

Significance
The ubiquitin proteasome components are often misregulated in numerous diseases, encouraging the search for drug targets and inhibitors. E3 ligases that specify ubiquitination targets are of particular interest. Multimeric Skp1–Cul1–F-box (SCF) E3 ligases constitute one of the largest E3 families connected to every cellular process and multiple diseases; however, their characterization as therapeutic targets is impeded by functional diversity and poor characterization of its members. Herein we describe a strategy to inhibit SCF E3 ligases using engineered ubiquitin-based binders. We identify a previously uncharacterized inhibitory site and design ubiquitin-based libraries targeting this site. Our strategy to target SCF E3 ligases with small-molecule–like agents will have broad applications for basic research and drug development relating to SCF E3 ligase function.
Keywords: Cul1 affinity, SCF inhibitors, Fbxw7, Fbxw11, β-Trcp
Abstract
Skp1–Cul1–F-box (SCF) E3 ligases play key roles in multiple cellular processes through ubiquitination and subsequent degradation of substrate proteins. Although Skp1 and Cul1 are invariant components of all SCF complexes, the 69 different human F-box proteins are variable substrate binding modules that determine specificity. SCF E3 ligases are activated in many cancers and inhibitors could have therapeutic potential. Here, we used phage display to develop specific ubiquitin-based inhibitors against two F-box proteins, Fbw7 and Fbw11. Unexpectedly, the ubiquitin variants bind at the interface of Skp1 and F-box proteins and inhibit ligase activity by preventing Cul1 binding to the same surface. Using structure-based design and phage display, we modified the initial inhibitors to generate broad-spectrum inhibitors that targeted many SCF ligases, or conversely, a highly specific inhibitor that discriminated between even the close homologs Fbw11 and Fbw1. We propose that most F-box proteins can be targeted by this approach for basic research and for potential cancer therapies.
The ubiquitin proteasome system (UPS) plays a central role in protein homeostasis through ubiquitination and degradation of substrate proteins. General inhibitors of the proteasome have proven effective in cancer therapy (1), and thus there is great interest in developing specific inhibitors of UPS enzymes to explore their biological functions and to provide paths to more specific therapeutics. The central player in the UPS is ubiquitin (Ub), a highly conserved 76-residue protein. Ub is covalently attached to protein substrates through sequential action of ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes. E3 ligases bind protein substrates and thus dictate specificity of ubiquitination.
E3 ligases constitute the largest class of UPS enzymes, with more than 600 members encoded by the human genome, and are divided into two major classes: a small, well-characterized class of ∼30 homologous to the E6AP carboxyl terminus (HECT) E3 ligases and a much larger, but less-characterized class of hundreds of RING E3 ligases and structurally related variants (2). HECT E3 ligases form transient thioester linkages with Ub before transferring it to substrates, whereas RING ligases serve as adaptors to recruit Ub-charged E2 enzymes to substrates for Ub transfer. The archetype for the RING class are the multisubunit Skp1–Cul1–F-box (SCF) complexes, which contains 69 members in humans (3). The SCF enzyme complexes are composed of constant Rbx1, Cul1, and Skp1 subunits, and a variable F-box protein that binds substrates and dictates specificity (Fig. 1B). Rbx1, the RING protein that recruits the E2 enzyme, binds the scaffold protein Cul1, which in turn binds Skp1, an adaptor for F-box proteins. F-box proteins are variable in domain composition but share a common F-box domain that binds Skp1. F-box proteins are subdivided into three subfamilies based on the structure of their substrate binding domains, including WD40, LRR, and other domains, referred to as the Fbw, Fbl, and Fbo subfamilies, respectively (3).
Fig. 1.

Ubvs selected for binding to the Skp1tr–Fbw7 complex. (B) Schematic of SCF E3 ligase. (A) Sequence alignment of selected Ubvs. Library 1 sequence is shown, where residue letters indicate the WT Ub sequence that was soft randomized and “X” denotes positions that were completely randomized. Only diversified positions are shown and residues in Ubvs conserved as WT Ub are indicated by dashes. Sequences showing conservation across selected Ubvs are highlighted in gray. (C) Binding of selected Ubvs to Skp1tr in complex with F-box–WD40Fbw7, F-boxFbw7, or F-boxFbw11. Ubv-phage binding was measured by ELISA with the indicated immobilized proteins. (D and E) Binding of purified Ubv.Fw7.1 or Ubv.Fw7.2 to Skp1tr–F-boxFbw7 as measured by ELISA. Data from a typical experiment are shown and the binding values are represented as mean ± SE of at least two experiments. (D) IC50 values were calculated by competitive ELISA as the concentration of Skp1tr–F-boxFbw7 in solution that blocked 50% of Ubv binding to immobilized Skp1tr–F-boxFbw7. (E) EC50 values were calculated by direct-binding ELISA as the concentration of Ubv at which 50% of the saturation signal is achieved for binding to immobilized Skp1tr–F-boxFbw7 complex.
Numerous F-box proteins are involved in processes relevant to tumorigenesis, including cell proliferation, cell cycle progression, and apoptosis, suggesting that these proteins may be targets for cancer treatment (4). Fbl1 (Skp2) eliminates the CDK inhibitor p27 and is a well-validated target for cancer treatment; several small-molecule inhibitors of Skp2 show activity in preclinical models (reviewed in ref. 5). However, given poorly defined roles for many F-box proteins and the functional complexity observed for those with characterized roles, further studies are required to gauge the therapeutic potential of this E3 family (4).
We have previously demonstrated that many UPS components can be targeted by Ub variants (Ubvs), which function by strengthening weak interactions between Ub and natural binding sites in UPS enzymes (6). Like small molecules, Ubvs can be used to assess the effects of enzyme inhibition and provide information applicable to the design of the mechanism based therapeutic inhibitors. Previously, Ubvs were developed against monomeric components of the UPS system, including E2 enzymes, E3 HECT enzymes, and deubiquitinases, which all contain well-defined Ub-binding sites (6). However, targeting F-box proteins poses a potentially greater challenge because these substrate receptors function as part of multimeric SCF complexes, show considerable diversity in the nature of their substrate binding domains (3) and, with the exception of several WD40 family members (7), are not known to interact directly with Ub.
Here, we describe the development and characterization of Ubv inhibitors targeting two well-characterized members of the Fbw subfamily, Fbw7 and Fbw11 (β-Trcp2). Fbw7 targets several critical oncoproteins, including Cyclin E, c-Myc, Notch 1, and Mcl1 (reviewed in ref. 8). Although Fbw7 is a tumor suppressor, its inhibition could be therapeutically beneficial under certain circumstances (9, 10). Fbw11 degrades targets in a multitude of pathways, including β-Catenin, Cdc25, Wee1, and IκB, and it is a potential therapeutic target in several cancers (reviewed in refs. 5 and 11). Fbw1 (β-Trcp1) shares 81% sequence identity with Fbw11 and the two proteins have overlapping functions (11). We used a phage-displayed Ubv library (6) to obtain inhibitors of Fbw7 expressed in complex with Skp1, and we solved the structure of one Ubv in complex with Skp1–Fbw7 to determine the binding mode and mechanism of inhibition. We used the structure to guide the design of second-generation libraries to obtain high specificity inhibitors of Fbw11. Finally, we demonstrated that the Ubvs function inside cells as inhibitors of their cognate enzymes. The strategy described in this study for Fbw7 and Fbw11 could be used to systematically develop Ubv inhibitors against the entire F-box family, with potential broad applications in basic research and drug development.
Results
Selection of Ubv Binders for the Skp1tr–Fbw7 Complex.
To investigate the potential of using Ubvs to target F-box family members, we used a naïve phage-displayed Ubv library (6) (Figs. 1A and Fig. S1) to perform binding selections against Fbw7 in complex with Skp1. To facilitate structural characterization, we used Fbw7 and Skp1 constructs that were previously used for structural studies but still contained all necessary functional elements required for E3 ligase activity. This included Fbw7 composed of F-box and WD40 domains (F-box–WD40Fbw7) (12) and Skp1 with truncations in two loops (Skp1tr) (13) (Table S1).
Fig. S1.

Ubv libraries. (A) Regions 1, 2, and 3 targeted in the library designs are shown. Only positions relevant to the library design are included and residues that differ from WT Ub sequence are highlighted in gray. Positions subjected to soft-randomization are boxed and positions subjected to complete randomization are indicated by an “X”. (B) Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex showing Ub positions diversified in the libraries. Regions 1, 2, and 3 are shown in separate panels and targeted positions are shown as salmon spheres and numbered. Skp1tr, F-boxFbw7, and Ubv are colored green, orange, or blue, respectively. Loop1 deleted in Skp1tr is labeled and colored magenta.
Table S1.
Protein expression vectors
| Constructs | Protein residues† | Vector | N terminus | Tag residues‡ |
| Bacterial vectors | ||||
| Fbw7 (F-box–WD40) | 263–708 | pPROEX HTb | His-TEV | MSYYHHHHHHDYDIPTT ENLYF*QGA |
| Fbw7 F-box | 263–367 | |||
| Fbw7 F-box (Structure) | 263–323 | |||
| Fbw2 F-box | 53–104 | |||
| Fbw5 F-box | 1–79 | |||
| Fbl1 F-box | 95–139 | |||
| Fbw12 F-box | 1–49 | |||
| Fbw1 F-box | 101–214 | |||
| Fbw11 F-box | 80–191 | |||
| Skp1 | Full length | pPROEX HTb | GST-TEV | GST-DYDIPTTENLYFQ*GA |
| Skp1 (Loop 1Δ) | 38–43Δ | |||
| Skp1 (Loop 2Δ) | 70–77Δ, | |||
| K78G E79S K80G R81G | ||||
| Skp1tr (ELISA/structure) | 38–43Δ, 70–77Δ, | |||
| K78G E79S K80G R81G | ||||
| Cul1 NTD§ | 1–410 | P11 | His-TEV-FLAG | MGSSHHHHHHSSGRENLYFQ*GHMDYKDDDDK |
| (V367D L371E) | ||||
| Ubv.Fw7.1 (structure) | 1–76 | p53DEST | His-TEV | MAHHHHHHVTSLYKKAG ENLYFQ*GSGS |
| Ubv (ELISA) | Full length | p53DEST | His-FLAG | MAHHHHHHVTSLYKKAG DYKDDDDK |
| Mammalian vectors | ||||
| FLAG-Ubv monomer | Full length | pcDNA3.1/ | FLAG-Linker-FLAG | MDYKDDDKGQGPDPSTNSADITSLYKKAGTMDYKDDDDK |
| nFLAGDEST | ||||
| FLAG-Ubv | Full length | pcDNA3.1/ | FLAG- | MDYKDDDKGQ RMKQLEDKIEELLSKIYHLENEIARLKKLIGERTSLYKKAGTMDYKDDDDK |
| Leu-zipper dimer | nFLAGDEST(+Leu-zipper) | Leu Zipper-FLAG | ||
| HA-Fbw7 | Full length | pcDNA3.1/ | HA | MYPYDVPDYAGQG PDPSTNSADITSLYKKAGST |
| HA-Fbw11 (Isoform B) | nHADEST | |||
| FLAG-Cul1 | Full length | pcDNA3.1/ | FLAG | MDYKDDDDKGQGPDPSTNSADITSLYKKAGT |
| FLAG-Fbw7 | nFLAGDEST | |||
| FLAG-Fbw11(isoform B) | ||||
| FLAG-Fbw1 | Full length | pcDNA3 | FLAG | MDYKDDDDK |
Residue limits of canonical human isoforms. Any amino acid substitutions relative to WT are indicated if present.
Tag residues are shown, except for GST tag. Residues that comprise functional elements are underlined and TEV cleavage site denoted by an asterisk. All tags are N-terminal.
Cul1 NTD was expressed with V367D and L371E substitutions that are necessary for solubility in the absence of the C-terminal domain as described previously (16).
The selections yielded four unique binding Ubvs that shared common mutations at several positions (Fig. 1A), suggesting that they all likely bind to a common site on the Skp1tr–Fbw7 complex. To determine the region targeted by the selected Ubvs, we performed phage ELISAs against Skp1tr complexed with F-box–WD40Fbw7, Fbw7 F-box domain (F-boxFbw7), or Fbw11 F-box domain (F-boxFbw11). Surprisingly, the Ubvs did not target the WD40 domain, which is known to interact with Ub (7) and small-molecule inhibitors (14), but rather specifically targeted F-boxFbw7 in complex with Skp1tr (Fig. 1C). Relative affinities of Ubv.Fw7.1 and Ubv.Fw7.2 were measured for purified proteins with ELISAs that determined half-maximum effective concentration of Ubv binding to immobilized Skp1tr–F-boxFbw7 (EC50) and half-maximum inhibitory concentration of Skp1tr–F-boxFbw7 in solution that inhibited binding of Ubv to immobilized Skp1tr–F-boxFbw7 (IC50). Because the IC50 value reflects the interaction between the two proteins in solution, it provides a good estimate of the affinity (15). Ubv.Fw7.1 exhibited the highest binding activity in both assay formats (IC50 = 70 nM and EC50 = 0.9 nM) and was chosen for further characterization (Fig. 1 D and E).
Structure of Ubv.Fw7.1 in Complex with Skp1tr–F-boxFbw7.
We crystallized Ubv.Fw7.1 in complex with Skp1tr–F-boxFbw7 and solved the structure at 2.5 Å resolution by molecular replacement (Fig. 2 A and B; see Table S2 for X-ray data collection and refinement statistics). Ubv.Fw7.1 makes extensive contacts with Skp1tr but also makes significant contacts with F-boxFbw7 (719 or 144 Å2 of Ubv accessible surface area buried, respectively). The structure of Skp1tr–F-boxFbw7 in the ternary complex aligns closely with the previously determined structure of the Skp1tr–(F-box–WD40)Fbw7 complex (12), suggesting that Ubv.Fw7.1 does not induce major conformational changes upon binding [RMSD of Skp1 = 0.93 Å and RMSD of Fbw7 (residues 279–313) = 1.07 Å].
Fig. 2.

Structural and mutational analysis of the interactions between Ubv.Fw7.1 and the Skp1tr–F-boxFbw7 complex. (A) Structure of Ubv.Fw7.1 in complex with Skp1tr–F-boxFbw7. Ubv regions (regions 1–3) that were diversified in Library 1 are labeled and colored dark blue, and other regions are colored light blue. Skp1tr and F-boxFbw7 are colored green or orange, respectively. (B) Details of the molecular interactions between Ubv.Fw7.1 and Skp1tr–F-boxFbw7 showing residues that are mutated relative to WT Ub and are critical for binding. Skp1tr and Fbw7 residues are denoted by “S” and “F” superscripts, respectively. Complex subunits are colored as in A and the location of Loop 1 deleted in Skp1tr is indicated in magenta. (C) Affinities of Ubv.Fw7.1 back-mutants for Skp1tr–F-boxFbw7. Ubv.Fw7.1Min lacks Ub tail (residues 75–78) and contains only six mutations relative to WT Ub (L8G, G10R, K11T, R42I, H68R, and L73F). “NB” indicates no detectable binding. (D) Superposition of Skp1tr–F-boxFbw7-Ubv.Fw7.1 complex with Skp1tr–F-boxFbl1-Cul1 complex (PDB ID code1LDK). Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex subunits are colored as in A and Skp1tr–F-boxFbl1-Cul1 complex subunits are colored as follows: Skp1tr, cyan; F-boxFbl1, purple; Cul1, red. (E) Comparison of the Ubv.Fw7.1-binding and predicted Cul1-binding surfaces on Skp1tr–F-boxFbw7. Skp1tr–F-boxFbw7 residues interacting with Ubv.Fw7.1 or predicted to interact with Cul1 (by comparison with the Skp1–F-boxFbl1–Cul1 complex) are shown as sticks and colored according to predicted interactions: magenta, interacts with Cul1 and Ubv.Fw7.1; red, interacts with Cul1 only; blue, interacts with Ubv.Fw7.1 only. Residues that were subjected to mutagenesis are labeled. (F) Effects of substitutions in Skp1tr or the F-boxFbw7 domain on the binding of Skp1tr–F-boxFbw7 to Ubv.Fw7.1 or Cul1 N-terminal domain (NTD).
Table S2.
Data collection and refinement statistics for the Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex
| Data collection and refinement | Statistics |
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 63.4, 98.0, 107.7 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 50.0–2.5 (2.56–2.50)* |
| Rmeas | 0.052 (0.752) |
| I/σI | 19.6 (1.3) |
| Completeness (%) | 99.4 (99.1) |
| Redundancy | 4.4 (3.9) |
| Refinement | |
| Resolution (Å) | 50.0–2.5 (2.56–2.50) |
| No. reflections | 23,612 (1,532) |
| Rwork/Rfree | 20.0/24.0 (30.9/39.4) |
| No. atoms | |
| Protein | 4,246 |
| Water | 38 |
| B-factors | |
| Protein | 81.1 |
| Water | 51.2 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.879 |
| Ramachandran | |
| Favored | 97.6 |
| Allowed | 2.4 |
| Outliers | 0 |
Values in parenthesis correspond to the highest shell.
Although Ubv.Fw7.1 contains 15 substitutions relative to WT Ub -and two additional C-terminal residues, back mutation analysis revealed that only six substitutions (L8G, G10R, K11T, R42I, H68R, and L73F) are responsible for most of the enhancement in binding to Skp1tr–Fbw7. A variant containing these six substitutions (Ubv.Fw7.1Min) bound to Skp1tr–F-boxFbw7 only ∼20-fold weaker than Ubv.Fw7.1, but further back mutation of any of the six substitutions greatly reduced or completely abrogated binding (Fig. 2C). Three of the six substitutions (L8G, G10R, and K11T) are located in region 1, a loop that contacts the Skp1–Fbw7 interface. The Arg-10 side-chain of the Ubv forms cation–pi interaction with the side-chain of Tyr-291Fbw7 and its aliphatic portion packs against the side-chains of Leu-288Fbw7 and Leu-116Skp1. Gly-8 and Thr-11 pack against Asn-108Skp1 and the side-chain NH2 of Asn-108Skp1 forms a hydrogen bond with the side-chain OH of Thr-11 (Fig. 2B). The other three substitutions (R42I, H68R, and L73F) contact Skp1 only. The Ile-42 side-chain engages in hydrophobic interactions with the side-chain of Leu-34Skp1 and the Phe-73 side-chain packs against Pro-46Skp1 and Pro-48Skp1. The Arg-68 side-chain forms cation–pi interaction with the side-chain of Tyr-109Skp1 and polar contacts with the side-chains of Thr-26Skp1 and Asp-111Skp1 (Fig. 2B).
Notably, the surface on Skp1tr–F-boxFbw7 for binding to Ubv.Fw7.1 largely overlaps with the previously elucidated surface on the analogous Skp1–F-boxFbl1 complex for binding to Cul1 (16) (Fig. 2 D and E). To compare the energetics of Ubv.Fw7.1 and Cul1 binding to Skp1tr–F-boxFbw7, we constructed a series of point mutants at positions within the common interface and measured the effects on binding to both ligands (Fig. 2F). Three of the substitutions (N108ASkp1, Y109ASkp1, and D111RSkp1), which reside in the center of binding surface, either abolished or significantly disrupted binding to both Ubv.Fw7.1 and Cul1 and most of the other substitutions also had significant effects on binding to both ligands. These results show that Ubv.Fw7.1 and Cul1 share a common structural and functional binding site on the Skp1tr–F-boxFbw7 complex.
To confirm that Ubv.Fw7.1 and Cul1 target overlapping sites on the Skp1–Fbw7 complex, we tested whether Ubv.Fw7.1 can inhibit Cul1 binding and SCFFbw7 ligase activity. Cul1 has been reported to bind to Skp1–Fbw7 in vitro with picomolar affinity (17). With surface plasmon resonance (SPR) analysis, we confirmed this tight interaction between Cul1 and Skp1–F-boxFbw7 (Fig. S2B) but we found that the interaction with Skp1tr–F-boxFbw7 was ∼1,000-fold weaker (Fig. S2 A, C, and D). Thus, we used in vitro assays with Skp1tr–Fbw7 to show that Ubv.Fw7.1 inhibits the polyubiquitination activity of SCFFbw7 (Fig. S2E) and Cul1 binding (Fig. S2F). We speculated that this mode of inhibition could be applied to other SCF ligases, prompting us to further characterize Ubv.Fw7.1 binding parameters with the ultimate goal of targeting other F-box proteins through the same mechanism.
Fig. S2.

Additional in vitro experiments. (A) Alignment of all Skp1 constructs tested in this study. Loop 1 (residues 38–43) and Loop 2 (residues 70–81) indicate residues deleted in truncated version of Skp1 (Skp1tr). (B and C) Binding of Cul1 NTD to Skp1–F-boxFbw7 and Skp1tr–F-boxFbw7 complexes demonstrates importance of Skp1 Loop 1 and Loop 2 residues for interaction with Cul1. Binding was measured by SPR analysis and obtained traces are shown for Cul1 NTD interaction with Skp1-\–F-boxFbw7 (B) and Skp1tr–F-boxFbw7 (C). Skp1–F-box complexes were immobilized and Cul1 NTD was injected at different concentrations. Colored lines represent the actual data (red, 100 nM Cul1; cyan, 33 nM Cul1; blue, 11 nM Cul1; green, 3.7 nM Cul1; and magenta, 1.2 nM Cul1), whereas black lines represent fits to a simple 1:1 Langmuir binding isotherm model. In the case of binding to Skp1–F-boxFbw7, only the three lowest Cul1 concentrations (11 nM Cul1, 3.7 nM Cul1, and 1.2 nM Cul1) were used to fit the data, because higher concentrations exceeded estimated affinity of the interaction >1,000 fold. Estimated kon, koff rates and Kd of the interactions are indicated. (D) Structure of Skp1tr–F-boxFbl1–Cul1 (PDB ID code 1LDK) complex highlighting positions of loops deleted in Skp1tr in relation to Cul1 binding surface. Skp1tr, F-boxFbl1, and Cul1are colored green, orange, or red, respectively. Location of loops deleted in Skp1tr are colored magenta and indicated. (E) Ubv.Fw7.1 blocks ubiquitination activity of SCFFbw7. A functional SCFFbw7complex was assembled from the Skp1tr–(F-box–WD40)Fbw7 and Cul1–Rbx1 complexes purified separately. Sic1, which is a natural substrate of yeast Fbw7 but is also recognized by human Fbw7, was used as the substrate. The ubiquitination reaction containing E1 (0.5 µM), E2 (14 µM), and SCFFbw7 (0.4 µM) was initiated by adding Ub (25 µM) to purified components in the absence and presence of Ubv.Fw7.1 (25 µM). The products of Sic1 (0.4 µM) ubiquitination were visualized by Western blotting at the indicated time points. (F) Ubv.Fw7.1 promotes dissociation of Cul1 from the SCFFbw7 complex. The SCFFbw7 complex containing GST-tagged Skp1tr on glutathionine Sepharose resin was incubated for 1 h with increasing amounts of Ubv.Fw7.1. Cul1 remaining bound to the resin was detected by Western blot. (G) Skp1 Loop 1 (residues 38–43) interferes with binding to Ubv.Fw7.1, whereas Skp1 Loop 2 (residues 70–81) has no effect on binding. Binding was tested by protein ELISA and dose–response curves of Ubv.Fw7.1 binding to different Skp1 constructs in complex with F-boxFbw7 domain are shown. (H) Structure of Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex highlighting positions of loops deleted in Skp1tr. Skp1tr, F-boxFbw7, and Ubv.Fw7.1 are colored green, orange, or blue, respectively. Location of loops deleted in Skp1tr are colored magenta and indicated. Positions that showed different preferences in Ubv selected against Skp1–F-boxFbw7 versus parental Ubv.Fw7.1 sequence are shown as salmon spheres. Position 75 was not defined in the structure and its projected location is shown.
Optimization of Ubvs for Binding to the Skp1–Fbw7 Complex.
Ubv.Fw7.1 was selected for binding to a Skp1tr–Fbw7 complex that contained a truncated form of Skp1 optimized for structural analysis. However, our ultimate goal was to develop inhibitors of endogenous SCF ligases, and Ubv.Fw7.1 bound only weakly to the Skp1–F-boxFbw7 complex containing full-length Skp1 (Fig. 3A), presumably because of unfavorable interactions with a negatively charged loop near the N terminus of Skp1 (Fig. S2 A, G, and H). To engineer Ubvs with enhanced affinity for the Skp1–F-boxFbw7 complex, we designed a second-generation library (Library 2) based on the sequence of Ubv.Fw7.1. Three residues involved in favorable contacts were held constant (Gly-8, Arg-10, Thr-11), whereas the remaining residues in contact with the Skp1tr–F-boxFbw7 complex were “soft randomized” using a mutagenesis strategy that favored the parental sequence but allowed for an ∼50% mutation frequency (Fig. S1). Following selections for binding to the Skp1–F-boxFbw7 complex, 14 unique Ubvs were purified and ELISAs showed dramatically improved affinities in comparison with Ubv.Fw7.1 (Fig. 3A).
Fig. 3.

Ubvs selected for binding to the F-boxFbw7 or F-boxFbw11 domain in complex with full-length Skp1. (A) Ubvs selected from Library 2 for binding to Skp1–F-boxFbw7. Positions that were soft-randomized in the library are shown and residues conserved as Ubv.Fw7.1 sequence are indicated by dashes. Positions that diverge from the Ubv.Fw7.1 sequence but show consensus among the selected sequences are boxed and conserved residues at these positions are shaded gray. (B) Affinities of Ubv.Fw7.5, Ubv.Fw11.1, and Ubv.Fw11.2 for different Skp1–F-box complexes. “NB” indicates no detectable binding and “WB” indicates weak binding for which IC50 values were >5,000 nM. Sequence of Ubv binding region (F-box residues located within 10 Å of Ubv in the structure of Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex) is shown for each F-box protein. Conserved positions are shaded gray and Fbw7 residues important for binding to Ubv.Fw7.1 (Fig. 2F) are boxed. (C) The sequences and affinities of Ubv.Fw11.1 and its derivatives selected for binding to Skp1–F-boxFbw11. Only the sequence in region 1 that differs from Ubv.Fw7.5 is shown, and residues conserved as Ubv.Fw11.1 sequence are indicated by dashes.
Many of the improved variants shared an A12G substitution and a preference for Arg at positions 49 and 75, and some also shared an I42R substitution (Fig. 3A). Although preference for Gly at position 12 is probably a result of optimization of Ubv interaction with the Skp1–Fbw7 interface, Arg substitutions at positions 42, 49, and 75 can be rationalized by the presence of a negatively charged loop in full-length Skp1, which should come in contact with residues at these positions and would thus favor the accumulation of positive charge in the Ubvs (Fig. S2H). Ubv.Fw7.5, the tightest binder to Skp1–F-boxFbw7, exhibited an IC50 of 45 nM and we focused on this variant for further characterization.
Ubv.Fw7.1 and its relatives bind to the Skp1–Fbw7 complex mainly through contacts with Skp1, raising the possibility that these Ubvs may exhibit cross-reactivity with at least some of the many different human Skp1–F-box complexes. Thus, we tested the binding of Ubv.Fw7.5 to six Skp1–F-box domain complexes and, compared with Fbw7, we observed weaker but significant binding to three of these (Fbw2, Fbl1, and Fbw5). The affinities correlated with the degree of sequence similarity with the Fbw7 Ubv-binding region (Fig. 3B). Fbw2, which shares the highest homology with Fbw7, exhibited an eightfold lower affinity, whereas Fbw5, which shows the least homology, exhibited more than 50-fold lower affinity. The three F-box domains that did not bind to Ubv.Fw7.5 (Fbw1, Fbw11, and Fbw12) showed the least homology with Fbw7.
Structure-Based Selection of Ubvs That Bind Specifically to the Skp1–F-boxFbw11 Complex.
Because contacts with F-boxFbw7 are mediated entirely by the region 1 loop of Ubv.Fw7.1, we wondered whether sequence and length diversity in this loop could be exploited to alter specificity in favor of particular Skp1–F-box complexes. To explore this possibility, we designed a phage-displayed library (Library 3) in which four residues in region 1 of Ubv.Fw7.5 were replaced by completely random sequences, ranging from 11 to 13 residues in length, to increase the size of the potential interaction interface with the F-box domain (Fig. S1). Library 3 was selected for binding to the Skp1–F-boxFbw11 complex to determine whether this approach could be used to alter the F-box domain preference of Ubv.Fw7.5. Sequencing of 44 binding clones revealed that 42 were identical and contained a 12-residue insertion in region 1 (Fig. 3C) (Ubv.Fw11.1). Remarkably, purified Ubv.Fw11.1 protein was highly specific for Skp1–F-boxFbw11, as it bound very weakly to Skp1 in complex with homolog F-boxFbw1(89% sequence identity) and did not bind detectably to any of the other five Skp1–F-box complexes that we tested (Fig. 3B). To further improve affinity, we designed a library (Library 4) in which region 1 of Ubv.Fw11.1 was soft-randomized, and binding selections yielded 16 unique Ubvs containing one to three substitutions (Fig. S3). Four of these variants exhibited enhanced affinities for the Skp1–F-boxFbw11 complex (Fig. 3C) and the best of these (Ubv.Fw11.2) retained high specificity (Fig. 3B).
Fig. S3.
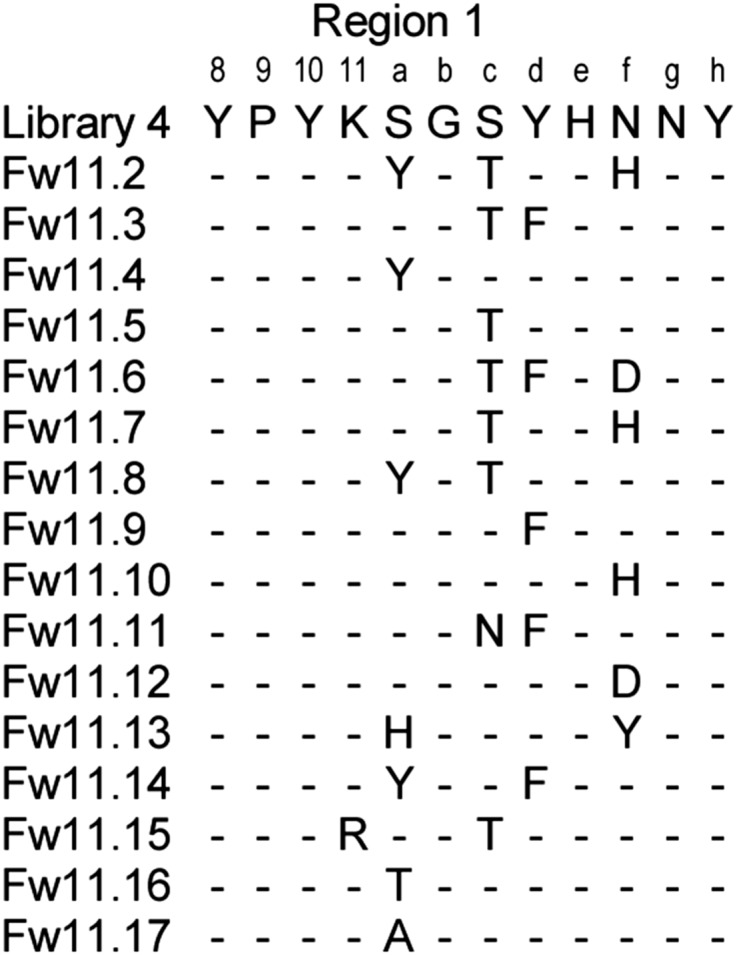
Ubvs selected for binding to the Skp1–F-boxFbw11 complex. Only positions in region 1 that were diversified in the library are shown and residues conserved as Ubv.Fw11.1 sequence are indicated by dashes.
Intracellular Activity of Ubvs Targeting Fbw7 and Fbw11 Complexes.
We transiently expressed Ubv.Fw7.5 or Ubv.Fw11.2 in HEK293T cells to ascertain whether these Ubvs were able to exert effects in live cells. Because Fbw7 and Fbw11 protein complexes function as dimers (18, 19), expression vectors were designed to express Ubvs either as monomers or as dimers held together by a homodimeric GCN4 leucine zipper to enhance effective affinities through avidity (Table S1) (20). To examine the interactions of Ubvs with endogenous proteins, Ubvs were immunoprecipitated, and coprecipitated proteins were identified by mass spectrometry (Fig. 4A). Consistent with the in vitro specificity profiles (Fig. 3B), Ubv.Fw7.5 coimmunoprecipitated Fbw7 and Skp1, and also several other F-box proteins, including Fbw2 and Fbl1. Fbw7 was detected with the lowest spectral counts among the F-box proteins, but this is likely a result of low expression levels of endogenous Fbw7. In support of this finding, a significant amount of Fbw7, but not Fbl1, coimmunoprecipitated with Ubv.Fw7.5 in cells overexpressing Fbw7 or Fbl1 (Fig. S4A). In contrast, Ubv.Fw11.2 was very specific for Fbw11, coimmunoprecipitating only Skp1, Fbw11, and small amounts of Fbw1. Similar levels of interacting proteins were detected, whether Ubvs were expressed as monomers or dimers, but Ubv dimers coimmunoprecipitated more nonspecific proteins involved in cell housekeeping functions (Table S3).
Fig. 4.

Biological activity of Ubvs in HEK293T cells. (A) Ubv interaction partners identified by mass spectrometry of FLAG-Ubv immunoprecipitates from cell lysates. Spectral counts refers to number of peptides corresponding to each identified protein. Only proteins relevant to SCF ligases are shown (see Table S3 for complete list of detected proteins). (B) Expression of Fw7.5 Ubv in monomer or dimer disrupts interaction of Fbw7 with Cul1. HA-Fbw7 immunoprecipitates were probed for FLAG-Cul1 and endogenous Skp1, in the absence or presence of FLAG-Ubv expression. (C) Expression of Ubv.Fw11.2 Ubv in dimer format, but not monomer format, disrupts interaction between Fbw11 and Cul1. Analysis performed as described in B. (D and E) Expression of Ubv.Fw7.5 (D) and Ubv.Fw11.2 (E) in monomer or dimer format stabilizes the SCFFbw7 (Cyclin E and c-Myc) and SCFFbw11 (Cdc25A and Wee1) substrates, respectively. Cells were transiently transfected with either siRNA molecules (positive control), empty vector (Vector), or vectors expressing FLAG-Ubv. Cells were treated with cycloheximide (CHX) for the indicated time points and cell lysates were probed with antibodies against the indicated proteins. Quantification of relative substrate levels was performed using ImageJ and represents average of two independent experiments (see D and Fig. S4C for c-Myc and Cyclin E, and E and Fig. S4D for Cdc25A and Wee1). (E) The effect of Fbw11 siRNA treatment and Ubv.Fw11.2 expression was assessed in the background of Fbw1 siRNA treatment.
Fig. S4.

Additional intracellular experiments. (A) Interaction between FLAG-Ubv.Fw7.5 and exogenously expressed HA-Fbw7 or HA-Fbl1 in cell lysates. FLAG immunoprecipitates were separated on gel electrophoresis and probed for the presence of the indicated HA-tagged F-box proteins. Ubv.Fw7.5 coimmunoprecipitates significant levels of Fbw7, whereas Fbl1 presence in immunoprecipitates could not be detected by this assay. It should be noted that the levels of Fbw7-HA in cell lysates are significantly greater in the presence of Ubv.Fw7.5 expression; this is the result of inhibition of Fbw7 auto-ubiquitination (19) by the Ubv.Fw7.5. (B) Validation of Fbw7, Fbw1, and Fbw11 siRNA. Cells expressing FLAG-Fbw7 (Upper) and FLAG-Fbw1, FLAG-Fbw11 and FLAG-Fbw1+HA-Fbw11 (Lower) were treated either with control siRNA (“C”) or the siRNAs directed against the indicated proteins. (C and D) Expression of Ubv.Fw7.5 (C) and Ubv.Fw11.2 (D) dimer or monomer selectively stabilizes substrates of SCFFbw7 and SCFFbw11, respectively., but not other SCF ligases. Cells were transiently transfected with either siRNA molecules (positive control), empty vector (vector), or vectors expressing FLAG-Ubv. Cells were treated with CHX for the indicated time points and cell lysates were probed with endogeneous antibodies against the indicated proteins [substrate (F-box protein)]. (E) Expression of Ubv.Fw11.2 does not affect stability of Cdc25A and Wee1 in the background of Fbw11 siRNA treatment. (F and G) Distribution of G1-phase, G2-phase, and S-phase populations in cells expressing Ubv.Fw7.5 (F) and Ubv.Fw11.2 (G). Cells were transiently transfected with either siRNA molecules (positive control), empty vector (vector), or vectors expressing FLAG-Ubv. Analysis of cell cycle kinetics was determined by Hoechst dye (nucleic acid stain) staining, followed by flow cytometry analysis. The graph combines data from three biological replicates and mean ± SE is shown. (G) The effect of Fbw11 siRNA treatment and Ubv.Fw11.2 expression was assessed in the background of Fbw1 siRNA treatment.
Table S3.
Mass spectrometry analysis of Ubv interactions in HEK293T cells
| ID* | Gene† | Description | V‡ | Ub‡ | Fw7.5 | Fw7.5 dimer | Fw11.2 | Fw11.2dimer |
| All Ubv | ||||||||
| P63208 | SKP1 | Skp1 | 0§ | 0 | 18 | 37 | 57 | 40 |
| P68363 | TUBA1B | Tubulin | 21 | 12 | 21 | 66 | 36 | 2 |
| Q09028 | RBBP4 | Histone binding | 2 | 0 | 0 | 12 | 0 | 0 |
| P53671 | LIMK2 | Histone kinase | 0 | 0 | 10 | 10 | 8 | 5 |
| Q9H479 | FN3K | Ketosamine-3-kinase | 0 | 0 | 9 | 0 | 4 | 0 |
| O15018 | PDZD2 | PDZ domain containing | 0 | 0 | 2 | 5 | 5 | 1 |
| Q8NI27 | THOC2 | mRNA processing | 0 | 0 | 5 | 5 | 0 | 3 |
| Monomer Ubv-specific | ||||||||
| O95479 | H6PD | Glucose dehydrogenase | 0 | 0 | 203 | 0 | 143 | 0 |
| Q9UIV8 | PI13 | Peptidase inhibitor | 0 | 0 | 146 | 0 | 120 | 0 |
| Q96QC0 | PPP1R10 | Phosphatase | 0 | 0 | 6 | 0 | 6 | 0 |
| Dimer Ubv-specific | ||||||||
| Q15233 | NONO | mRNA processing | 0 | 0 | 0 | 118 | 3 | 114 |
| Q8WXF1 | PSPC1 | mRNA processing | 3 | 0 | 0 | 56 | 0 | 51 |
| P23246 | SFPQ | mRNA processing | 5 | 2 | 0 | 60 | 2 | 70 |
| Q15691 | MAPRE1 | Microtubule binding | 0 | 0 | 0 | 58 | 0 | 48 |
| P17980 | PSMC3 | 26S proteasome subunit | 0 | 0 | 0 | 39 | 0 | 37 |
| Q9P2E9 | RRBP1 | Ribosome receptor | 0 | 0 | 0 | 13 | 0 | 28 |
| Q9Y490 | TLN1 | Cytoskeletal component | 0 | 0 | 2 | 38 | 0 | 10 |
| P42566 | EPS15 | EGFR substrate | 0 | 0 | 0 | 20 | 0 | 27 |
| Q9UII2 | ATPIF1 | ATPase inhibitor | 0 | 0 | 0 | 27 | 0 | 19 |
| O00233 | PSMD9 | 26S proteasome subunit | 0 | 0 | 0 | 14 | 0 | 21 |
| P05787 | KRT8 | Keratin | 0 | 0 | 0 | 20 | 0 | 18 |
| Q16204 | CCDC6 | Unknown | 0 | 0 | 0 | 18 | 0 | 13 |
| Q15019 | SEPT2 | Cytoskeletal GTPase | 0 | 0 | 0 | 15 | 0 | 10 |
| Q14203 | DCTN1 | Microtubule transport | 0 | 0 | 0 | 9 | 0 | 17 |
| P46736 | BRCC3 | Lys-63 deubiquitinase | 0 | 0 | 0 | 15 | 0 | 16 |
| P80303 | NUCB2 | Calcium homeostasis | 0 | 0 | 0 | 8 | 0 | 14 |
| Q07065 | CKAP4 | Cytoskeleton associated | 0 | 0 | 0 | 14 | 0 | 12 |
| P31146 | CORO1A | Cytoskeletal component | 0 | 0 | 0 | 12 | 0 | 7 |
| O60271 | SPAG9 | Scaffold protein | 0 | 0 | 0 | 9 | 0 | 13 |
| Q9BW19 | KIFC1 | Microtubule transport | 0 | 0 | 0 | 5 | 0 | 13 |
| P16220 | CREB1 | Transcription factor | 0 | 0 | 0 | 8 | 0 | 10 |
| Q13625 | TP53BP2 | p53 regulator | 0 | 0 | 0 | 5 | 0 | 8 |
| O00139 | KIF2A | Microtubule transport | 0 | 0 | 0 | 9 | 0 | 7 |
| O43293 | DAPK3 | Serine/Threonine kinase | 0 | 0 | 0 | 9 | 0 | 6 |
| Q9NVA2 | SEPT 11 | Cytoskeletal GTPase | 0 | 0 | 0 | 8 | 0 | 9 |
| Q13976 | PRKG1 | Serine/Threonine kinase | 0 | 0 | 0 | 7 | 0 | 9 |
| Q14141 | SEPT6 | Cytoskeletal GTPase | 0 | 0 | 0 | 4 | 0 | 8 |
| Q9P0K7 | RAI14 | Actin associated | 0 | 0 | 0 | 2 | 0 | 7 |
| Q16181 | SEPT7 | Cytoskeletal GTPase | 0 | 0 | 0 | 10 | 0 | 10 |
| Q14980 | NUMA1 | Nuclear matrix | 0 | 0 | 0 | 4 | 0 | 10 |
| Q96CN9 | GCC1 | Golgi associated | 0 | 0 | 0 | 4 | 0 | 7 |
| P40222 | TXLNA | Vesicle traffic | 0 | 0 | 0 | 3 | 0 | 7 |
| O95396 | MOCS3 | tRNA biosynthesis | 0 | 0 | 0 | 2 | 0 | 7 |
| Q9UPY8 | MAPRE3 | Microtubule associated | 0 | 0 | 0 | 2 | 0 | 7 |
| P62195 | PSMC5 | 26S proteasome subunit | 0 | 0 | 0 | 6 | 0 | 3 |
| Q8N302 | AGGF1 | Angiogenic factor | 0 | 0 | 0 | 2 | 0 | 6 |
| Q15390 | MTFR1 | Mitochondrial fission | 0 | 0 | 0 | 0 | 0 | 6 |
| O75146 | HIP1R | Clathrin associated | 0 | 0 | 0 | 5 | 0 | 3 |
| P53621 | COPA | Golgi-to-ER transport | 0 | 0 | 0 | 5 | 0 | 2 |
| O60308 | KIAA0562 | Centrosomal protein | 0 | 0 | 0 | 5 | 0 | 2 |
| Q969V6 | MKL1 | Transcription factor | 0 | 0 | 0 | 4 | 0 | 5 |
| Q15007 | WTAP | mRNA processing | 0 | 0 | 0 | 2 | 0 | 5 |
| Q8TBA6 | GOLGA5 | Golgi formation | 0 | 0 | 0 | 2 | 0 | 5 |
| Q4VCS5 | AMOT | Tight junction maintenance | 0 | 0 | 0 | 0 | 0 | 5 |
| Q9H6D7 | HAUS4 | Spindle assembly | 0 | 0 | 0 | 0 | 0 | 5 |
| Ubv.Fw11.2 specific | ||||||||
| Q9U.K.B1 | FBXW11 | F-box | 0 | 0 | 0 | 0 | 125 | 85 |
| Q9Y297 | FBXW1 | F-box | 0 | 0 | 0 | 0 | 10 | 4 |
| P0C0L4 | C4A | Complement comp. | 0 | 0 | 0 | 0 | 13 | 3 |
| Q06203 | PPAT | Ribosyl transferase | 0 | 0 | 0 | 0 | 13 | 3 |
| P04632 | CAPNS1 | Protease subunit | 0 | 0 | 0 | 0 | 12 | 0 |
| Q96K76 | USP47¶ | Ubiquitin protease | 0 | 0 | 0 | 0 | 3 | 7 |
| Q01664 | TFAP4 | Transcription factor | 0 | 0 | 0 | 0 | 6 | 0 |
| Ubv.Fw7.5 specific | ||||||||
| Q9U.K.T8 | FBXW2 | F-box | 0 | 0 | 24 | 18 | 0 | 0 |
| P19838 | NFKB1 | Transcription factor | 0 | 0 | 22 | 0 | 0 | 0 |
| Q13309 | SKP2 | F-box | 0 | 0 | 12 | 7 | 0 | 0 |
| Q7Z6M2 | FBXO33 | F-box | 0 | 0 | 3 | 7 | 0 | 0 |
| Q9NVF7 | FBXO28 | F-box | 0 | 0 | 0 | 7 | 0 | 0 |
| Q969H0 | FBXW7 | F-box | 0 | 0 | 5 | 0 | 0 | 0 |
| O94952 | FBXO21 | F-box | 0 | 0 | 3 | 5 | 0 | 0 |
ID for identified proteins showing >twofold enrichment relative to control and five peptides or more in any of the Ubv samples.
Gene name for identified proteins showing >twofold enrichment relative to control and five peptides or more in any of the Ubv samples.
Controls correspond to HEK293T cells transfected with empty vector (V) or Ub Δ75G76G (Ub).
Numbers of endogenous peptides corresponding to the identified protein.
USP47 is a known interactor of Fbw1 and Fbw11 (30).
To determine whether Ubvs are able to disrupt interactions between Cul1 and Skp1–F-box complexes in cells, exogenously expressed Fbw7 or Fbw11 was immunoprecipitated in the absence or presence of Ubv. Expression of Ubv.Fw7.5 monomer or dimer significantly reduced or completely abrogated the coimmunoprecipitation of Cul1 with Fbw7, respectively, but did not affect coimmunoprecipitation of Skp1 (Fig. 4B). In the case of Ubv.Fw11.2, expression of the dimer, but not the monomer, caused significant reduction in the amount of Cul1 (but not Skp1) that coimmunoprecipitated with Fbw11, and this was consistent with the fact that the dimer but not the monomer coimmunoprecipitated with Fbw11 (Fig. 4C). Thus, coimmunoprecipitation assays show that both Ubv.Fw7.5 and Ubv.Fw11.2 interfere with the interactions between Skp1–F-box complexes and Cul1 in cells but do not affect interactions between Skp1 and F-box proteins, although dimerization is required to observe this effect in the case of Ubv.Fw11.2.
To determine whether cellular expression of Ubv.Fw7.5 or Ubv.Fw11.2 led to inhibition of their corresponding ligases, we analyzed the stability of ligase substrates. Expression of Ubv.Fw7.5 in either monomeric or dimeric format increased protein levels and decreased degradation rate of the SCFFbw7 substrates Cyclin E and c-Myc to levels comparable with those observed upon expression of an siRNA targeting Fbw7 but had no effect on substrates of other SCF ligases, demonstrating that the observed inhibition was specific (Fig. 4D and Fig. S4C). In the case of Ubv.Fw11.2, assays were performed in the presence of an siRNA targeting Fbw1 to reduce levels of SCFFbw1 (Fig. S4B), which shares substrates with SCFFbw11. Expression of the Ubv.Fw11.2 dimer and monomer increased the abundance and decreased the degradation rate of the SCFFbw11 substrates Cdc25A and Wee1, which was similar to stabilization observed upon expression of an siRNA targeting Fbw11 (Fig. 4E). Expression of the Ubv.Fw11.2 monomer had a smaller effect, consistent with the dimer being much more effective than the monomer in disruption of the interaction between Cul1 and the Skp1–Fbw11 complex (Fig. 4C). The inhibitory effect of Ubv.Fw11.2 was specific to SCFFbw11, as it did not affect substrates of other SCF ligases (Fig. S4D) and it did not stabilize substrates of Fbw1/11 in the background of Fbw11 siRNA treatment (Fig. S4E). Because Fbw7 and Fbw11 are involved in cell cycle progression (4), we also tested whether inhibition of these E3 ligases by Ubvs exerts any effect on cell cycle. Although we did not detect any large effects, the small changes that were observed (decrease in G1 population for Ubv.Fw7.5 and increase in G2/M population for Ubv.Fw11.2) (Fig. S4 F and G) were similar to those obtained with siRNA treatment and consistent with the previously reported effects of Fbw7 (21) and Fbw11 inhibition (22). Taken together, these data show that engineered Ubvs interact with endogenous Skp1–F-box complexes in cells and cause displacement of Cul1 and consequent inhibition of specific SCF E3 ligases.
Discussion
Designing specific inhibitors of the SCF E3 ligases has been challenging because of their multisubunit nature and the absence of distinct catalytic sites. Small-molecule inhibitors have been developed, which function by disrupting substrate binding directly or through allosteric mechanisms, or by disrupting the interaction between the F-box protein and Skp1 (reviewed in ref. 1). Our study suggests a novel method of specifically inhibiting SCF ligases by targeting the Cul1 binding surface on the Skp1–F-box interface. The interaction between Skp1–F-box and Cul1 in vitro has been reported (17) and confirmed here (Fig. S2 A–D) to be extremely tight. However, despite these high affinities, our data show clearly that Ubvs are able to disrupt the interactions between Cul1 and Skp1–F-box complexes in cells (Fig. 4 B and C). It is possible that the inhibitory activity of Ubvs in cells may be enhanced by endogenous cellular factors, such as the Cand1 protein, which binds Cul1 and promotes its dissociation from Skp1–F-box complexes. (17).
A major advantage of inhibiting SCF ligases by targeting the F-box domain is that the entire F-box family (Fig. S5) may be inhibited in a systematic manner without knowledge of F-box–substrate interactions, which are poorly characterized for most members of the family. Furthermore, Skp1–F-box domain complexes are easier to purify and are more amenable to structure determination than their full-length counterparts, and this should facilitate the search for inhibitors with our approach. We have shown that Ubvs selected for binding to Skp1–F-box domain complexes are biologically active as inhibitors of SCF function that act by disrupting binding of Cul1 (Fig. 4). This opens avenues for the use of these Ubv inhibitors as tools to validate potential drug targets and to aid the development of small-molecule inhibitors.
Fig. S5.

Sequence alignment of human F-box domains. Ubv binding region was defined by alignment to Fbw7 residues that fall within 10 Å of Ubv.Fw7.1 in the structure of Skp1tr–Fbw7F-box–Ubv.Fw7.1 complex. Sequences were clustered according to sequence identity in the Ubv binding region using average distance method. Fbw7 and Fbw11 are highlighted by red boxes.
It is intriguing to speculate that Ubvs described in this study target a natural Ub binding site [as observed for Ubvs targeting deubiquitinases (6)], which is relevant to a natural mechanism for regulation of SCF function. In particular, it is striking that only six mutations were sufficient to generate a high affinity binder to the Skp1tr–F-boxFbw7 complex (Fig. 2C), suggesting that this surface may be predisposed for binding to Ub. To explore this possibility, we looked for binding of monomeric Ub to the Skp1–F-boxFbw7 complex using NMR spectroscopy, but we did not find any evidence of interaction (Fig. S6). However, it is possible that the Skp1–F-box interface is involved in a regulatory interaction with more complex Ub structures such as covalently attached Ub chains. For example, inhibition of Cul1 binding by growing Ub chains on the substrate might signal the termination of the ubiquitination reaction or Ub chains attached to the F-box protein itself might function to accelerate the exchange of F-box subunits in the SCF complex, in a manner analogous to the effects of Cand1 binding to Cul1 (17). Further experiments aimed at studying the interaction of more complex Ub structures with the binding surface identified in this study might uncover new mechanisms regulating SCF function.
Fig. S6.

NMR analysis of N15 labeled ubiquitin in the absence and presence of unlabeled Skp1–F-boxFbw7 protein. Heteronuclear single quantum correlation spectra of 150 µM N15-labeled Ub (red), 150 µM N15-labeled Ub + 240 µM unlabeled Skp1–F-boxFbw7 added (black), and overlay of the two spectra. Absence of chemical shift changes in Ub N15 spectra upon addition of Skp1–F-boxFbw7 indicates no interaction in the micromolar range.
In summary, we have discovered a previously unidentified mechanism for inhibition of SCF ligases using engineered Ubvs that target the Skp1–F-box interface and inhibit Cul1 binding. We demonstrate that high specificity is attainable by this method, as exemplified by Fbw11 inhibitors that can discriminate against even the close homolog Fbw1. However, the ability to engineer inhibitors with broader specificities could also be useful, as it could be exploited to inhibit groups of SCF ligases containing similar F-box proteins. We anticipate that the Ubv inhibitors described here will be useful as tools for studying the function of SCF enzymes and for facilitating the discovery of small-molecule inhibitors of these enzymes through target validation, displacement screens, and structure-based design.
Materials and Methods
Protein Purification and Structure Determination.
His-tagged proteins were expressed in Escherichia coli BL21 and purified by Ni-NTA chromatography using standard techniques. See Table S1 for detailed list of all expression constructs. Refer to SI Materials and Methods for further details. The structure of the Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex was deposited in the Protein Data Bank with PDB ID code 5IBK.
Phage-Displayed Ubv Library Construction, Binding Selections, and in Vitro Binding Assays.
Previously described methods were used for the construction of phage-displayed Ubv libraries, for binding selections, for the isolation of individual binding Ubv-phage clones, and for phage and protein ELISAs to estimate affinities (6). Refer to SI Materials and Methods for specific details describing library construction (Table S4), phage selections, ELISAs, and SPR analysis.
Table S4.
Oligonucleotides used for construction of Ubv Libraries
| Oligo | Sequence* | Library |
| oMG210 | ATG CAG ATT TTC GTG (5) (5) (5) (5) (6) (6) GGT (7) (6) (5) CGT ACC (7) (6) (5) ATC ACC CTC GAG GT | 2 |
| oMG212 | AAG ATC CAG GAT AAG (7) (5) (5) GGA ATT CCT CCT GAT CAG CAG (5) (8) (8) CTG (5) (1) (1) TTT (8) (6) (6) (6) (7) (8) (5) (5) (7) (6) (8) (6) CTG GAA GAT GGA CGT | 2 |
| oMG214 | ATT CAA AAG GAG TCT (5) (6) (8) CTT (6) (7) (8) CTT (7) (8) (7) (8) (8) (7) (N5) (8) (8) (8) (8) (8) (6) (7) (8) (7) (7) (8) (7) (7) (8) GGC GGT GGC GGA TCC | 2 |
| oMG281 | ATT TTC GTG AAA ACCNNK NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK ACC ATC ACC CTC GAG | 3 |
| oMG282 | ATT TTC GTG AAA ACC NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK ACC ATC ACC CTC GAG | 3 |
| oMG283 | ATT TTC GTG AAA ACC NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK NNK ACC ATC ACC CTC GAG | 3 |
| oMG289 | ATTTTCGTGAAAACC (8) (5) (9) (6) (6) (6) (8) (5) (9) (5) (5) (5) (8) (6) (9) (7) (7) (8) (8) (6) (9) (8) (5) (9) (6) (5) (9) (5) (5) (9) (5) (5) (9) (8) (5) (9) ACCATCACCCTCGAG | 4 |
Numbers denote specific nucleotide mixtures: 5 = 70% A and 10% other nucleotides; 6 = 70% C and 10% other nucleotides; 7 = 70% G and 10% other nucleotides; 8 = 70% T and 10% other nucleotides; 9 = 90% T and 10% G. “N” denotes an equimolar mixture of all four nucleotides. “K” denotes an equimolar mixture of G and T.
Cell-Based Assays.
Genes encoding for FLAG-tagged Ubvs were cloned into pcDNA3.1/nFLAG-Dest vector for monomer expression or into the same vector modified to encode a GCN4 leucine zipper dimerization sequence (RMKQLEDKIEELLSKIYHLENEIARLKKLIGER) inserted in place of vector nucleotides 944–976 for dimer expression. Cul1, Fbw11, Fbw1, and Fbw7 were expressed from pcDNA3.1 based-vectors (see Table S1 for additional details). See SI Materials and Methods for additional details on mass spectrometry analysis, flow cytometry analysis, coimmunoprecipitation, and functional assays.
SI Materials and Methods
Phage-Displayed Ubv Library Design and Construction.
Library 1 in this study is the same as Library 2 in a previous study (6). Libraries 2, 3, and 4 in this study were constructed using methods described previously (23). For the construction of Library 2, a phagemid designed for the phage display of Ub (6) was subjected to site-directed mutagenesis with degenerate oligonucleotides to simultaneously mutate three regions in the gene encoding for Ub. Positions were diversified with a “soft randomization” strategy (24), in which the nucleotide ratio at degenerate positions was adjusted to 70% of the WT nucleotide and 10% of each of the other nucleotides. See Fig. S1 for original sequence and positions targeted for diversification and Table S4 for oligonucleotides used for library construction. For the construction of Libraries 3 and 4, a phagemid was designed for the display of an Ub variant in which positions 1–35 were WT sequence and positions 42–76 were the sequence of Ubv.Fw7.5. For the construction of Library 3, a set of mutagenic oligonucleotides was used to replace Ub positions 8–11 with completely random sequences containing 11–13 residues (Fig. S1 and Table S4). For the construction of Library 4, a mutagenic oligonucleotide was used to replace positions 8–11, with a soft-randomized sequence corresponding to the sequence of Ubv.Fw11.1 (Fig. S1 and Table S4). The diversities of the constructed libraries were as follows: Library 2, 2.2 × 109; Library 3, 5.0 × 109; Library 4, 1.5 × 109.
Selection of Ubv Variants.
GST-tagged target proteins (GST-Skp1:His-F-box) were coated on 96-well MaxiSorp plates (Thermoscientific 12565135) by adding 100 µL of 1 µM proteins and incubating overnight at 4 °C. Five rounds of binding selections with phage library pools were performed against immobilized proteins, as described previously (23). To eliminate Ubv-phage that bound nonspecifically, input phage pools were either mixed with nontarget proteins (round 1) or preincubated on plates coated with nontarget proteins (rounds 2–5). The nontarget proteins were GST for selections with Libraries 1 and 2 or a mix of nontarget Skp1-F-box complexes for selections with Libraries 3 and 4.
ELISAs.
GST-tagged target proteins were immobilized on 384-well MaxiSorp plates (Thermoscientific 12665347) by adding 30 µL of 1 µM proteins for overnight incubation at 4 °C or for 2-h incubation at room temperature. Phage and protein ELISA against immobilized proteins were performed as described previously (23), except that three washes were performed for all wash steps and volumes were scaled down from 100 µL to 30 µL to accommodate the 384-well format. Binding of phage was detected using anti–M13-HRP antibody (1:5,000 dilution; GE Healthcare 27-9421-01) and binding of FLAG-tagged ligands (Ubv or Cul1) was detected using anti-FLAG-HRP antibody (1:5,000 dilution; Sigma A8592). To measure protein ELISA EC50 values, the concentration of ligand proteins (Ubv or Cul1) was varied, whereas the concentration of target proteins (GST-Skp1:His–F-box) immobilized on the plate remained constant. EC50 values were calculated by fitting the obtained binding curves to four parameter logistic nonlinear regression model and corresponded to ligand concentration (curve inflection point) at which 50% of binding was observed. To measure protein ELISA IC50 values, the concentration of target in solution (Skp1–F-box) was varied, whereas the concentration of target immobilized on the plate (GST-Skp1:His–F-box) and concentration of ligand (Ubv) in solution remained constant. IC50 values, which corresponded to the concentration of target in solution which inhibited 50% of ligand binding to the immobilized target, were calculated by fitting the data as described for EC50 values.
SPR Analysis.
SPR measurements were performed at 25 °C using ProteOn XPR36 instrument (Bio-Rad). Skp1tr–F-boxFbw7 and Skp1–F-boxFbw7 ligands were immobilized by amine coupling to GLC sensor chip surface. Cul1 N-terminal domain (NTD) was diluted into PBT buffer (PBS, 0.05% Tween, and 0.5% BSA) and injected for 360 s at 50 µL/min. Dissociation was monitored for 1,200 s in PBT buffer. Sensorgrams were fitted to 1:1 Langmuir model using ProteOn Manager Software (Bio-Rad).
Protein Expression and Purification.
His-tagged Fbw7 and GST-tagged Skp1 were coexpressed from dicistronic mRNA. Ubvs and Cul1 NTD were expressed with His-FLAG tag. See Table S1 for detailed list of all expression constructs. All proteins were expressed in Escherichia coli BL21 (pLys) cells, which were grown to OD600 0.6–1.0 and induced with 1 mM IPTG either overnight at 16 °C (for Skp1–F-box complexes and Cul1 NTD) or for 3 h at 37 °C (for Ubvs). Cells were lysed and proteins were purified by Ni-NTA chromatography using standard techniques. Because expression of Skp1 was higher than expression of F-box proteins, purifying Skp1–F-box complexes through His-tagged F-box proteins ensured that only complexes were purified. Eluted proteins were dialyzed into 50 mM Hepes, pH 7.5, 500 mM NaCl, 10% (vol/vol) glycerol, 1 mM DDT buffer, and stored at 4 °C or frozen at –80 °C for further applications.
Protein Purification for Crystallization and Structure Determination.
Complex consisting of GST-tagged Skp1tr and His-tagged F-boxFbw7 was purified on Ni-NTA resin from cells coexpressing both proteins (see Table S1 for constructs used). To remove GST and His purification tags [containing tobacco etch virus (TEV) protease cleavage sites] the obtained complex was first bound to glutathionine resin, next eluted from the resin by TEV cleavage of GST tag, and finally repurified on Ni-NTA resin to remove His-tagged TEV protease and other impurities. Similarly, His-tagged Ubv.Fw7.1 was purified on Ni-NTA resin, His tag (containing a TEV protease cleavage site) was removed by TEV protease and cleaved Ubv was repurified on Ni-NTA resin. Cleaved Skp1tr–F-boxFbw7 complex was mixed with excess of cleaved Ubv.Fw7.1 and subjected to gel-filtration chromatography. A single peak corresponding to Skp1tr–F-boxFbw7–Ubv.Fw7.1 complex was collected, exchanged into 20 mM Hepes pH 7.5, 200 mM NaCl, 1 mM DTT buffer, and concentrated to 19 mg/mL. Crystals were grown by mixing equal volumes of Skp1–F-boxFbw7–Ubv.Fw7.1 solution with the reservoir solution [100 mM acetate pH 4.5, 12% (wt/vol) PEG 4000, 15% (vol/vol) glycerol] and incubating at 20 °C. The crystals were cryoprotected by soaking in reservoir solution with a final glycerol concentration of 20% (vol/vol). Data were collected at NE-CAT 24 ID-C (Advanced Photon Source, Chicago, IL) and processed with HKL2000 (25). The structure was solved by molecular replacement with Phaser (26) using structures of Skp1tr–Fbw7 (PDB ID code 2OVR) and ubiquitin (PDB ID code 1UBQ) as search models. The model was rebuilt using Coot (27) and refined to 2.5 Å with a working Rvalue of 20.0% and Rfree of 25.0% using PHENIX (28).
Cell-Based Functional Assays.
On day 0, six-well plates were seeded with 4 × 105 HEK293T cells. On day 1, cells were transfected with 2 µg of plasmid DNA (empty vector, various FLAG-Ubv, or various FLAG-F-box constructs) using the X-tremeGENE transfection reagent (Roche 06365809001), according to the manufacturer’s protocol. After 6 h, media was removed and replaced with fresh media, and cells were subjected to a second round of transfection with 10 nM siRNA (Control, Fbw1, Fbw11, Fbw1+Fbw11, or Fbw7) using Lipofectamine RNAiMAX (Invitrogen 13778-075), according to the manufacturer’s instructions. siRNA included the following: Silencer Select Negative Control #1 (Invitrogen 4390843); Fbw1: CGGAAGAGUUUUUCGACUAtt (Invitrogen 17110); Fbw11: GGUUGUUAGUGGAUCAUCAtt (Invitrogen s23485); Fbw7: CGGGUGAAUUUAUUCGAAAtt (Invitrogen s30665).
On day 2, media was replaced with fresh media. On day 3, CHX (100 µg/mL) was added for 0–6 h. Cells were lysed in lysis buffer (Cell Signaling 9803) and cell lysates were subjected to Western blot analysis with antibodies against endogenous proteins [Cdc25A (Upstate 05–743), Wee1 (Cell Signaling 4936), c-Myc (Santa Cruz SC-40), Cyclin E (ABCAM 3927), p27 (BD Transduction Laborotories 610241), Cry2 (Abcam 93802), GADPH (Cell Signaling 2118L)], or FLAG-tagged proteins (Sigma-Aldrich A8592).
Flow Cytometry Analysis.
HEK293T cells were treated as described in cell-based functional assays section. Two days posttransfection, cells were resuspended in PBS, fixed by addition of 70% (vol/vol) ethanol, and stored at −20 °C. Immediately before flow cytometry analysis, fixed cells were washed in PBS, resuspended in 500 µL PBS at concentration of 1 × 106 cells/mL, and stained by addition of Hoechst 33342 (Life Technologies H3570) dye to a final concentration of 4 µg/mL Stained cells were analyzed by UV excitation at 355 nm on a BD LSRFortessa X-20 cell analyzer and detected using a 450/50-nm bandpass filter. The data acquired was analyzed by FlowJo10 software.
Immunoprecipitation Assays.
HEK293T cells were grown to 70–80% confluency on 10-cm plates and transfected with 10 µg total plasmid DNA (HA-F-box, FLAG-Ubv, FLAG-Cullin1, empty vector or various combinations) using the X-tremeGENE 9 transfection reagent (Roche 06365809001) according to the manufacturer’s instructions. Cells were harvested 2 d posttransfection and cell pellets were frozen for further applications. For coimmunoprecipitation analysis, cells were resuspended in 1 mL lysis buffer [25 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 5% (vol/vol) glycerol, protease inhibitor mixture (Sigma S8830)], and after 1 h incubation at 4 °C, cell lysate was clarified by centrifugation at 21,130 × g for 10 min. Next, 800 µg of total cell lysate was incubated with 3 µg of anti-FLAG (Sigma F1804) or anti-HA (Sigma 3663) antibody overnight at 4 °C. Immunoprecipitations were performed using Protein A/G Agarose (Thermo Scientific Pierce 20422) according to the manufacturer’s protocol. Immunoprecipitated proteins were visualized by Western blot using anti–FLAG-HRP (Sigma A8592), anti-HA-HRP (Sigma H6533), and anti-Skp1 (Abcam 10536) antibodies.
Mass Spectrometry Analysis.
Frozen cell pellets were thawed into 1 mL High Salt AFC buffer [10 mM Tris⋅HCl pH7.9, 420 mM NaCl, 0.1% Nonidet P-40, 1 mM sodium orthovanadate, 2 mM sodium pyrophosphate, 10 mM NaF, protease inhibitor mixture (Sigma S8830)]. Cell suspensions were then were subjected to three freeze-thaw cycles, sonicated (five cycles of 0.3-s on and 0.7-s off) and incubated for 30 min at 4 °C with 12.5–25 U of benzonase nuclease (Sigma E1014). The samples were centrifuged at 15,871 × g for 30 min at 4 °C and 10-µL slurry of M2 anti-Flag beads (Sigma 8823) was added for overnight incubation. Beads were washed two times with low salt AFC buffer (10 mM Tris⋅HCl, pH7.9, 100 mM NaCl, 0.1% Nonidet P-40) and three times with low salt AFC buffer without detergent. Immunoprecipitated proteins were eluted from the beads with 0.5 M ammonium hydroxide, 4 × 50 µL for a total of 200 µL. Samples were snap-frozen in liquid nitrogen and dried. Further preparation of samples, mass spectrometry, and analysis of obtained data were performed as previously described (29).
Acknowledgments
This work was supported by the Canadian Institutes of Health Research Operating Grants MOP-136956 (to S.S.S.), MOP-126129 (to M.T. and F.S.), Foundation Grant (to F.S.), and postdoctoral Fellowship (to M.G.); and an innovation grant from the Canadian Cancer Society Research Institute (to M.T. and F.S.). This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by Award GM103403 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the US Department of Energy, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 5IBK).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1519389113/-/DCSupplemental.
References
- 1.Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. J Clin Invest. 2014;124(1):6–12. doi: 10.1172/JCI71602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhowmick P, Pancsa R, Guharoy M, Tompa P. Functional diversity and structural disorder in the human ubiquitination pathway. PLoS One. 2013;8(5):e65443. doi: 10.1371/journal.pone.0065443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin J, et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18(21):2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14(4):233–247. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, et al. Targeting the ubiquitin pathway for cancer treatment. Biochim Biophys Acta. 2015;1855(1):50–60. doi: 10.1016/j.bbcan.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ernst A, et al. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339(6119):590–595. doi: 10.1126/science.1230161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pashkova N, et al. WD40 repeat propellers define a ubiquitin-binding domain that regulates turnover of F box proteins. Mol Cell. 2010;40(3):433–443. doi: 10.1016/j.molcel.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: Mechanisms and opportunities. Cancer Cell. 2014;26(4):455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busino L, et al. Fbxw7α- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14(4):375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeishi S, et al. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23(3):347–361. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 11.Lau AW, Fukushima H, Wei W. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci (Landmark Ed) 2012;17:2197–2212. doi: 10.2741/4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: Multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26(1):131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 13.Schulman BA, et al. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature. 2000;408(6810):381–386. doi: 10.1038/35042620. [DOI] [PubMed] [Google Scholar]
- 14.Orlicky S, et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol. 2010;28(7):733–737. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CV, et al. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol. 2004;340(5):1073–1093. doi: 10.1016/j.jmb.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 16.Zheng N, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416(6882):703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 17.Pierce NW, et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell. 2013;153(1):206–215. doi: 10.1016/j.cell.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki H, et al. Homodimer of two F-box proteins betaTrCP1 or betaTrCP2 binds to IkappaBalpha for signal-dependent ubiquitination. J Biol Chem. 2000;275(4):2877–2884. doi: 10.1074/jbc.275.4.2877. [DOI] [PubMed] [Google Scholar]
- 19.Welcker M, et al. Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes Dev. 2013;27(23):2531–2536. doi: 10.1101/gad.229195.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262(5138):1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 21.Wu XZ, et al. MiR-27a-3p promotes esophageal cancer cell proliferation via F-box and WD repeat domain-containing 7 (FBXW7) suppression. Int J Clin Exp Med. 2015;8(9):15556–15562. [PMC free article] [PubMed] [Google Scholar]
- 22.Guardavaccaro D, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4(6):799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 23.Fellouse FA, Sidhu SS. Making antibodies in bacteria. In: Howard GC, Kaser MS, editors. Making and Using Antibodies. CRC Press; Boca Raton, FL: 2007. pp. 157–180. [Google Scholar]
- 24.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcon E, et al. Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation. Cell Reports. 2014;8(1):297–310. doi: 10.1016/j.celrep.2014.05.050. [DOI] [PubMed] [Google Scholar]
- 30.Peschiaroli A, Skaar JR, Pagano M, Melino G. The ubiquitin-specific protease USP47 is a novel beta-TRCP interactor regulating cell survival. Oncogene. 2010;29(9):1384–1393. doi: 10.1038/onc.2009.430. [DOI] [PMC free article] [PubMed] [Google Scholar]