pathogens is of particular importance for predicting whether wildlife populations can adapt to novel infections. The amphibian disease chytridiomycosis, caused by the fungus Batrachochytrium dendrobatidis (Bd), has resulted in the decline or extinction of hundreds of species worldwide [3–5]. Amphibian species demonstrate a wide range of responses to chytridiomycosis [6] that are largely driven by environment, ecology, and life history [7–10]. Controlled laboratory experiments show that host immunological responses also contribute to Bd resistance [11–15], but it has proved difficult to quantify differences in susceptibility among species or natural populations because of the confounding effects of environment, pathogen dynamics, and host demographic factors contributing to disease [16–18]. Thus, the potential evolution of host resistance in response to this emergent disease remains largely unexplored in natural populations.
Amphibian immune systems are structurally and functionally similar to other vertebrates in possessing innate and acquired immune pathways [19]. One important host immune component contributing to Bd responses is the major histocompatibility complex (MHC), a family of immune-related genes conserved across vertebrates [20]. Class I and II MHC molecules bind pathogen molecules on their peptide-binding regions (PBRs) and present them to T-cells to initiate an acquired immune response [21]. This central role in initiating immunity creates strong selection on MHC loci for numerous polymorphisms and gene copies, thereby maximizing the array of pathogens that can be recognized [22,23]. Class II MHC genes are expressed on immune surveillance cells in amphibian skin [19,24] and typically recognize bacterial and fungal pathogens, whereas class I molecules are involved primarily in viral immunity and self-discrimination [25]. Class II loci are, therefore, ideal targets for the study of immunogenetic responses to chytridiomycosis, a fungal disease that infects amphibian epidermal cells [3].
Natural wildlife populations show correlations between MHC polymorphism and disease susceptibility [22]. Four non-exclusive evolutionary mechanisms potentially explain MHC allele distributions after pathogen-imposed selection in populations. First, overdominance may arise if MHC heterozygotes are able to bind a wider inventory of antigens [26], resulting in higher fitness compared with homozygotes [27]. Second, directional selection may occur if a specific allelic lineage that confers resistance to a common pathogen increases in frequency over successive generations [28,29]. Third, frequency-dependent selection may occur when pathogens become adapted to the most common host genotype and rare MHC alleles confer a selective advantage until they become common [30–32]. Finally, diversifying selection for numerous resistance-conferring alleles within a spatially heterogeneous selective landscape [33] may cause balanced MHC polymorphism, a pattern that is indistinguishable from frequency-dependent selection [22,34]. Each of these mechanisms have probably shaped MHC diversity over the history of natural populations; thus, teasing apart the specific immunogenetic consequences of Bd-imposed selection will require multiple lines of evidence and the ability to distinguish historical versus recent selective events.
In anurans, the MHC genomic region has been characterized in two model species, Xenopus laevis [35,36] and Silurana tropicalis [37]. Both species have the ancestral tetrapod MHC gene organization [38,39] and diverged early in the anuran phylogeny [40]. Experimental studies in Silurana find that under some conditions, Bd infection activates innate immune defences [41] or minimal immune responses [42], while under other conditions, acquired immunity is induced [14]. Interestingly, the Bd-susceptible species Rana muscosa and R. sierrae, are similar to Silurana in that they show no evidence of a robust immune response [42]. By contrast, the highly susceptible Atelopus zeteki mounts both innate and acquired immune defences against Bd in challenge experiments, but these efforts are not protective and previous Bd exposure does not increase survival [43]. In other species, exposure to Bd increases subsequent immunity; previous Bd exposure in Osteopilus septentrionalis decreased pathogen burden and increased lymphocyte proliferation and survival [14]. Bd also potentially suppresses effective acquired immune responses. Anuran T- and B-cells are killed by Bd in vitro [13], and expression of T-cell pathway genes are suppressed in experimentally Bd infected individuals compared with controls in four frog species [44]. Uncertainty thus remains over the necessary immune system components, antigenic targets, and particularly the gene by environment interactions that lead to an effective immune response against Bd.
Variation in MHC genes has been characterized in natural amphibian populations that differ in susceptibility to non-fungal pathogens [28,45,46] as well as Bd [12,15]. In Bufo calamita, class II genotype frequencies varied in a pattern consistent with directional selection in response to pathogen prevalence among populations [12], and a comparison of class II diversity across nine amphibian genera with Bd susceptibility data found that more resistant species and populations have common amino acids in peptide-binding pockets [15]. Combined, these studies indicate a functional role for MHC genes in natural chytridiomycosis dynamics.
Lithobates yavapaiensis is a North American frog that has declined due to seasonal chytridiomycosis outbreaks since at least 1990 [12,47]. Our earlier experimental Bd infections of laboratory-reared L. yavapaiensis from five natural populations identified specific class II MHC genotypes that were associated with survival within and among populations [11]. Both MHC heterozygotes and individuals bearing MHC allele Q had significantly higher probabilities of surviving Bd infection [48]. Bataille et al. [15] subsequently extended these findings with experimental Bd infections of the Australian tree frog Litoria verreauxii alpina, and survival was significantly associated with MHC alleles with amino acid substitutions in the same region where we detected positive selection acting on allele Q. Here, we test the generality of our experimental infection results in natural populations, and ask whether the same signatures of selection and immunogenetic predictors of susceptibility can be found among individuals from populations that differ in their demographic responses to Bd. We characterize class II MHC in field-sampled frogs from eight populations currently infected with Bd, and interpret genetic variation at this locus in the light of our published multi-year seasonal field estimates of population and individual Bd susceptibilities [11,16]. We also compare neutral genetic markers with immunogenetic genotypes to identify significant signals of natural selection in response to chytridiomycosis. We extend the experimental finding that immunogenetic variation determines Bd susceptibility by elucidating the mechanisms of evolutionary response to disease across a variable ecological and environmental landscape, predicting the potential for evolution of resistance in natural populations.
2. Material and methods
(a). Field surveys
We surveyed eight L. yavapaiensis populations for Bd and chytridiomycosis during winter months (January–February) of 2007–2011 [11,16], the time of year when Bd mortalities occur in this species [16]. Using standardized protocols [49], we swabbed the epidermis of all individuals and used quantitative PCR to measure infection intensity (the number of Bd genome equivalents (GE) recovered per swab). We categorized each dead individual that tested positive for Bd infection as a chytridiomycosis mortality event. Additionally, we collected any individual with signs of chytridiomycosis (skin redness, lethargy, failure to seek shelter, and loss of righting ability) for overnight observation, and also categorized these as chytridiomycosis-induced mortality events if death occurred within 24 h and the individual tested positive for Bd. All eight populations were surveyed greater than or equal to two times per winter, and because L. yavapaiensis is a stream-dwelling species, inhabiting shallow flowing water and small plunge pools, the sites could be exhaustively screened for dead and dying individuals. Populations could, therefore, be definitively characterized for Bd susceptibility based on the number of frogs found dead or dying with Bd infection. By contrast, the ultimate fate of individuals sampled alive could not be determined for the five populations with mortality (i.e. they could hypothetically develop chytrydiomycosis at a later time point). Our analyses thus measure genetic correlates of Bd-susceptible frogs rather than directly assessing Bd survival. We estimated Bd and chytridiomycosis mortality prevalence and 95% Clopper–Pearson binomial confidence intervals [50] for each L. yavapaiensis population.
(b). Microsatellite genotyping
We previously genotyped 128 L. yavapaiensis individuals at 14 unlinked microsatellite loci using published protocols [16,17,51]. We used GENEPOP v. 3.4 [48] to calculate observed and expected heterozygosity and test for deviations from Hardy–Weinberg equilibrium at each locus and population locality using a Monte-Carlo chain method (1 000 dememorizations, 100 batches, 1 000 iterations) [52] with Bonferroni correction for multiple tests (adjusted p = 0.00022).
(c). Major histocompatibility complex amplification, cloning, and sequencing
We extracted genomic DNA from ethanol-preserved toe clips from the same 128 individuals that were microsatellite genotyped. The majority (108) of individuals were sampled in winter, but for populations with prohibitively small winter sample sizes (CIC, TV, and WC), we sampled 20 additional frogs collected in the summers of 2006–2007. We used a degenerate forward primer [45] and a ranid frog intron-specific reverse primer [11,46] to amplify 249 base pairs (bp) of exon 2 (which encompasses the peptide-binding region) and 189 bp of adjacent 3′-flanking intron of an expressed MHC class II locus. We used previously published protocols to amplify, clone, and sequence MHC alleles [11,46]. We screened each MHC sequence and only retained those obtained from at least two clones. For each individual, sequences recovered only once with less than or equal to two nucleotide differences to other cloned sequences were attributed to PCR/cloning errors and discarded. After excluding these sequences, no more than two unique MHC alleles were recovered from any individual. To assess the frequency of artefactual alleles arising from cloning, we also used the same MHC primers with 454 adapters added to the 5′ ends (fusion primers) to perform amplicon sequencing on a GS Junior (Roche) using two different PCR amplifications and more than 40× depth of sequencing for a subset of our sampled individuals (N = 29 frogs from five populations). We multiplexed individuals by using a suite of 8 bp unique sequence tags added to the 454 fusion primers that differed from each other in at least three positions to minimize misassignment. PCR amplifications were performed in 20 µl, including 3 µl of each primer, 3 µl of DNA, and 7.5 µl of HotStar PCR Master Mix (Qiagen). We pooled equimolar quantities of PCR products amplified with distinct fusion primers, purified DNA with a MinElute PCR Purification Kit (Qiagen) and sequenced the pool of amplicon samples from all individuals on a GS Junior run (Roche). All individuals were amplified and sequenced at least twice to ensure we did not generate any artificial alleles resulting from PCR or sequencing error. We used Newbler v. 2.6 (Roche) to remove adapter sequences and assemble MHC amplicons into contigs. All raw MHC amplicon reads were analysed with jMHC v. 1.0 to demultiplex samples and assign genotypes. Only reads that had two complete barcodes and a perfect match to the primer barcode were retained and assigned to their respective sample and repetition number, and only alleles that were found in at least two separate samples or two separate repetitions with a minimum of 15 reads were considered real. We aligned all MHC alleles using Sequencher (Gene Codes Corporation) with adjustment by eye and compared cloning-derived with 454-derived alleles across individuals.
(d). Genealogy reconstruction
We tested the MHC alignment for evidence of recombination using the single breakpoint method [53] before performing a Bayesian analysis to reconstruct genealogical relationships among alleles. We used class II exon 2 sequences from X. laevis and S. tropicalis (GenBank accession numbers NM_001114771 and NM_001045794) as outgroups. Model parameters were determined using the Akaike information criteria in jModeltest [54]. We used the best-fit model (general time reversible (GTR) model with invariable sites plus gamma (I + γ) distribution (GTR + I + G)) to estimate a 95% credible set of rooted MHC genealogies in the software MrBayes 3.1 [55]. We ran two separate analyses in MrBayes for 1 × 107 generations and sampled every 500th generation of the Markov chain. We used Tracer v. 1.4 to assess stationarity of model parameters, convergence of model parameters between runs, the number of burn-in samples, and the effective sample sizes for each parameter.
(e). Tests of selection
We ran tests of selection using HyPhy [56] with the Bayesian genealogy as our input tree, excluding outgroup sequences. We used PARRIS to test for positive selection in the entire alignment [57], evolutionary fingerprinting to infer the number of positive selection rate classes and the intensity of selection in each rate class [58], and the most conservative maximum-likelihood method (SLAC) to test for residue-specific positive selection across lineages [59].
(f). Major histocompatibility complex supertyping
To collapse MHC alleles into functional supertypes, we extracted the 13 codon positions in our MHC alignment known to affect peptide-binding capabilities of human class II alleles [15,21] and then characterized each site based on five physio-chemical descriptor variables: z1 (hydrophobicity), z2 (steric bulk), z3 (polarity), z4 and z5 (electronic effects) [60]. We used discriminant analysis of principle components to define functional genetic clusters using the adegenet 1.4-0 package in R [61], which implements a k-means clustering algorithm using the Bayesian information criterion (BIC). The optimal number of clusters was determined using ΔBIC ≤ 2, and alleles within clusters were collapsed into a single MHC supertype.
(g). Selection and genetic differentiation among populations
We used software for the measurement of genetic diversity (SMOGD) [62] to estimate D [63] across all population pairs for (i) 14-locus microsatellite genotypes, (ii) MHC exon 2 genotypes, and (iii) MHC supertypes. We also calculated observed and expected heterozygosity, nucleotide diversity (π), and theta (θ), and performed Ewens–Watterson (E-W) tests on MHC exon and intron genotypes using Arlequin v. 3.5 [64]. We used the lm function in R [65] to perform linear regression on all pairwise population measures of D from MHC supertype versus microsatellite genotypes. Significant outliers were identified as data points with both Cook's D > 4/n and leverage values > 3/n, where n is the number of observations [66].
(h). Statistical analyses
Differences in Bd infection intensity and nucleotide diversity across populations were assessed using Student's t-tests implemented in JMP software, v. 9.0 (SAS). Differences in supertype frequencies across individuals that were alive versus dead at the time of sampling were inferred using 95% Clopper–Pearson binomial confidence intervals. We used a previously published protocol [11] to calculate the relative risk (RR) for MHC supertypes and for the three alleles recovered in both the field and laboratory study.
3. Results
(a). Batrachochytrium dendrobatidis infection dynamics vary within and among populations
Lithobates yavapaiensis individuals varied within and among the eight field-sampled populations in Bd infection prevalence (figure 1a), associated mortality (figure 1b), and infection intensity (figure 1c) [16]. Three populations were Bd tolerant, with no winter mortality (HR, SM, and SS) despite high winter Bd infection prevalence and intensity. For the five populations with both laboratory and field data, mortality was consistently higher in our experimental infection study (41–100% mortality [11]) than in the field (figure 1b). Within populations, frogs that died had significantly higher infection intensities than frogs that survived (two-tailed paired Student's t-test, p = 0.046).
Figure 1.

Batrachochytrium dendrobatidis (Bd) infection dynamics and immunogenetics among eight Lithobates yavapaiensis populations sampled in winter. Asterisks represent populations with 100% mortality and circles represent populations with less than 100% mortality in previous controlled laboratory infections [11]. (a) Proportion of individuals infected with Bd, (b) proportion of individuals found dead versus alive, (c) average Bd infection intensity among Bd-positive frogs, and (d) MHC supertype frequencies. Circle sizes are proportional to sample size, mean infection intensity, and allele frequency, respectively. AC, Aravaipa Canyon; CIC, Cienega Creek; HR, Hassayampa River; MR, Muleshoe Ranch; SM, Santa Maria River; SS, Seven Springs; TV, Tanque Verde Canyon; WC, Willow Creek. (Online version in colour.)
(b). Major histocompatibility complex variation is associated with Batrachochytrium dendrobatidis susceptibility in the wild
We identified 84 unique MHC class II PBR alleles among the eight sampled populations (figure 2). Three PBR alleles were recovered at high frequency (alleles A, N, and Q) and are the same three alleles that were common in our experimental infection study [11]. Sixty-six of the 84 alleles were recovered from a single individual. Owing to the high proportion of singletons and knowledge that a subset of codon sites within exon 2 are responsible for peptide-binding capabilities of the MHC molecule [15,21], we converted alleles into four MHC supertypes based on physio-chemical binding properties (figure 1d). For the subset of individuals genotyped using both cloning and 454 amplicon sequencing, MHC alleles from 26/29 individuals (90%) were identical between methods. Of the individuals that were not identical, one individual was mismatched for one allele only, and two individuals were mismatched for both alleles (electronic supplementary material, S1). After converting alleles into four functional supertypes (electronic supplementary material, S2), none of these five mismatched alleles resulted in differences in MHC supertype assignments. Therefore, we conclude that only a small proportion of cloned alleles in this study are artefactual, and that any undetected error derived from cloning is minimal once analysed as MHC supertypes.
Figure 2.

Maximum-likelihood genealogy of 84 recovered MHC alleles from eight L. yavapaiensis populations comprising four functional MHC supertypes (ST1–ST4). Branches with significantly elevated non-synonymous substitution rates for codon 46 are shown in bold. Posterior probabilities (PP) > 70% are shown, population name abbreviations follow figure 1, and circle size is proportional to allele frequency. Dashed lines indicate spacing for presentation purposes and are not branches.
The MHC supertypes ST1 and ST4 show a phylogenetic signal, with ST1 comprising all alleles in the clade including allele A, and ST4 comprising all alleles in a clade unique to population SS (figure 2). By contrast, ST2 includes most of the clade with alleles N and Q, but ST3 and ST4 render it paraphyletic, and ST3 is distributed broadly throughout the genealogy. Only MHC ST1 was significantly associated with susceptibility (figure 3; electronic supplementary material, S3); individuals with ST1 had nearly a threefold increased risk of death (RR = 2.8, p = 0.004). Notably, allele A (the dominant allele within ST1) was also significantly associated with mortality both in our field-collected samples (RR = 3.2, p < 0.0001) and in our experimental infection study [11]. ST4, which was only present in six frogs sampled alive from population SS, showed a trend towards a reduced risk of mortality (RR = 0, p = 0.06). ST2 and ST3 were not significantly associated with susceptibility or survival, nor were ST heterozygotes, despite a significant heterozygote survival advantage in our experimental infection study [11]. Allele Q was significantly associated with survival both in our field-collected samples (RR = 0.23, p = 0.008) and in our earlier experimental infection study [11].
Figure 3.
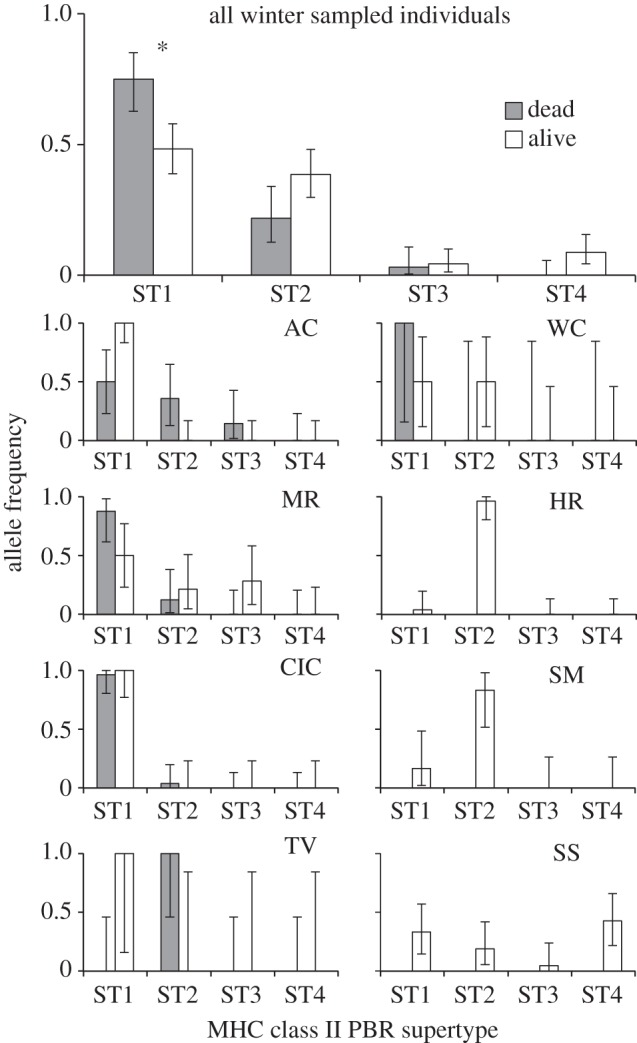
MHC supertype allele distributions for individuals sampled alive (white bars) versus dead (grey bars) across all individuals sampled in winter (upper panel) and within each population (lower panels). Error bars represent 95% binomial confidence intervals for proportions. Significantly different supertype frequencies between alive versus dead frogs are show with asterisks. Population name abbreviations follow figure 1.
(c). Positive selection drives major histocompatibility complex evolution at a peptide-binding codon
Evolutionary models that included positive selection (a ratio of non-synonymous nucleotide substitutions per non-synonymous sites (dN) to synonymous substitutions per synonymous sites (dS), or ω, > 1) fit the alignment of PBR sequences significantly better than models excluding positive selection (evolutionary fingerprinting, log likelihood = −1951.54, p < 0.001). The best-fit model included three nucleotide substitution rate classes, and found that 65% of codons experienced negative selection (ω < 1), 30% experienced moderate positive selection (ω = 1.28), and 5% experienced strong positive selection (ω = 1.95). In codon-specific tests of selection, we detected positive selection acting on one codon: position 46 of the MHC alignment (p = 0.05, normalized dN–dS = 4.08; bold branches in figure 2), which is among the 13 codon positions that determine human MHC peptide-binding [21]. Codon 46 is also the only residue where a significant signal of positive selection was detected in our experimental infection study [11].
(d). Selection and not demography drives major histocompatibility complex population differentiation
To disentangle signatures of selection from demography, we compared MHC supertype frequencies with 14 putatively neutral microsatellite loci (figure 4). The average population genetic differentiation (D) was significantly higher when measured from microsatellites (D = 0.54) compared with MHC supertypes (D = 0.39; two-tailed paired Student's t-test, p = 0.025). Population pairwise estimates of D inferred from microsatellites were not significantly correlated to those from MHC supertypes (figure 4a), indicating discordance between MHC and neutral genetic differentiation. Three comparisons between TV and other populations were identified as significant regression outliers (figure 3a, dashed circles), and removing these three data points produced a significant positive correlation between D measured from microsatellites and MHC supertypes (figure 4b).
Figure 4.

Population differentiation (D) inferred from MHC supertypes versus 14 microsatellite markers demonstrates that both selective and demographic processes shape MHC evolution. Regression of MHC and microsatellite pairwise population D values for (a) all pairwise comparisons and (b) after removing three significant outliers. Outliers are encompassed within dashed circles in a, and the comparisons they represent are shown using population abbreviations from figure 1.
Genetic diversity was significantly higher for MHC exons compared with introns when measured either as nucleotide diversity (π; two-tailed paired Student's t-test, p = 0.014) or the number of segregating sites (θ; two-tailed paired Student's t-test, p = 0.0097; electronic supplementary material, S4). We detected significant signatures of directional selection from MHC heterozygosity but not from microsatellite heterozygosity in populations CIC (20% mortality) and TV (60% mortality), indicating directional selection acts on MHC in these two populations. By contrast, AC (8% mortality), HR (0% mortality), and SM (0% mortality) had significant signatures of directional selection from both MHC and microsatellite heterozygosities, indicating recent demographic expansion rather than selection may shape contemporary MHC evolution in those populations. Neither locus type produced a significant signature of directional or balancing selection in populations SS, WC, or MR (electronic supplementary material, S4).
4. Discussion
Whether natural populations can rapidly adapt to novel pathogens remains a critically important question for evolutionary biology in an era of emerging infectious diseases. L. yavapaiensis has declined in recent decades due to chytridiomycosis outbreaks, habitat loss, and invasive species [47,67]. The seasonal selective pressure imposed by Bd each winter [16,17] means that populations unable to adapt will probably become extirpated. Here, we find that some populations may be adapting to Bd via standing MHC variation [68] that confers survival (namely, allele Q and ST4), while other populations lacking these MHC variants may not have the immunogenetic variation necessary to adapt. Positive selection was only detected along lineages leading to survival-associated or neutral alleles and supertypes (figure 2), but not the susceptibility-associated ST1. Thus, at the time of initial chytridiomycosis outbreaks, populations with standing MHC variation that included survival alleles have evolved partial (AC, 8% mortality) or complete (HR, SM, SS, no mortality) Bd tolerance. By contrast, populations with high frequencies of the susceptibility supertype ST1 (CIC, MR, TV, WC) were probably decimated by initial chytridiomycosis outbreaks and now have small population sizes, limited genetic diversity, and struggle to persist in the face of repeated bouts of Bd mortality [16,17]. MR is an exception, maintaining high MHC diversity (including allele Q) and high mortality (figure 3). However, this pattern is consistent with previous analyses of this population, which is adjacent to a thermal spring. Here, warm water protects the source population from extirpation but also prevents selection from acting to increase the frequency of survival alleles, creating an ongoing source-sink dynamic when juveniles migrate out from the thermal spring pools and develop chytridiomycosis [16,17]. Finally, in addition to selection acting on standing genetic variants, novel MHC variants providing a Bd survival advantage may have arisen in one population. SS had no observed Bd mortality and a low frequency of allele Q, but is the only population with individuals bearing ST4, a recently diversified clade with a significant signature of positive selection (figure 2). This pattern suggests that adaptation to Bd may have evolved rapidly in population SS from de novo genetic mutations, a less frequently documented phenomenon [69].
By using field data to validate a controlled experimental study, our analyses collectively show that selection caused by chytridiomycosis has contributed to the evolution of an expressed class II MHC locus in L. yavapaiensis. Our sampling from wild populations confirms three of the immunogenetic disease association patterns that were previously recovered in our experimental infection study [11]: individuals with ST1/allele A had a significantly increased risk of Bd mortality, those with allele Q had a significantly reduced risk of mortality (figure 3), and we found a significant signature of selection acting on the same peptide-interacting codon in both studies (figure 2). By contrast, here we found no evidence of heterozygote advantage when examining MHC genotypes in natural population samples, despite a strong survival advantage of heterozygotes in our experimental infection study [11]. Because we could only definitively assess mortality events, whereas Bd survivors may have occasionally been mis-identified among individuals developing chytridiomycosis exceptionally late in the season after surveys took place, MHC associations to susceptibility are stronger than associations to tolerance. Thus, an MHC-based heterozygote advantage may exist and could be uncovered with finer-scale field sampling or additional experimental infection studies. Alternately, because the previous laboratory study used full- and half-sibling clutches to represent populations, the apparent signal of overdominance may have been driven by the allelic composition of heterozygous individuals; all survivors with allele Q were heterozygotes. Indeed, experimental infection studies examining resistance to single pathogens rather than general pathogen-fighting ability find evidence for survival based on particular MHC alleles rather than heterozygotes [70].
Both directional [71] and diversifying [72] selection on MHC alleles are commonly observed evolutionary responses to pathogen pressure in natural populations. Given that Bd imposes strong selective pressure, we might therefore expect directional selection for one or a subset of MHC alleles to predominate in our system. Alternately, selection from other pathogens or different evolutionary forces such as sexual selection may contribute to MHC variation and produce an overall pattern of balancing selection. In our study, we found evidence for directional selection in two populations with high Bd mortality (CIC and TV), demographic expansion that has probably resulted from directional selection for allele Q and ST4 in two populations with infection but no Bd mortality (HR and SS), and no support for balancing selection acting to equalize frequencies of MHC supertypes in any population (figure 3). Continued population monitoring in future generations may provide direct evidence for the benefits of particular supertypes if the expected changes in Bd susceptibility occur. Among our sampled populations, AC has the highest likelihood of evolving future disease resistance, as this population currently harbours allele Q at low frequency, experiences moderate Bd mortality, and showed the highest proportion of Bd survival in previous laboratory trials [11]. Interestingly, all three population differentiation outliers involved population TV, which was undifferentiated from 0% mortality populations based on MHC supertypes but exceptionally distinct from another high mortality population CIC (figure 4). Further, frogs sampled alive from TV all had susceptibility ST1 and those found dead all had ST2, despite the overall survival disadvantage we found for ST1. MHC dynamics are therefore highly unusual in this population, highlighting that allelic advantages may be unique within populations due to eco-immunological differences in the host or the pathogen under distinct environmental regimes [73]. Excluding these three outlier comparisons involving TV, concordance between MHC- and microsatellite-based differentiation measures demonstrate that demographic processes have played a significant role in shaping MHC evolution. Recent positive selection on MHC codons and strong MHC allelic associations with Bd survival may therefore be modest drivers of overall population genetic change compared with forces such as drift and population bottlenecks in a species facing ongoing habitat loss and competition from invasive species [67].
Global declines caused by chytridiomycosis have had catastrophic consequences for amphibian diversity [4] and Bd continues to spread to new regions and hosts [74]. Emerging fungal diseases of wildlife are also on the rise, including white-nose syndrome in bats, fungal skin disease in snakes, and honeybee colony collapse disorder [1,2]. Our study highlights the importance of examining fine-scale demographic, epidemiological, and genetic patterns if we are to elucidate the key processes underlying infectious disease dynamics and evolution of resistance in free-living wildlife populations. Identifying immunogenetic correlates of chytridiomycosis outcomes provides a mechanism to explain variable host susceptibility among individuals, populations, and species. Analyses of Bd infection dynamics within and across amphibian species, life-history traits, and geographical regions [7–9], may also benefit from the incorporation of data on host genetic variation [17]. Finally, identifying immunogenetic hallmarks of Bd resistance in natural populations is a critical step towards species recovery, as the global spread and persistence of Bd means that wild populations must ultimately evolve disease resistance to achieve long-term species survival.
Supplementary Material
Acknowledgements
J.Q. Richmond, K. Kiemnec-Tyburczy, and M. Lenker provided laboratory support, D. Caldwell, M. Sredl, M. Haberstitch, R. Rogers, and M. Lawrence provided field support, T. Torres provided GIS support, K.P. Mulder and R. Fleischer at the Smithsonian's Center for Conservation and Evolutionary Genetics provided 454 MHC amplicon sequencing, and members of the Zamudio lab provided helpful discussions and comments.
Ethics
Sampling in Arizona was done under permit SP704506 issued by the Arizona Game and Fish Department. This research was conducted under Cornell University's Institutional Animal Care and Use Committee permit number 2007-0105.
Data accessibility
DNA sequences: GenBank accession numbers KU877031-KU877108. Microsatellite genotyping data for Lithobates yavapaiensis populations available from Dryad Digital Repository http://dx.doi.org/10.5061/dryad.3qk53 and also from [17].
Authors' contributions
A.E.S. and K.R.Z. wrote the manuscript. A.E.S. planned the research, executed the research, and collected field data. A.E.S. analysed the data; A.E.S. and K.R.Z. interpreted the results; A.E.S. and K.R.Z. obtained funding.
Competing interests
We have no competing interests.
Funding
This research was supported by a Population and Evolutionary Process NSF Grant (DEB-0815315) to K.R.Z., an NSF Doctoral Dissertation Improvement Grant (DEB-0909013) to A.E.S., and a National Geographic Society Young Explorer Grant to A.E.S.
References
- 1.Daszak P, Cunningham AA, Hyatt AD. 2000. Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287, 443–449. ( 10.1126/science.287.5452.443) [DOI] [PubMed] [Google Scholar]
- 2.Tompkins DM, Carver S, Jones ME, Krkošek M, Skerratt LF. 2015. Emerging infectious diseases of wildlife: a critical perspective. Trends Parasitol. 31, 149–159. ( 10.1016/j.pt.2015.01.007) [DOI] [PubMed] [Google Scholar]
- 3.Berger L, et al. 1998. Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc. Natl Acad. Sci. USA 95, 9031–9036. ( 10.1073/pnas.95.15.9031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skerratt LF, Berger L, Speare R, Cashins S, McDonald KR, Phillott AD, Hines HB, Kenyon N. 2007. Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth 4, 125–134. ( 10.1007/s10393-007-0093-5) [DOI] [Google Scholar]
- 5.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484, 186–194. ( 10.1038/nature10947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW. 2004. Status and trends of amphibian declines and extinctions worldwide. Science 306, 1783–1786. ( 10.1126/science.1103538) [DOI] [PubMed] [Google Scholar]
- 7.Vredenburg VT, Briggs CJ, Tunstall TS, Knapp RA. 2010. Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc. Natl Acad. Sci. USA 107, 9689–9694. ( 10.1073/pnas.0914111107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray KA, Rosauer D, McCallum H, Skerratt LF. 2011. Integrating species traits with extrinsic threats: closing the gap between predicting and preventing species declines. Proc. R. Soc. B 278, 1515–1523. ( 10.1098/rspb.2010.1872) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohr JR, Halstead NT, Raffel TR. 2011. Modelling the future distribution of the amphibian chytrid fungus: the influence of climate and human-associated factors. J. Appl. Ecol. 48, 174–176. ( 10.1111/j.1365-2664.2010.01891.x) [DOI] [Google Scholar]
- 10.Searle CL, Biga LM, Spatafora JW, Blaustein AR. 2011. A dilution effect in the emerging amphibian pathogen Batrachochytrium dendrobatidis. Proc. Natl Acad. Sci. USA 108, 16 322–16 326. ( 10.1073/pnas.1108490108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savage AE, Zamudio KR. 2011. MHC genotypes associate with resistance to a frog-killing fungus. Proc. Natl Acad. Sci. USA 108, 16 705–16 710. ( 10.1073/pnas.1106893108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.May S, Zeisset I, Beebee TJC. 2011. Larval fitness and immunogenetic diversity in chytrid-infected and uninfected natterjack toad (Bufo calamita) populations. Conserv. Genet. 12, 805–811. ( 10.1007/s10592-011-0187-z) [DOI] [Google Scholar]
- 13.Fites JS, et al. 2013. The invasive chytrid fungus of amphibians paralyzes lymphocyte responses. Science 342, 366–369. ( 10.1126/science.1243316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon TA, et al. 2014. Amphibians acquire resistance to live and dead fungus overcoming fungal immunosuppression. Nature 511, 224–227. ( 10.1038/nature13491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bataille A, et al. 2015. Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc. R. Soc. B 282, 20143127 ( 10.1098/rspb.2014.3127) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savage AE, Sredl MJ, Zamudio KR. 2011. Disease dynamics vary spatially and temporally in a North American amphibian. Biol. Conserv. 144, 1910–1915. ( 10.1016/j.biocon.2011.03.018) [DOI] [Google Scholar]
- 17.Savage AE, Becker CG, Zamudio KR. 2015. Linking genetic and environmental factors in amphibian disease risk. Evol. Appl. 8, 560–572. ( 10.1111/eva.12264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohr JR, Raffela TR, Romansica JM, McCallum H, Hudson PJ. 2008. Evaluating the links between climate, disease spread, and amphibian declines. Proc. Natl Acad. Sci. USA 105, 17 436–17 441. ( 10.1073/pnas.0806368105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du Pasquier L, Schwager J, Flajnik MF. 1989. The immune system of Xenopus. Ann. Rev. Immunol. 7, 251–275. ( 10.1146/annurev.iy.07.040189.001343) [DOI] [PubMed] [Google Scholar]
- 20.Ohta Y, Okamura K, McKinney E, Bartl S, Hashimoto K, Flajnik MF. 2000. Primitive synteny of vertebrate major histocompatibility complex class I and class II genes. Proc. Natl Acad. Sci. USA 97, 4712–4717. ( 10.1073/pnas.97.9.4712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones EY, Fugger L, Strominger JL, Siebold C. 2006. MHC class II proteins and disease: a structural perspective. Nat. Rev. Immunol. 6, 271–282. ( 10.1038/nri1805) [DOI] [PubMed] [Google Scholar]
- 22.Bernatchez L, Landry C. 2003. MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J. Evol. Biol. 16, 363–377. ( 10.1046/j.1420-9101.2003.00531.x) [DOI] [PubMed] [Google Scholar]
- 23.Shiina T, Inoko H, Kulski JK. 2004. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Ant. 64, 631–649. ( 10.1111/j.1399-0039.2004.00327.x) [DOI] [PubMed] [Google Scholar]
- 24.Carillo-Farga J, Castell A, Perez A, Rondan A. 1990. Langerhans-like cells in amphibian epidermis. J. Anat. 172, 39–45. [PMC free article] [PubMed] [Google Scholar]
- 25.Bevan MJ. 1987. Class discrimination in the world of immunology. Nature 325, 192–194. ( 10.1038/325192b0) [DOI] [PubMed] [Google Scholar]
- 26.Hughes AL, Nei M. 1989. Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc. Natl Acad. Sci. USA 86, 948–962. ( 10.1073/pnas.86.3.958) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doherty P, Zinkernagel R. 1975. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256, 50–52. ( 10.1038/256050a0) [DOI] [PubMed] [Google Scholar]
- 28.Teacher AGF, Garner TWJ, Nichols RA. 2009. Evidence for directional selection at a novel major histocompatibility class I marker in wild common frogs (Rana temporaria) exposed to a viral pathogen (Ranavirus). PLoS ONE 4, e4616 ( 10.1371/journal.pone.0004616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vassilakos D, Natoli A, Dahlheim M, Hoelzel AR. 2009. Balancing and directional selection at exon-2 of the MHC DQB1 locus among populations of odontocete cetaceans. Mol. Biol. Evol. 26, 681–689. ( 10.1093/molbev/msn296) [DOI] [PubMed] [Google Scholar]
- 30.Ladle R. 1992. Parasites and sex: catching the Red Queen. Trends Ecol. Evol. 7, 405–408. ( 10.1016/0169-5347(92)90021-3) [DOI] [PubMed] [Google Scholar]
- 31.Clarke B, Kirby D. 1966. Maintenance of histocompatibility polymorphisms. Nature 211, 999–1000. ( 10.1038/211999a0) [DOI] [PubMed] [Google Scholar]
- 32.Meyer D, Thomson G. 2001. How selection shapes variation of the human major histocompatibility complex: a review. Ann. Hum. Genet. 65, 1–26. ( 10.1046/j.1469-1809.2001.6510001.x) [DOI] [PubMed] [Google Scholar]
- 33.Hill AVS. 1998. The immunogenetics of human infectious diseases. Ann. Rev. Immunol. 16, 593–617. ( 10.1146/annurev.immunol.16.1.593) [DOI] [PubMed] [Google Scholar]
- 34.Hedrick P. 2002. Pathogen resistance and genetic variation at MHC loci. Evolution 56, 1902–1908. ( 10.1111/j.0014-3820.2002.tb00116.x) [DOI] [PubMed] [Google Scholar]
- 35.Kaufman JF, Flajnik MF, Du Pasquier L, Riegert P. 1985. Xenopus MHC class II molecules. I. Identification and structural characterization. J. Immunol. 134, 3248–3257. [PubMed] [Google Scholar]
- 36.Flajnik MF, Du Pasquier L. 1990. The major histocompatibility complex of frogs. Immunol. Rev. 113, 47–63. ( 10.1111/j.1600-065X.1990.tb00036.x) [DOI] [PubMed] [Google Scholar]
- 37.Ohta Y, Goetz W, Hossain MZ, Nonaka M, Flajnik MF. 2006. Ancestral organization of the MHC revealed in the amphibian Xenopus. J. Immunol. 176, 3674–3685. ( 10.4049/jimmunol.176.6.3674) [DOI] [PubMed] [Google Scholar]
- 38.Nonaka M, Namikawa C, Kato Y, Sasaki M, Salter-Cid L, Flajnik MF. 1997. Major histocompatibility complex gene mapping in the amphibian Xenopus implies a primordial organization. Proc. Natl Acad. Sci. USA 94, 5789–5791. ( 10.1073/pnas.94.11.5789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flajnik MF, Ohta Y, Namikawa-Yamada C, Nonaka M. 1999. Insight into the primordial MHC from studies in ectothermic vertebrates. Immunol. Rev. 167, 59–67. ( 10.1111/j.1600-065X.1999.tb01382.x) [DOI] [PubMed] [Google Scholar]
- 40.Pyron RA, Wiens JJ. 2011. A large-scale phylogeny of Amphibia including over 2,800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylog. Evol. 61, 543–583. ( 10.1016/j.ympev.2011.06.012) [DOI] [PubMed] [Google Scholar]
- 41.Ribas L, et al. 2009. Expression profiling the temperature-dependent amphibian response to infection by Batrachochytrium dendrobatidis. PLoS ONE 4, e8408 ( 10.1371/journal.pone.0008408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenblum ET, Poorten TJ, Settles M, Murdoch GK. 2012. Only skin deep: shared genetic response to the deadly chytrid fungus in susceptible frog species. Mol. Ecol. 21, 2110–3120. ( 10.1111/j.1365-294X.2012.05481.x) [DOI] [PubMed] [Google Scholar]
- 43.Ellison AR, Savage AE, DiRenzo GV, Langhammer P, Lips KR, Zamudio KR. 2014. Fighting a losing battle: vigorous immune response countered by pathogen suppression of host defenses in a chytridiomycosis-susceptible frog. G3 4, 1275–1289. ( 10.1534/g3.114.010744) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellison AR, et al. 2015. More than skin deep: functional genomic basis for resistance to amphibian chytridiomycosis. Genome Biol. Evol. 7, 286–298. ( 10.1093/gbe/evu285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hauswaldt JS, Stuckas H, Pfautsch S, Tiedemann R. 2007. Molecular characterization of MHC class II in a nonmodel anuran species, the fire-bellied toad Bombina bombina. Immunogenetics 59, 479–491. ( 10.1007/s00251-007-0210-1) [DOI] [PubMed] [Google Scholar]
- 46.Kiemnec-Tyburczy KM, Richmond JQ, Savage AE, Zamudio KR. 2010. Selection, trans-species polymorphism and locus identification of major histocompatibility complex class IIβ alleles of New World ranid frogs. Immunogenetics 62, 741–751. ( 10.1007/s00251-010-0476-6) [DOI] [PubMed] [Google Scholar]
- 47.Bradley G, Rosen P, Sredl M, Jones T, Longcore J. 2002. Chytridiomycosis in native Arizona frogs. J. Wildl. Dis. 38, 206–212. ( 10.7589/0090-3558-38.1.206) [DOI] [PubMed] [Google Scholar]
- 48.Raymond M, Rousset F. 1995. GENEPOP: population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249. [Google Scholar]
- 49.Hyatt AD, et al. 2007. Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis. Aquat. Org. 73, 175–192. ( 10.3354/dao073175) [DOI] [PubMed] [Google Scholar]
- 50.Clopper C, Pearson S. 1934. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 26, 404–413. ( 10.1093/biomet/26.4.404) [DOI] [Google Scholar]
- 51.Savage AE, Jaeger JR. 2009. Isolation and characterization of microsatellite markers in the lowland leopard frog (Rana yavapaiensis) and the relict leopard frog (R. onca), two declining frogs of the North American desert southwest. Mol. Ecol. Res. 9, 199–202. ( 10.1111/j.1755-0998.2008.02343.x) [DOI] [PubMed] [Google Scholar]
- 52.Guo SW, Thompson EA. 1992. Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48, 361–372. ( 10.2307/2532296) [DOI] [PubMed] [Google Scholar]
- 53.Kosakovsky Pond SL, Posada D, Gravenort MB, Woelk CH, Frost SDW. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901. ( 10.1093/molbev/msl051) [DOI] [PubMed] [Google Scholar]
- 54.Posada D. 2008. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. ( 10.1093/molbev/msn083) [DOI] [PubMed] [Google Scholar]
- 55.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. ( 10.1093/bioinformatics/btg180) [DOI] [PubMed] [Google Scholar]
- 56.Kosakovsky Pond SL, Frost SDW, Muse SV. 2005. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 21, 6760–6779. ( 10.1093/bioinformatics/bti079) [DOI] [PubMed] [Google Scholar]
- 57.Scheffler K, Martin DP, Seoighe C. 2006. Robust inference of positive selection from recombining coding sequences. Bioinformatics 22, 2493–2499. ( 10.1093/bioinformatics/btl427) [DOI] [PubMed] [Google Scholar]
- 58.Kosakovsky Pond SL, Scheffler K, Gravenor MB, Poon AFY, Frost SDW. 2010. Evolutionary fingerprinting of genes. Mol. Biol. Evol. 27, 520–536. ( 10.1093/molbev/msp260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kosakovsky Pond SL, Frost SDW. 2005. Not so different after all: comparison of various methods for detecting amino-acid sites under selection. Mol. Biol. Evol. 22, 1208–1222. ( 10.1093/molbev/msi105) [DOI] [PubMed] [Google Scholar]
- 60.Jombart T, Devillard S, Balloux F. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 ( 10.1186/1471-2156-11-94) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sandberg M, Eriksson L, Jonsson J, Sjostrom M, Wold S. 1998. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 41, 2481–2491. ( 10.1021/jm9700575) [DOI] [PubMed] [Google Scholar]
- 62.Crawford NG. 2010. SMOGD: Software for the measurement of genetic diversity. Mol. Ecol. Res. 10, 556–557. ( 10.1111/j.1755-0998.2009.02801.x) [DOI] [PubMed] [Google Scholar]
- 63.Jost LOU. 2008. GST and its relatives do not measure differentiation. Mol. Ecol. 17, 4015–4026. ( 10.1111/j.1365-294X.2008.03887.x) [DOI] [PubMed] [Google Scholar]
- 64.Excoffier L, Lischer HEL. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 10, 564–567. ( 10.1111/j.1755-0998.2010.02847.x) [DOI] [PubMed] [Google Scholar]
- 65.R Core Team. 2012. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 66.Cook RD. 1979. Influential observations in linear regression. J. Am. Stat. Assoc. 74, 169–174. ( 10.2307/2286747) [DOI] [Google Scholar]
- 67.Witte CL, Sredl MJ, Kane AS, Hungerford LL. 2008. Epidemiologic analysis of factors associated with local disappearances of native ranid frogs in Arizona. Conserv. Biol. 22, 375–383. ( 10.1111/j.1523-1739.2007.00878.x) [DOI] [PubMed] [Google Scholar]
- 68.MacDonald KS, et al. 2000. Influence of HLA supertypes on susceptibility and resistance to human immunodeficiency virus type 1 infection. J. Infect. Dis. 181, 1581–1589. ( 10.1086/315472) [DOI] [PubMed] [Google Scholar]
- 69.Linnen CR, Kingsley EP, Jensen JD, Hoekstra HE. 2009. On the origin and spread of an adaptive allele in deer mice. Science 325, 1095–1098. ( 10.1126/science.1175826) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eizaguirre C, Lenz TL, Kalbe M, Milinski M. 2012. Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat. Comm. 3, 621 ( 10.1038/ncomms1632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harf R, Sommer S. 2005. Association between MHC Class II DRB alleles and parasite load in the hairy-footed gerbil, Gerbillurus paeba, in the Southern Kalahari. Mol. Ecol. 14, 85–91. ( 10.1111/j.1365-294X.2004.02402.x) [DOI] [PubMed] [Google Scholar]
- 72.Paterson S, Wilson K, Pemberton J. 1998. Major histocompatibility complex variation associated with juvenile survival and parasite resistance in a large unmanaged ungulate population (Ovis aries L.). Proc. Natl Acad. Sci. USA 95, 3714–3719. ( 10.1073/pnas.95.7.3714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rollins-Smith LA, Ramsey JP, Pask JD, Woodhams DC. 2011. Amphibian immune defenses against chytridiomycosis: impacts of changing environments. Int. Comp. Biol. 51, 552–562. ( 10.1093/icb/icr095) [DOI] [PubMed] [Google Scholar]
- 74.Bletz MC, et al. 2015. Widespread presence of the pathogenic fungus Batrachochytrium dendrobatidis in wild amphibian communities in Madagascar. Sci. Rep. 5, 8633 ( 10.1038/srep08633) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences: GenBank accession numbers KU877031-KU877108. Microsatellite genotyping data for Lithobates yavapaiensis populations available from Dryad Digital Repository http://dx.doi.org/10.5061/dryad.3qk53 and also from [17].