

Abstract
Significance: The pulmonary circulation is a low-pressure, low-resistance, highly compliant vasculature. In contrast to the systemic circulation, it is not primarily regulated by a central nervous control mechanism. The regulation of resting membrane potential due to ion channels is of integral importance in the physiology and pathophysiology of the pulmonary vasculature. Recent Advances: Redox-driven ion conductance changes initiated by direct oxidation, nitration, and S-nitrosylation of the cysteine thiols and indirect phosphorylation of the threonine and serine residues directly affect pulmonary vascular tone. Critical Issues: Molecular mechanisms of changes in ion channel conductance, especially the identification of the sites of action, are still not fully elucidated. Future Directions: Further investigation of the interaction between redox status and ion channel gating, especially the physiological significance of S-glutathionylation and S-nitrosylation, could result in a better understanding of the physiological and pathophysiological importance of these mediators in general and the implications of such modifications in cellular functions and related diseases and their importance for targeted treatment strategies. Antioxid. Redox Signal. 22, 465–485.
Introduction
“Why grass is green or why our blood is red, are mysteries which none have reach'd unto.” John Donne (1571–1631).
The interaction between the air, the lungs, and the blood has fascinated philosophers and physiologists for centuries. In 1669, an Oxford physician, Richard Lower, reported that it was exposure to air in the lungs that caused the change from dark venous blood to bright arterial blood. However, more than another 200 years elapsed before Bradford and Dean described constriction of the pulmonary arteries (PA) caused by asphyxia (20). The recognition of the involvement of ion channels in hypoxic pulmonary vasoconstriction (HPV) has been relatively recent; of L-type calcium channels in 1976 (86) and of potassium (K+) channels in 1992 (118). The suggestion that redox changes might play a role in the mechanism of HPV was initially made in 1986 (9) and focused on ion channels in 1993 (4). Subsequently, many laboratories have found changes in reactive oxygen species (ROS) and the redox couples GSH/GSSG and NAD(P)H/NAD(P) in the course of HPV and of chronic hypoxic pulmonary hypertension (PH).
Besides ROS, small signaling chemical species such as nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) molecules participate in the regulation of pulmonary vascular function. Independent of transporters, membrane receptors, or second messenger systems, they freely diffuse through cell membranes and elicit various responses. Oxidative stress is postulated to play a prominent role in the etiology of vascular and ventricular dysfunction that is associated with cardiovascular disease (53, 129, 134, 185). In the pulmonary vasculature and in the heart, changes in the ROS levels result in changes in the redox state of proteins, some of which can be reversed. In part, reversible protein oxidation involves the free thiol (-SH) side chain of cysteine residues that can undergo a number of redox-mediated molecular modifications which may elicit positive or negative changes in protein function (34, 185). Thus, under conditions where ROS levels are markedly elevated and/or oxidoreductase systems are impaired, there is significant alteration in the physiological function of cells, mediated by direct protein oxidation or changes in protein interaction with redox molecules. Although redox-target specification is, no doubt, dictated by the innate susceptibility of the target, additional measures are in place within the cell to prevent accumulation of ROS to toxic levels. This regulation is facilitated, in part, by the discrete subcellular compartmentalization of ROS production, the restricted availability of NADPH oxidase (Nox) activating and regulatory subunits, and the ubiquitous presence of cytosolic scavenging and neutralizing enzymes such as Cu/Zn superoxide dismutases (SODs), glutathione peroxidase (GPx), and peroxiredoxins.
The role of redox regulation in the pulmonary circulation emerged, in part, because the pulmonary vasculature constricts in response to hypoxia, while the ductus arteriosus (DA) and systemic vessels, such as the renal arteries, dilate. It is possible that redox control of ion channels might provide a mechanism which could explain these disparate responses. It is clear that the same redox signal can cause opposite changes of the gating of K+ channels in the resistance PAs and the DA (102). For instance, the reducing agent dithiothreitol (DTT) inhibits K+ current and causes depolarization and contraction in the smooth muscle cells (SMCs) of the PA (PASMC), while it activates K+ current and causes hyperpolarization and relaxation of DASMCs. The oxidizing agent 5′,5′-dithio-bis(2-nitrobenzoic) acid (DTNB) has the opposite effects on K+ current, membrane polarization, and tone in PA and DA SMCs. Similar contrary actions have been described in response to changes in endogenous ROS in PA and renal artery SMCs (89). The fact that the role of redox signaling in the control of ion channels and tone in the PA is not yet agreed probably derives from the variety of ROS generated and methods of their inactivation, variation in ion channel expression in different vessels and different stages of maturation, and the use of a number of different experimental techniques. Are the relevant ROS produced in the mitochondria, by Noxs, in the plasma membrane electron transport (PMET) system, or in all of these places? Which ROS is key, superoxide anion, H2O2 or another radical (170)? Given that ROS may rise and fall in different places in the PASMCs during hypoxia, the location of changes is likely to be critical (168). ROS can have different physiologic and pathologic roles, and the overlap of these effects can cause confusion. In this regard, when exogenous ROS or anti-oxidant enzymes are added to cells in culture, attention should be paid as to whether the concentrations used and the sites of administration are physiological or pathological. Furthermore, since ion channels and calcium-handling proteins act as effectors, differences in the ion channels expressed in resistance and conduit vessels and in fetal and adult vessels also have to be considered. Consequently, much of the work discussed in this review is relevant to our understanding of the interaction of redox status, ion channel control, and tone in the pulmonary vasculature.
Redox Buffer Systems
Glutathione
The glutathione system includes reduced (GSH) and oxidized (GSSG) forms of glutathione, the enzymes required for its synthesis and recycling, such as gamma-glutamate cysteine ligase (γ-GCL), glutathione synthetase (GS), glutathione reductase (GR), and gamma-glutamyl transpeptidase (γ-GGT); also, the enzymes required for its use in metabolism and in mechanisms of defense against free radical-induced oxidative damage, such as glutathione s-transferases (GSTs) and GPxs. The glutathione system is one of the most important antioxidant systems for most tissues in vivo, and the ratio of oxidized to reduced glutathione (GSSG/GSH) is a widely used indicator of oxidative stress within different cell types. The ratio of reduced glutathione (GSH) to glutathione disulfide (GSSG; oxidized form) is >10 (43). Under normal circumstances, GSH levels within the cell are maintained via recycling of GSSG by the NADPH-dependent enzyme GR (Fig. 1). However, under pathological conditions, the GSH/GSSG ratio can decrease significantly as shown during ischemia/reperfusion in the right ventricle (109).
FIG. 1.
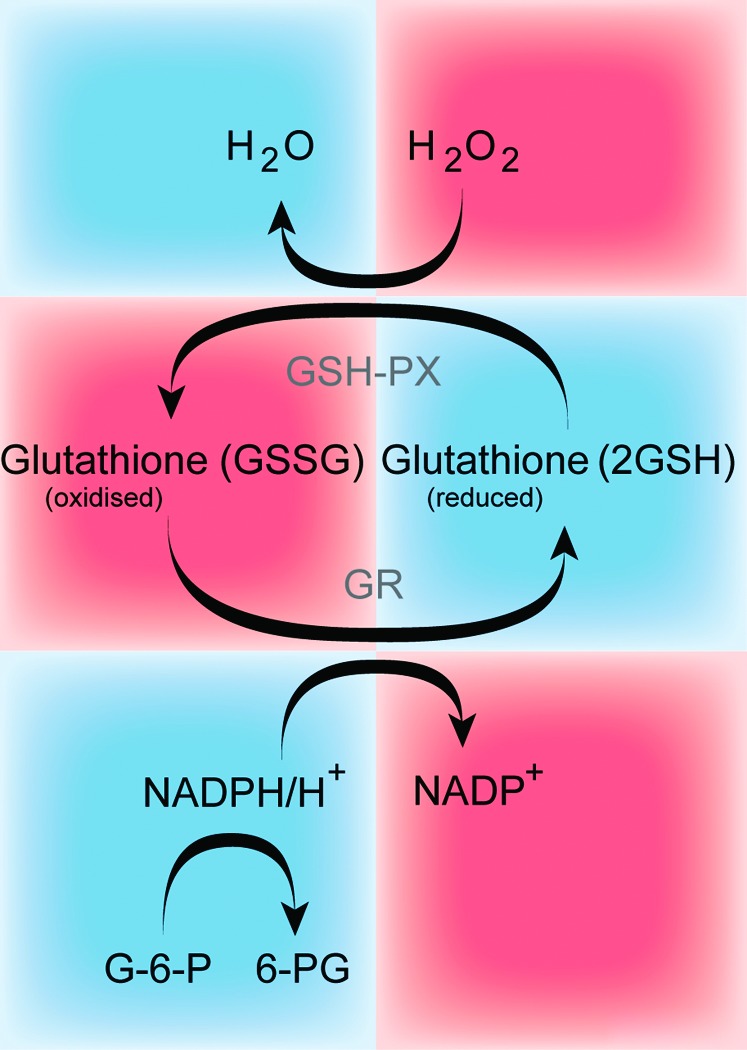
The schematic of the glutathione system. GSH is synthesized from amino acids by the action of γ-glutamylcysteine synthetase and glutamyl synthase. GSH undergoes the GSH-PX coupled reaction, thereby detoxifying ROS such as H2O2. During this reaction, GSH is oxidized and generates GSSG, which is recycled back to GSH by the action of GR at the expense of reduced nicotinamide (NADPH/H+), thus forming the redox cycle. GSH, glutathione; GSH-PX, glutathione-peroxidase; GR, glutathione reductase; H2O2, hydrogen peroxide; ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
S-glutathionylation is a redox-dependent post-translational modification with growing recognition in signal transduction. Protein S-glutathionylation (P-SSG), the reversible modification of cysteine thiols by glutathione, is elevated in response to oxidative stress. Initially, the function of S-glutathionylation was thought to be the protection of cysteine residues against over-oxidation to sulfenic (RSOH), sulfinic (RSO2H), or sulfonic (RSO3H) acids, which can result in protein inactivation. Now, S-glutathionylation is considered a real regulatory event in “redox signaling” and it is involved in several pathways, often cross-talking with each other (180). Chen et al. have shown in an elegant study that oxidized glutathione (GSSG) induces dose-dependent S-glutathionylation of endothelial nitric oxide synthase (eNOS) in human endothelial cells (EC), with loss of NO and gain of superoxide (O2•−) generation, which is associated with impaired endothelium-dependent vasodilatation in the systemic circulation (23). In the lung, bleomycin induces pulmonary fibrosis and pulmonary vascular remodeling in animal models. Day et al. showed, more than 10 years ago, that bleomycin up-regulates GSH and γ-glutamylcysteine synthetase after only 3 h of treatment, indicating the early involvement of the system in the development of the pulmonary vascular remodeling (26). In pulmonary arterial myocytes, the reducing agents, such as GSH, DTT, and NADH, inhibit the large conductance calcium-dependent potassium channel (BKCa) activity; whereas oxidizing agents, such as 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) and GSSG, stimulate it (55). Although the role of ATP-sensitive K+ channel (KATP) in the PAs is almost unknown, it is worthwhile to introduce a recent study by Yang et al. showing that H2O2 or pyridine disulfides, pharmacological tools which react with protein cysteine residues similar to S-glutathionylation, markedly inhibit KATP current carried out by the potassium inwardly rectifying channel (Kir)6.1 subunit. Simulation modeling of Kir6.1 S-glutathionylation suggested that after incorporation to residue 176, the channel remains in its closed state (181). Finally, Lock et al. have utilized the thiol-oxidant diamide to investigate the consequences of protein S-glutathionylation on the calcium handling protein inositol trisphosphate receptor (IP3R) in aortic EC. The authors found that Ca2+-induced Ca2+-release (CICR) via the IP3R is enhanced by diamide, which reacts with GSH and, therefore, promotes P-SSG formation (78).
Thioredoxin
Thioredoxin (Trx), Trx peroxidase, and thioredoxin reductase (TrxR) comprise the thioredoxin system that exists in nearly all living cells (Fig. 2). The Trx system reduces oxidized cysteine groups on proteins through an interaction with the redox-active center of TRX (Cys-Gly-Pro-Cys) and forms a disulfide bond, which, in turn, can be reduced by Trx reductase and NADPH. It functions in thiol-dependent thiol-disulfide exchange reactions and activation of transcription factors (such as NF-κB and AP-1) that are crucial to control of the reduced intracellular redox environment, cellular growth, defense against oxidative stress, and control of apoptosis. In line with these observations, both Trx and TrxR are already regarded as interesting targets for chemotherapy, and they represent novel therapeutic options for pulmonary vascular and/or right heart remodeling.
FIG. 2.
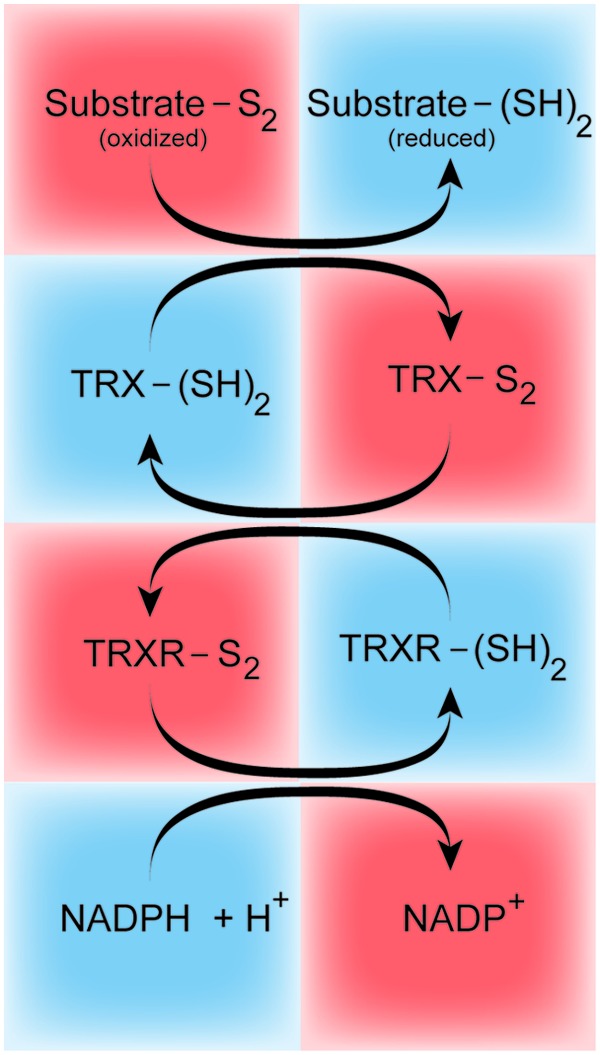
The thioredoxin system. The TRX in the oxidized (TRX-S2) or reduced [TRX-(SH)2] state. The oxidized thioredoxin is converted by the TRXR and NADPH to its reduced state, where TRX directly reduces disulfides in oxidized substrate proteins (Substrate-S2). TRX, thioredoxin; TRXR, thioredoxin reductase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Trx is ubiquitously expressed in EC (36) and in vascular smooth muscle cells (VSMC) of normal arteries (22). While treatment with H2O2 increases Trx expression in EC, suggesting that Trx is an ROS-inducible protein in EC, the expression of Trx in VSMC is not regulated by ROS. It appears that Trx acts to reduce cellular oxidative stress by scavenging ROS, and might, therefore, attenuate the pathology associated with vascular disease. Trx and TrxR are present in distinct isoforms that are predominantly cytosolic (Trx1 or TrxR1) or mitochondrial (Trx2 and TrxR2). While the Trx system has been extensively investigated in lung diseases and in epithelial cells, its role in pulmonary vascular cells and in right heart diseases is largely unknown. In the pulmonary vasculature, transient-receptor potential cation (TRPC) channels are widely expressed. Xu et al. have detected Trx as a novel type of ion channel agonist that acts through its reduced form to break a restraining intra-subunit disulfide bridge between cysteine residues in TRPC5, thereby stimulating the channel and resulting in an increased calcium influx, either as a homomeric assembly or as a hetermultimer with TRPC1. A transduction mechanism is, therefore, revealed that can directly couple ion channel activity to extracellular reduced thioredoxin (179).
NAD(P)H/NAD(P)+
The two specialized pyridine nucleotide systems (NAD+/NADH and NADP+/NADPH) are optimized for the ready exchange of electrons and for recognition by specialized nucleotide-binding domains located at the active sites of>200 different oxidoreductases (60). Moreover, these enzymes also can distinguish between reduced and oxidized states of the nucleotide and by specifically recognizing the phosphorylation state of the ribose ring, between NAD+ and NADP+. NADP(H) is recognized by enzymes that are primarily involved in anabolic (reductive) reactions, such as lipid or cholesterol synthesis or fatty acid chain elongation; whereas NAD(H) is recognized by enzymes which catalyze catabolic reactions of glycolysis and by components of the electron transport chain (ETC) and, therefore, play an important role in (pulmonary) SMCs.
Due to the low glycolytic flux and the ability of mitochondria to oxidize cytosolic NADH by the malate-aspartate and glycerol phosphate shuttles, under normal aerobic conditions, cytosolic nicotinamide-adenine dinucleotides are mostly oxidized (i.e., present as NAD+) with an NADH/NAD+ ratio<0.05 (16, 75). However, during ischemia, for example, the cytosolic NADH/NAD+ ratio can increase approximately 30 times (112). The ratio of NADH/NAD+ fluctuates in response to changes in metabolism. The key enzyme that regulates cytosolic NADH/NAD+ levels is glyceraldehydes(s) 3-phosphate dehydrogenase (GAPDH) which is located in the sarcoplasmic reticulum (SR) membrane (178). Thus, changes in activity of this enzyme may result in more significant fluctuations of local NADH/NAD+ levels in close proximity to SR-bound proteins (such as the ryanodine receptor [RyR] and sarcoplasmic/endoplasmic reticulum calcium ATPase [SERCA]) which might also explain the high compartmentalization of redox states within cytosolic subregions.
In PASMCs, changes in potassium conductance directly regulate membrane potential and thus vascular tone. Both KCa and the voltage-gated potassium channels (Kvs) are regulated by pyridine nucleotides (110, 113, 154, 157) (Fig. 3). The regulatory β-subunits of the Kv1.5 bind to pyridine nucleotides with a high affinity, enabling the Kvβ to respond to a wide range of metabolic conditions by being sensitive to changes in both NADP(H) and NAD(H) levels (77). Several observations indicate that NAD(P)+ binding induces a specific conformational change that prevents Kvβ-induced inactivation, supporting K+ conductance; whereas NADP(H) binding preserves or promotes inactivation (110, 111, 155–157, 175). In contrast, the Kv2.1 protein is not associated with a pyridine-binding subunit, such as Kvβ, and direct binding of pyridine nucleotide to Kv2.1 has not yet been demonstrated. Furthermore, recent work has shown that NAD+ modifies the activity of the transient-receptor potential cation channel, subfamily M (TRPM), member 2 channels and, thus, regulates calcium homeostasis. Data from whole-cell patch-clamp experiments show that intracellular dialysis with NAD+ evokes a large inward current in cells expressing TRPM2 channels. In HEK-293 cells, intracellular application of NAD+ results in instantaneous activation of the channel, suggesting that NAD+ directly activates the channel without the involvement of cytoplasmic or membrane components (63).
FIG. 3.

Regulation of potassium channels and the RyR/Ca2+ release channel (RyR2) by pyridine nucleotides. Hypothetical scheme showing the regulation of plasma membrane and intracellular ion channels by NAD(P)(H). Individual cells may or may not express the entire complement of ion channels shown in the figure. RyR, ryanodine receptor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
NO and S-nitrosylation
The radical gas NO diffuses freely from its site of production in the pulmonary EC to its target, soluble guanylate cyclase (sGC) in the smooth muscle, and is involved in a number of roles within the endothelium, regulating vascular tone, vascular growth, platelet aggregation, and modulation of inflammation (24). As such, decreased bioavailable NO results in endothelial dysfunction, which is a characteristic feature of many disease states.
NO is generated by the three isoforms of NO synthase (NOS): eNOS, neuronal (nNOS), and inducible (iNOS) isoenzyme. The classical NO signaling pathway is the activation of sGC enhancing cyclic guanosine monophosphate (cGMP) production, which, in turn, mediates vasodilatation. The alternative NO signaling pathway is S-nitrosylation (SNO; see next paragraph). NO has a modest role as a vasodilator in the pulmonary circulation under most basal physiological conditions. However, endogenous NO seems to suppress the full expression of HPV (8, 69, 70). In addition, since NO inhibits proliferation of VSMCs, it may also function to prevent the remodeling associated with the development of PH (142). Fagan et al. evaluated the importance of each NOS isoform in the maintenance of pulmonary vasomotor tone using isolated-perfused lungs from mice deficient in each NOS isoform. The investigators reported that under basal conditions, nNOS and iNOS have little or no role in the maintenance of low pulmonary tone in contrast to eNOS (32, 33). In addition, eNOS activity was an important determinant of pulmonary vascular responsiveness to chronic hypoxia, in vivo (32, 33, 143). The effect of eNOS-derived NO appeared to be most prominent under modest hypoxic stress when HPV was augmented in eNOS−/− mice (32, 33). The latter finding is, however, controversial (121, 144). Finally, a recent elegant study by Seimetz et al. highlighted the relevance of iNOS activity in bone marrow-derived cells for the development of PH in vivo (135). The reaction of NO with sGC results in a several 100-fold increase in production of cGMP from guanosintriphosphat (GTP). In VSMCs, the effects of cGMP are mediated through activation of its effector proteins, the cGMP-dependent protein kinase, cGMP-gated ion channels, such as calcium-activated potassium channels, and cGMP-regulated phosphodiesterases.
Recent investigations show that many effects of NO in the vasculature are mediated by S-nitrosylation, the covalent modification of a protein cysteine thiol by an NO group to generate an S-nitrosothiol as recently reviewed in detail (48, 74). The composition of SNO-based signaling complexes also includes NOSs; NO group donors, including S-nitrosoglutathione (GSNO) and other SNO-proteins that can participate in transnitrosylation reactions; and denitrosylases which curtail the signals. Ion channels and transporters participating in calcium homeostasis have been reported to be modulated by S-nitrosylation. Among Kvs, a recent report has shown that S-nitrosylation of the potassium voltage-gated channel, KQT-like subfamily, member 1 (KCNQ1) subunit facilitates the slowly activating component of the delayed rectifier K+ current (10); whereas S-nitrosylation exerts an inhibitory influence on Kv4.3 and thus the transient outward potassium current (39). In addition, Nunez et al. have shown that NO inhibits hKv1.5 channels by a cGMP-dependent mechanism and by the direct S-nitrosylation of the protein (100). Sun et al. as well as Poteser et al. have reported that S-nitrosylation of the α1C subunit of the L-type calcium channel inhibits the L-type calcium current, perhaps due to eNOS-induced NO production, which is adjacent to the calcium channels in the sarcolemmal membrane caveolae (120, 147). Finally, in cardiomyocytes, the RyR/Ca2+ release channel (RyR2) has been shown to be activated by S-nitrosylation (40, 148) (Fig. 4). The emergence of SNOs as second messengers and of S-nitrosylation as the pre-eminent NO-based signal may represent a new era in pulmonary vascular physiology, as a disruption of the SNO/redox balance in myocytes, for example, in pulmonary vascular remodeling can occur. Consequently, the restoration of this equilibrium might provide a fruitful approach to restoring pulmonary vascular and/or right ventricular performance.
FIG. 4.

S-nitrosylation of ion channels in myocytes. S-nitrosylation modulates function of different ion channels expressed either in the cell membrane (sarcolemma) or in the sarcoplasmic reticulum of myocytes. Arrows indicate activation, whereas dotted line indicates inhibition. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Reactive oxygen species
The cellular mechanisms responsible for HPV have been debated since the description of this physiological mechanism by Euler and Liljestrand (30). Since the 1970s, there has been a keen interest to identify specific ROS altered during hypoxia and their generating source(s). Thus, in the pulmonary circulation, Noxs and mitochondria have been identified as the major sources producing ROS and initiating ROS signaling. They seem to be interdependent, at least in acute HPV. However, there is still a major controversy as to whether ROS go up or down during hypoxia. Such a discrepancy is likely related to differences in preparations and detection methods (each of which has its own severe, specific limitations) as summarized in Table 1 [modified from Olschewski and Weir (104)]. Since the changes in ROS in the pulmonary circulation are not the focus of this review, we would like to highlight recent reviews dealing extensively with the topic (62, 96, 132, 150).
Table 1.
Changes in the Reactive Oxygen Species Production Under Hypoxia in Different Preparations
| Tissue/preparation | Hypoxia | [ROS]i | Technique of detection | References |
|---|---|---|---|---|
| Isolated rabbit lung | Minutes | ↓ | Lucigenin | Paky et al. (108) |
| Isolated rat lungs, pulmonary artery ring, PASMCs | Minutes | ↓ | Lucigenin, amplex red, dihydroethidium, H2DCF | Archer et al. (6) |
| Isolated rabbit lung | Minutes | ↓ | Lucigenin | Weissmann et al. (174) |
| Calf pulmonary arteries | Minutes | ↓ | Lucigenin | Mohazzab and Wolin (93) |
| Rat pulmonary artery | Minutes | ↓ | Lucigenin, amplex red, H2DCF | Michelakis et al. (89) |
| Rat pulmonary artery | Weeks | ↓ | L-012 (luminol analogue) amplex red | Bonnet et al. (19) |
| Human PASMCs | Hours | ↓ | Lucigenin, amplexred, dihydroethidium, H2DCF | Mehta et al. (88) |
| Human PASMCs | Minutes | ↓ | Dihydroethidium | Wu et al. (176) |
| Isolated mouse lung | Minutes | ↑ | Electron spin resonance | Weissmann et al. (173) |
| Rat pulmonary arteries | Minutes | ↑ | H2DCF | Jernigan et al. (56, 57) |
| Weeks | ↑ | |||
| Rat pulmonary arteries | Hours | ↑ | Dihydroethidium | Wang et al. (162) |
| Mouse pulmonary arteries, rabbit PASMCs | Minutes | ↑ | H2DCF | Paddenberg et al. (107) |
| Porcine pulmonary arteries | Minutes | ↑ | Electron paramagnetic resonance, H2DCF, lucigenin | Liu et al. (76) |
| Weeks | ↑ | |||
| Calf PASMCs | Minutes | ↑ | Lucigenin | Marshall et al. (85) |
| Rat PASMCs | Minutes | ↑ | H2DCF | Killilea et al. (61) |
| Rat PASMCs | Minutes | ↑ | Fluorescence resonance energy transfer probe, H2DCF | Waypa et al. (165, 166) |
| Mouse PASMCs | Minutes | ↑ | Cytochrome c reduction assay, H2DCF | Rathore et al. (122, 123) |
| Mouse PASMCs | Minutes | ↑ | H2DCF, lucigenin, Redox Sensor Red CC-1 | Wang et al. (161) |
| Human PASMCs | Hours | ↑ | Dihydroethidium | Wu et al. (176) |
Modified from Olschewski and Weir (104).
H2DCF, dichlorodihydrofluorescein; PASMC, pulmonary artery smooth muscle cell; ROS, reactive oxygen species.
A number of publications suggest that the hypoxic increase or decrease in intracellular ROS concentration can affect the activity of ion channels, resulting in a large increase in intracellular Ca2+ concentration in PASMCs (7, 80, 81, 94, 128). Although most of these studies applied the measurement of the global intracellular calcium concentration as one of the major read-outs, unfortunately, there are a few studies specifically targeting Nox (homologues) or mitochondria under acute or chronic hypoxia (or hyperoxia) in the pulmonary circulation in order to investigate the direct interaction between the ion channels and ROS. One of the potential mechanisms by which ROS are able to regulate pulmonary vascular tone involving ion channels is via the control of the cellular redox potential. According to the redox theory, acute hypoxia decreases the release of a mitochondrial-derived ROS (hydrogen peroxide) and/or increases the NAD(P)H/NAD(P) ratio, resulting in contraction through the reduction of thiol groups of the channel and causing PASMC membrane depolarization as a result of closure of cell membrane potassium channels (4, 9, 95). Although in vitro experiments almost universally demonstrate an elevation in intracellular calcium concentration during hypoxia and similar mechanisms as for potassium channels indicated earlier might be also true for calcium channels, alternative pathways of HPV focus mainly on the source of ROS or on second messengers (cyclic adenosine diphosphate [cADP]-ribose, diacylglycerol [DAG] or calmodulin). Thus, our knowledge about Nox or mitochondria-produced ROS-driven direct regulation of calcium-handling proteins expressed in the pulmonary circulation is still very limited (136–138, 141).
NADPH oxidase
Many studies have investigated the role of the Nox in the pulmonary circulation during more than the last 20 years; however, only recently reports link their findings to Nox subtypes. The Nox family consists of seven catalytic homologues, four of which (Nox1, Nox2, Nox4, and Nox5) are found in the vasculature. These enzymes transfer electrons from NADPH to molecular oxygen, thus producing superoxide. The enzyme complex in phagocytes comprises at least five components: two cytosolic subunits p47phox and p67phox, a cell membrane-bound cytochrome b558, which consists of gp91phox (renamed Nox2) and p22phox, and a small G protein Rac. Under physiological conditions, Nox proteins (Nox1, Nox2, Nox4, and Nox5) and their products superoxide and hydrogen peroxide participate in cardiovascular homeostasis by contributing to cell differentiation, repair of damaged tissue, and regulation of vascular tone and, thus, blood pressure (68).
Acute HPV
The TWIK-related acid-sensitive potassium channel 1 (TASK-1), which is commonly expressed in oxygen-sensing cells and conveys a background K+ current, is inactivated by hypoxia. However, TASK-1 per se cannot sense oxygen and may require a regulatory protein that can do so. Lee et al. proposed that Nox4 functions as an oxygen-sensing partner and that it modulates the oxygen sensitivity of TASK-1. In their study, the oxygen sensitivity of TASK-1 was abolished by Nox4 siRNA and Nox inhibitors, suggesting a novel function for Nox4 in the oxygen-dependent regulation of TASK-1 activity (71).
Chronic hypoxic PH
The chronic effects of ROS on ion channels are mediated via transcription factors. In a very recent study, Mittal et al. provided evidence for the regulation of the Kv1.5 channels by Nox4-derived ROS and for the development of pulmonary vascular remodeling, showing that sustained hypoxia decreases Kv channel currents by a direct effect of an Nox4-derived increase in ROS (92). Support for their findings was provided by Sturrock et al., reporting the cross-talk between transforming growth factor (TGF)-β1 and Nox4, and thus implications for mechanisms of pulmonary vascular remodeling. They have suggested that TGF-β1 may facilitate proliferation by up-regulating Nox4 and ROS production, with transient oxidative inactivation of phosphatases and augmentation of growth signaling cascades (145). There is accumulating evidence that the Nox enzymes, and in particular Nox4, play an important role in chronic responses of the pulmonary vasculature to changes in oxygen tension. These exciting findings may facilitate our understanding of the pathophysiology of PH secondary to chronic hypoxia. However, elucidation of the role of redox-regulated ion channels in chronic hypoxia-driven pulmonary vascular remodeling needs further combined efforts.
Mitochondria
The mitochondria play an important role in normal vascular physiology, for HPV and potentially for the development of PH, which has been extensively reviewed during recent years (2, 28, 115, 138, 140, 164, 167, 168). In the mitochondria, ROS are produced by the ETC at complexes I, II, and III. The non-charged H2O2 diffuses through the outer membrane to access the cytosol, superoxide reaches the cytosol through the trans-plasma-membrane electron transport protein, the voltage-dependent anion channels. During the last decade, large numbers of studies on mitochondria have been carried out, focusing on the role of the ETC as an oxygen sensor for hypoxia and HPV. Investigations providing evidence for the direct regulation of calcium-handling proteins and particularly of K+ channels by the ETC, however, are limited. In addition, the identification of the main site for ROS production within the mitochondria is still a matter of debate.
Acute hypoxia-induced changes initiated by ROS production by mitochondria
In PASMCs and in SMCs from systemic mesenteric artery (MA), Firth et al. investigated differences in functional coupling between mitochondria and K+ channels (35). They found that mitochondria are located significantly closer to the plasmalemmal membrane in PASMCs compared with MASMCs. Consistent with these findings, the effects of mitochondrial inhibitors on K+ current were significantly more potent in PASMCs than in MASMCs, suggesting a greater structural and functional coupling between mitochondria and K+ channels in PASMCs. In SMCs from human DA, ETC inhibitors (rotenone and antimycin) mimicked hypoxia, increasing K+ current and reversing constriction to oxygen, as shown by Michelakis et al. (90). They concluded that normoxia controls human DA tone by modulating the function of the mitochondrial ETC, thereby varying mitochondrial membrane potential and the production of H2O2, which regulates DASMC K+ channel activity and DA tone.
Effects of chronic hypoxia
In human PASMC, chronic hypoxia activates mitochondrial KATP channels, resulting in depolarization of mitochondrial membrane potential, increase of cytochrome C accumulation, and stimulation of mitochondrial H2O2 overproduction (54). However, in Fawn Hooded rats, Bonnet et al. have shown that mitochondrial dysfunction results in hyperpolarized mitochondria due to deficiency in components of several electron transport complexes, particularly complex I and SOD2. The net result of these abnormalities was reduced to total ROS production mimicking chronic hypoxia and causing normoxic activation of hypoxia-inducible factor (HIF)-1α, which then inhibited expression of oxygen-sensitive, voltage-gated Kv1.5, postulating a novel pathway of Kv channel regulation by mitochondrial membrane potential changes (19).
Trans-plasma membrane electron transport
The trans-plasma membrane electron transport (t-PMET) has been established with the discovery of the transmembraneous NADPH dehydrogenase (25, 42, 59). T-PMET enables reduction of extracellular oxidants at the expense of intracellular reducing equivalents. Thus, the cell can respond to changes in the redox microenvironment, regulating a variety of biological functions, including proton pumping and cell metabolism, cell growth, or even activity of ion channels (27). The t-PMET is a balanced interplay of several components that have been identified during the last two decades. Some are ubiquitously expressed, and others are only present in certain cell types. Some utilize only a subset of electron donors or acceptors, whereas others are less specific (83). The key components of t-PMET are intracellular electron donors [e.g., NAD(P)H] and either enzyme-mediated and/or shuttle-based electron transfer such as NAD(P):quinone oxidoreductase 1 (NQO1), disulfide-thiol exchanger (ENOX), members of the NOX family, and voltage-gated anionic channels also called porins or Coenzyme Q (CoQ); the last acts as a mobile electron shuttle (Fig. 5). Different strategies can be applied to identify the components that are responsible for the electron transport and to measure their activity, including measuring oxygen consumption, monitoring the disappearance of the electron donor (NAD(P)H), measuring the reduction of artificial electron acceptors, determining the amount of superoxide formation, and measuring ubiquinone reduction or hydroquinone oxidation (27).
FIG. 5.

Schematic representation of the t-PMET. At the cytosolic side of the plasma membrane, a quinone reductase is present that reduces CoQ to ubiquinol (CoQH2). CoQH2 shuttles the electrons to the NOX that is able to reduce extracellular ascorbyl radicals (Asc•) produce superoxide (O2•−), reduce O2 to water, and reduce protein disulfides. CoQ, Coenzyme Q; NOX, NADPH oxidase; t-PMET, trans-plasma membrane electron transport. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
A role for the t-PMET system and in particular the central function of Nox-driven superoxide in cardiovascular diseases, including PH and right heart failure, is very likely as discussed in detail earlier (see NADPH Oxidase section). However, most of the studies about the role of the other components have been carried out with a focus on the systemic circulation. The NQO1 in the lung has been investigated in several studies (11, 15, 16). Recently, Bongard et al. have shown that alterations in CoQ1 redox metabolism, either by addition of the complex I inhibitor rotenone or exposure of the animal to hyperoxia, can reveal a decrease in mitochondrial complex I activity (18). In addition, by using bovine pulmonary arterial EC, they have demonstrated that the cytoplasmic redox status, as reflected in the NADH/NAD+ and NADPH/NADP+ ratios, affects t-PMET reduction of thiazine compounds and NQO1-mediated quinone reduction (16). Ion channels in the pulmonary circulation are also affected by t-PMET. Our group described the effect of CoQ10 and duroquinone on K+ channels in PASMCs and showed that both inhibited the whole-cell K+ channel current, resulting in depolarization of the SMC membrane (126). Thus, CoQ10 and duroquinone, similar to hypoxia or ETC inhibition, shift the K+ channels to the more reduced state, resulting in the closing of the channels. On balance, cellular metabolism generates excess reducing equivalents that the t-PMET system exports to oxygen. Hypothetically, hypoxia may result in a greater concentration of reducing equivalents at the cell membrane, where they can modulate the activity of the ion channels.
Redox-Sensitive Enzymes
Hydrogen peroxide and superoxide can alter the function of proteins by oxidizing aromatic residues such as histidine, tyrosine, tryptophan, and phenylalanine; sulfur-containing amino acids such as cysteine and methionine or by resulting in carbonylation of catalytic active sites containing lysine, arginine, proline, or threonine; or the racemization of lysine, arginine ,or aspartate. Thus, ROS fine tune the duration and amplification of the phosphorylation signal. The oxidation of cysteine has been the focus of the majority of redox-signaling investigations as a result of the widely acknowledged role of this redox reaction in mediating intracellular signal transduction due to forming a disulfide bond (S-S) (29, 67). From a signaling perspective, it is the reversible cysteine modification that is most relevant, because hyperoxidation tends to occur predominantly during conditions of oxidative stress, promoting enzyme inactivation and degradation [for detailed reviews, see Refs. (62, 124)].
Receptor and non-receptor TK
The level of protein tyrosine phosphorylation is the function of opposing actions of protein tyrosine kinases and protein tyrosine phosphatases (PTPs). All PTPs contain a catalytic cysteine residue in the active site, and oxidation of this residue results in the inactivation of the PTP activity (158), which is recognized as a major mechanism by which ROS regulate the level of protein tyrosine phosphorylation. However, a number of studies suggest that the proto-oncogene tyrosine-protein kinase Src (src as a short for sarcoma) also plays an important role in the cellular response to ROS, because Src specific inhibitors and dominant negative Src mutants strongly attenuate the cellular response to ROS (87).
Receptor tyrosine kinases (RTKs) such as the insulin receptor, epidermal growth factor receptor, platelet-derived growth factor (PDGF) receptor, and the protein kinase receptor c-ret have been reported to undergo direct oxidation (98). We have recently proposed that hypoxia and PDGF-BB up-regulate protease-activated receptor-2 (PAR-2) expression in PASMC (66). This effect was reversed by HIF-1α depletion or the RTK inhibitor Imatinib. The PAR-2 expression observed in SMCs of pulmonary vessels of mice exposed to hypoxia was attenuated by Imatinib treatment, strengthening the link between hypoxia and RTK in PASMCs. Although there are increasing numbers of clinical studies investigating the role of RTK inhibition for the treatment of PH (38, 153), experimental evidence for the redox regulation of RTK in the pulmonary circulation is largely missing.
The non-RTK Src contains regulatory structures such as a myristoylation motif, a unique region, an Src homology (SH)3 domain, an SH2 domain, and a regulatory tail in addition to the catalytic domain. Src activity is regulated directly by reversible phosphorylation on multiple tyrosine (tyr) and serine (ser) residues, which are also targets for redox control. We recently reported that Src controls K+ channel function in human PASMCs in response to changes in oxygen tension (Fig. 6) (97a). We showed that moderate hypoxia decreased the level of phospho-SrcTK (at tyr419) and reduced the co-localisation of the TASK-1 channel and phospho-SrcTK, resulting in closure of the channel. Our results indicate that the modulation of SrcTK is a crucial factor controlling K+ channels, acting under normoxic conditions as a cofactor to set a negative resting membrane potential in human PASMCs and low resting pulmonary vascular tone.
FIG. 6.

Schematic representation of the t-PMET. Scheme of the proposed interplay between potassium channels and c-Src in human PASMCs. Under normoxia, the phospho-Src (active-Src, phosphorylated at Tyr419) binds to TASK-1 channels, resulting in functional TASK-1 channels. Active TASK-1 channels maintain negative resting potential in hPASMCs. In hypoxia, the phospho-Src (active-Src) is decreased. Closed potassium channels result in depolarization and increased intracellular calcium levels. (+) Indicate increase and (−) indicate decrease. Em, resting membrane potential; KCa, calcium-dependent potassium channel; Kv, voltage-gated potassium channel; PASMC, pulmonary artery smooth muscle cells; TASK-1, TWIK-related acid-sensitive potassium channel-1. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Redox Modulation of Ion Channels and Transporters in the Pulmonary Circulation
Redox modulation of ion channel activity seems to be an important regulatory mechanism under physiological conditions for several vasomotor functions, including not only regulation of cell membrane potential and vascular tone but also activation of transcription factors required for the expression of genes, apoptosis and necrosis, and cellular protective mechanisms against ischemic or hypoxic insults. Acute contraction of PASMCs is activated, in part, by a potassium (K+) inhibition-induced membrane depolarization and subsequent Ca2+ entry through nifedipine-sensitive L-type Ca2+ channels (Fig. 7). The global Ca2+ release is the result of different localized elementary Ca2+ release events, termed receptor- and store-operated Ca2+ influx pathways (64). Extrusion of Ca2+ through the plasma membrane is mediated by the plasma membrane Ca2+-Mg2+ ATPase, or Ca2+ pump, and the Na+/Ca2+ exchanger (NCX). In addition, the inward transportation of Ca2+ through the reverse model of NCX is important for maintaining a high cytosolic Ca2+ concentration and refilling Ca2+ into the SR/ER via SERCA, when cytosolic Na+ concentration is increased locally by increased Na+ influx through transient-receptor potential (TRP) channels (72).
FIG. 7.

Regulation of pulmonary vascular tone by potassium channels. Acute contraction of PASMCs is activated, in part, by a potassium (K+) inhibition-induced membrane depolarization and subsequent Ca2+ entry through nifedipine-sensitive L-type Ca2+ channels. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Potassium (K+) channels
Different types of K+ channels are sensitive to redox modulation. In the endothelial (porcine aortic endothelial cell [PAEC]) and in the PASMCs, the K+ current is an ensemble, reflecting the activity of many different channels. At least four classes of K+ channels have been identified in the cells of the wall of the PAs: voltage-gated K+ channels (Kv) (31, 117, 183), calcium-dependent K+ channels (KCa) (1, 114), KATPs (99), and the more recently identified family of two-pore domain K+ (K2P) channels (103). Different types of Kv channels have been shown to be expressed in the pulmonary vasculature (5, 7, 183). At the molecular level, Kv channels are homo- or heteromultimeric tetramers that are composed of two structurally distinct subunits: the pore-forming α-subunits and the regulatory β-subunits (52). Nine families of Kv channels α-subunits are recognized from cloning studies (Kv1–9) (52, 116), each with subtypes (e.g., Kv1.1–1.6). To date, 12 different Kv channel proteins have been described. We have extensively reviewed the response of the K+ channels in PASMCs to acute and chronic hypoxia (171) and also described in our earlier work the graded response of the K+ current to hypoxia (101).
A recent elegant report by Svoboda et al. shows that oxidative stress using the organic hydroperoxide tertiary butyl hydroperoxide (tBOOH) can directly result in internalization and protein degradation of Kv1.5 and that sulfenic acid modification of COOH-terminal C581 alone is sufficient to trigger these events (149). Thus, sulfenic acid modification to proteins can regulate Kv1.5 channel surface expression and stability under oxidative stress and divert channels from a recycling pathway to degradation. Redox modulation of Kv1.5 current is associated with numerous pathophysiological states that involve loss of channel protein, including PH (2). Therefore, this report might provide a novel molecular mechanism linking oxidative stress and down-regulation of channel expression observed in cardiovascular diseases. Earlier, we have reported that the same redox signal can elicit opposite effects on K+ current, resting membrane potential, cytosolic calcium, and vascular tone in resistance PASMCs and the SMCs obtained from the DA Botalli (102). These observations support the concept that redox changes could signal the opposite effects of oxygen in the PA and DA. The thiol oxidant diamide inhibits HPV (172). Schach et al. have shown that this pulmonary vasodilation is markedly inhibited when the transmembrane K+ driving force is reduced or vessels are constricted with high potassium, indicating that the thiol oxidation induces pulmonary vasodilation by opening K+ channels (131). The role of the Nox for the ROS-induced changes in Kv current has been recently studied by Mittal et al. They have shown that Kv1.5 and NOX4 are colocalized in isolated PASMC. Furthermore, the Nox inhibitor and ROS scavenger apocynin as well as NOX4 siRNA reversed the hypoxia-induced decrease in Kv current density, whereas the protein levels of the channels remain unaffected by siNOX4 treatment. Determination of cysteine oxidation revealed increased NOX4-mediated Kv1.5 channel oxidation. Their observation suggests that the decreased Kv current was a direct effect of an NOX4-derived increase in ROS (92). This finding complements the report that a normoxic increase in endogenous H2O2 in SMCs of the DA inhibits K+ channels and causes constriction (125). However, the similar observations in PA and DA are contrary to the opposite redox effects with diamide and other redox agents described earlier.
Oxidative modification of Kv channels has been extensively investigated by Sahoo et al. (130). They identified four critical cysteine residues and not only revealed the major target sites of oxidative modification in potassium voltage-gated channel subfamily H member 1 (KCNH1), also known as Kv10.1, but also provided new insights into the dynamic interplay of functional channel domains under redox modulation. The study provides solid evidence that the cysteine residues C145/C214 in the N-linker are responsible for fast modification of the ion channels, and the C532/C562 in the C-linker are responsible for slow modification of the ion channels. The authors hypothesize that in physiological levels of ROS, mild oxidative stress may act through a positive feedback by reducing Kv channel activity. Another recent experimental study has identified oxidative stress as a key contributor to Kv4 channel up-regulation due to chronic impairment of the Trx system (152). The results of Tang et al. suggest that expression of cardiac Kv channels is redox regulated. They have found that in myocytes from the ventricle after myocardial infarction, TrxR is markedly suppressed. This chronic impairment of the Trx system contributed to Kv4 up-regulation through sustained activation of ASK1-JNK-p38 signaling in a TrxR-dependent manner thus highlighting the Trx system as a novel therapeutic target.
Modification of Kv channels by pyridine nucleotides has been extensively reviewed recently by Kilfoil et al. (60). According to earlier reports, the Kv1 and Kv4 channels seem to interact with pyridine nucleotide-binding β-subunits. Binding of NADP+ to Kvβ subunit removes N-type inactivation of Kv currents, whereas NADPH stabilizes channel inactivation. PAs sense and respond to environmental changes such as hypoxia. Therefore, linking K+ transport to the metabolic state of the cell is important. In small resistance arteries, oxygen-sensitive changes in K+ channel activity mediate HPV and decreased Kv1.5 activity may result in PH (84). It has been recently reported that in vitro the C-terminal peptide of Kv1.5 interacts avidly with NADPH (155). Thus, the pyridine nucleotide-binding Kvβ proteins with their ability to regulate Kv current could, in principle, impart oxygen sensitivity to Kv1.5 channels. However, the findings of the studies carried out in different species and in different cell systems are contradictory. Although the differences might be explained by differences in Kvβ expression, further work is required to determine the role of the pyridine regulation of Kv channels in HPV.
The resting potential depends on a background K+ current comprising voltage-dependent and -independent components (46, 106) in PASMC. TASK-1 channels, described by Gurney et al. and also by our group (47, 103), appear to mediate the latter (46, 47). The biophysical and pharmacological properties of the voltage-dependent component were noted to bear a striking resemblance to the neuronal M-current (31). The channels responsible for the M-current are encoded by genes of the KCNQ (Kv7) family (127, 184). Along with the finding that KCNQ channel blockers are potent pulmonary vasoconstrictors (57a), the similarity of IKN to the M-current led the authors to consider the possibility that KCNQ channels mediate the voltage-dependent component of IKN (58). A triple cysteine pocket within neuronal M-channel subunits (Kv7.2–7.5) can be oxidatively modified by H2O2 (37, 105) and O2•− (75a), both of which cause augmentation of M-current. Here, they identified a reactive redox module within M-channels that is responsible for dynamic M-current modulation. Furthermore, emerging evidence demonstrates that protein S-nitrosylation is an important NO-mediated regulatory mechanism of various classes of proteins, including ion channels (146, 159). In a recent study of Asada et al., the authors demonstrated that an NO donor induces S-nitrosylation at Cys445 in the C terminus of the pore-forming α-subunit KCNQ1. They provide convincing evidence which shows that the redox motif flanking Cys445 is required for the site-specific S-nitrosylation of Cys445. S-nitrosylation at Cys445 of the KCNQ1 channel functionally regulates the KCNQ1/potassium voltage-gated channel subfamily E member 1 (KCNE1) complex channel. Since KCNQ1 contributes to the slowly activating delayed rectifier potassium channel current, this work may provide a molecular basis of NO-mediated phenomena in smooth muscle electrophysiology (10).
The large conductance Ca2+-dependent potassium channel (BKCa) plays a key role in the oxygen-sensing function of the carotid body chemoreceptors. Li et al. have recently reported that the H2S donor, NaHS, plays a crucial role in mediating the response of carotid body chemoreceptors to hypoxia via modulating the BKCa channels (73). In these cells, the BKCa currents were inhibited by hypoxia and such inhibition was mimicked by NaHS and diminished by inhibitors of cystathionine beta-synthetase (CBS), which produces H2S from L-cysteine. Finally, mice hyperventilated in response to hypoxia, which was prevented by the CBS inhibitors amino oxyacetic acid (AOAA) and hydroxylamine. Almost 20 years ago, in PASMC and SMCs from systemic arteries, such as ear arterial smooth muscle cells (EASMC), BKCa have been reported to be redox sensitive (113). Reducing agents (DDT, GSH, and NADH) decreased PASMC, but not EASMC, KCa channel activity. However, oxidizing agents (DTNB, GSSG, and NAD) increased KCa channel activity in both PASMC and EASMC. The increased activity due to oxidizing agents was diminished by applying reducing agents. From these results, the authors suggest that the basal redox state of the PASMC KCa channel is more oxidized than that of the EASMC channel, as the response of KCa channels of the PASMC to intracellular reducing agents differs from that of the EASMC. In contrast, in isolated SMC from large conduit PAs, the KCa channel activity was unaffected by NAD and GSSG, or NADH and GSH (154). This suggests that the change in the intracellular redox state, which occurs during acute hypoxia, does not alter the activity of KCa channels in SMC from large conduit PAs. The different behavior of these channels in response to redox changes could be explained by the heterogeneity of the KCa in different tissues and may be related to the different responses of PASMC and EASMC KCa channels to hypoxia Archer et al. (5). Furthermore, redox-dependent modulation of activity of Na+−K+ ATPase (65) and Na+−H+(177) exchange has been reported.
Calcium handling proteins
In the pulmonary circulation, the vascular tone and blood vessel diameter is determined to be due to the regulation of cytosolic/intracellular Ca2+, mainly in the PASMCs of the resistance arteries. The steep (∼10,000-fold) concentration gradient between the intracellular and the extracellular Ca2+ concentration already indicates the importance of constant and tight control of cellular Ca2+ homeostasis due to a complex machinery of (i) Ca2+-permeable channels, enabling Ca2+ to flux through the membrane based on its electrochemical gradient; (ii) the Ca2+ pumps (e.g., Ca2+/Mg2+ ATPase or Ca2+ pump) transporting Ca2+ against its concentration gradient; and (iii) the Ca2+ exchangers (e.g., NCX). The calcium-selective ion channels in the PASMC and EC include (i) voltage-dependent Ca2+ channels (VDCC) that are opened by membrane depolarization, (ii) receptor-operated Ca2+ channels (ROC) which are opened by DAG when membrane receptors are activated by ligands, and (iii) store-operated Ca2+ channels (SOC) that are opened when Ca2+ in the intracellular Ca2+ stores (e.g., SR/ER) is depleted by, for example, activation of IP3R/RyR and inhibition of SERCA on the SR/ER membrane [for a detailed review, see also Song et al. (141)]. In the pulmonary circulation, the investigations have focussed on measurements of ROS, pharmacological and functional studies, and mechanisms that control Ca2+ handling under hypoxia without determining the exact molecular site of action of the major players (138, 141) (Fig. 8).
FIG. 8.

Regulation of calcium selective ion channels by redox. The oxidizing agent H2O2 or NO directly activates calcium selective channels. In contrast, DTT or extracellular reduced Trx can increase ion current through TRPC5 homotetrameric and TRPC5/TRPC1 heterotetrameric channels. DTT, dithiothreitol; NO, nitric oxide; TRPC, transient-receptor potential cation. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Oxidants have been found to regulate voltage-gated Ca2+ channel activity on different levels, affecting their expression, trafficking, open time, and open probability. Some of these studies were able to demonstrate that the pore-forming α1-subunit, which is rich in reactive cysteines, is the molecular target for ROS. Although almost all studies have been carried out on myocytes, the mode of action of the redox modulation can be translated into VDCC in PASMCs. However, the findings are contradictory. While several reports have shown that oxidation increases the activity of voltage-gated Ca2+ channels, others reported inhibition of channel activity by oxidizing agents [for a more detailed review, see Hidalgo and Donoso (51)]. This might be explained either by the concentrations of ROS/RNS involved or by different effects on ion transport mechanisms in different cell types. Thus, thiol oxidizing agents (e.g., thimerosal) irreversibly decreased current through the human α1C-subunit, which was reversed by DTT, expressed in HEK-293 cells (91, 133). In contrast, ROS such as H2O2 and O2•− were shown to stimulate Ca2+ entry through L-type voltage-gated channels in VSMC (151) and in cardiomyocytes (160). Oxidative stress also plays a major role in the pathogenesis of heart failure, and this condition involves L-type Ca2+ channels. Zheng et al. have very recently shown that the redox state of Ca2+ channels is controlled by the thioredoxin system which plays a key role in Ca2+ channel remodeling of the failing heart (187). Furthermore, NO also modulates VDCC by a direct redox-dependent stimulation. In ventricular myocytes, S-nitrosothiols (CysNO and GSNO) increased the amplitude of Ca2+ current as a result of S-nitrosylation of extracellular SH groups of the L-type Ca2+ channel, independently of the activity of kinases, phosphatases, or SR Ca2+ release (21, 120).
ROC and SOC provide alternative ways for Ca2+ influx into PAEC and/or PASMCs. Since TRP proteins are believed to underlie the molecular structure of many ROC entry channels and at least some SOC entry channels, it is logical to question whether the TRP proteins expressed in the pulmonary circulation are sensitive to regulation by redox. More than 10 TRP isoforms are expressed in vascular smooth muscle and EC, including TRPC1–7, TRPV1–4, TRPP2, TRPM2–4, -7, and -8. However, it needs to be noted that almost all investigations discussed next were carried out in heterologous expression systems or in silico modulations, which provides access for targeted studies of the channels. We will only briefly summarize the redox modulation of these channels, as several reviews recently discussed the existing data (138, 141, 163, 164).
Even though there are still some uncertainties as to which compounds act directly and which act indirectly to gate the TRPM2 channel, it is clear that TRPM2 is modulated by redox changes. Using NAD+ and ADP-ribose, Wehage et al. reported activation of the cationic current in TRPM2-expressing HEK cells as the consequence of redox regulation (50, 169). We have recently detected the mechano-sensitive TRPM4 in human PAs (unpublished observation). Simon et al. reported that H2O2 induces sustained activity of TRPM4, showing that oxidative modulation of the C-terminally located cysteine C1093 is crucial for the H2O2 effect (139). The redox regulation of TRPC3/TRPC4 channels has been extensively investigated by the Groschner group. They have shown that TRPC3 proteins are the basis of the redox-activated cation conductance of PAECs, as they are activated by H2O2. They identified the TRPC3/TRPC4 complex as subunits of native endothelial cation channels that are governed by the cellular redox state (12, 119). The mechanisms of the redox activation of the complex, unfortunately, remains elusive. Groschner et al. have described it as an “oxidant-induced disruption of cholesterol-rich lipid rafts,” a cross-talk between TRPC channels and signaling molecules which reside in caveolae (or lipid rafts) and further speculate that membrane cholesterol oxidation might actually be the signaling molecule that activates TRPC3 (44, 119). TRPC5 homotetrameric channels and TRPC5/TRPC1 heterotetrameric channels have been reported to be activated by DTT or extracellular reduced Trx. Both break a disulfide bond in the extracellular loop adjacent to the ion selectivity filter of TRPC5 and increase the ion conductivity of the channel (179). Similar to TRPC3 or TRPM4, TRPC6 is activated by H2O2 via modification of thiol groups of intracellular proteins. This cysteine oxidation-dependent pathway not only stimulates the TRPC6 channel by itself but also sensitizes the channels to DAG and promotes TRPC6 trafficking to the cell surface (41). TRPC5 and 7 are regulated by cysteine S-nitrosylation by NO (182). NO or the NO donor SNAP induces Ca2+ entry into cells, an effect that is partially reversed by additional application of the reducing agent DTT. In contrast to the other TRP channels, the mechanism is well described. The effect is specifically mediated by cysteine S-nitrosylation of the cytoplasmically accessible Cys553 and Cys558, to open the intracellular activation gate. The membrane-permeable thiol-reactive compound MTSEA mimicks this effect, but not the membrane-impermeable MTSET.
The signaling cascade underlying store-operated Ca2+ entry spans from receptor-mediated activation of PLC with subsequent IP3 production to IP3R-mediated depletion of ER Ca2+ stores, which, in turn, results in a conformational change, oligomerization, and translocation of stromal interaction molecule 1 (STIM1) to plasma membrane near regions of the ER, where STIM1 activates calcium release-activated calcium channel (Orai) through a direct interaction with its C-terminal region (13). The molecular identity of store-operated Ca2+ influx was described first in 2006: isoforms of Orai (1, 2, and 3), as essential pore-forming units and the Ca2+-sensing ER-proteins STIM1 and STIM2 (186). In addition, TRPC1 and TRPC3 have been reported to participate in the STIM-mediated Ca2+ entry pathway. STIM1 proteins possess several cysteine residues that could be targets for modification. Similarly, all three Orai isoforms possess predicted extracellular and intracellular cysteines. For a detailed review of the redox modulation of SOC, see Bogeski et al. (13). Here, we focus only on recent studies reporting modification of the newly discovered components of the SOC pathway (STIM and Orai) by redox. Recently, using electrophysiology, Ca2+ imaging, and site-directed mutagenesis, Bogeski et al. have found that Orai1 is inhibited by preincubation with H2O2 and that cysteines 195, 126, and 143 contribute to redox sensitivity (14). Hawkins et al. studied redox-mediated activation of STIM1 and found that oxidative stress results in gluthationylation of STIM1's cysteine 56, triggering STIM1 oligomerization, and, thus, they were able to activate Orai channels and induce Ca2+entry (49). In contrast, Grupe et al. have shown that H2O2 directly activates Ca2+ entry in an STIM-dependent manner (45). Thus, the limited number of these studies and the potential discrepancies in the recent findings clearly indicate the need for further detailed investigations of these complex signaling events. Activation of IP3 receptors and RyRs enables Ca2+ release from the SR/ER to the cytosol, thus resulting in an increase in intracellular Ca2+ concentration and vasoconstriction in myocytes. In bovine and human aortic ECs, glutathionylation by a chemical oxidant, diamide, which selectively converts reduced glutathione (GSH) to its disulfide (GSSG), dramatically increased both the rate and magnitude of the thapsigargin-induced Ca2+ transient. These findings suggest that CICR via the IP3R is enhanced by the formation of protein–glutathione (P-SSG) mixed disulfide, that is, glutathionylation. Similar to diamide, H2O2 increased the sensitivity of HAECs to both histamine and thapsigargin. The biochemical studies have shown that glutathionylation of native IP3R1 is increased in cells challenged with H2O2, indicating that thiol-oxidizing agents primarily increased the sensitivity of the IP3R to Ca2+ (78). Through ATP hydrolysis, SERCA and plasma membrane calcium (PMCA) pump actively clear Ca2+ from the cytoplasm and, thus, play a key role by maintaining cytoplasmic Ca2+ homeostasis. The different SERCA isoforms contain between 22 and 28 cysteine residues, and, therefore, provide a target for redox modification. According to the data available so far, only one or two of them are essential (97). SERCA inhibition in vitro through modification of sulfhydryl groups by either ROS or RNS has been reported in several cell types, mainly in coronary EC and in cardiomyocytes (51). Finally, the PMCA pump has also been reported to be reversibly inhibited by H2O2, O2•−, •OH, and ONOO−/ONOOH (51). It is very likely that residues Tyr589, Met622, and Met831 are the redox targets (82).
Conclusion
The pulmonary circulation is a target for a number of redox-active signaling factors, both circulating and locally produced. Under physiological circumstances, the concerted action of vasodilators and vasoconstrictors maintains the physiological structure and relaxed tone of the PAs. Since redox changes trigger signaling pathways to ion channels that may result in changes in ion fluxes, contraction, cell proliferation, and perhaps to vascular remodeling, to elucidate the molecular mechanisms of changes in ion channel conductance and the identification of the sites of action is essential. In the past decade, significant advancement in the understanding of protein thiol modification by pro- and antioxidants has been made. Further investigation of the interaction of redox status and ion channel gating, especially the significance of S-glutathionylation, S-nitrosylation, and protein kinase-mediated phosphorylation, will result in a better understanding of the physiological and pathophysiological importance of these mediators in general. Thus, the implications of such modifications in cellular functions and related diseases will guide our endeavour in further altering the ion channel for therapeutic purposes.
Abbreviations Used
- γ-GCL
gamma-glutamate cysteine ligase
- γ-GGT
gamma-glutamyl transpeptidase
- AOAA
amino oxyacetic acid
- BKCa
calcium-dependent potassium channel
- Ca2+
calcium
- cADP
cyclic adenosine diphosphate
- CBS
cystathionine beta-synthetase
- cGMP
cyclic guanosine monophosphate
- CICR
Ca2+-induced Ca2+-release
- CO
carbon monoxide
- CoQ
Coenzyme Q
- DA
ductus arteriosus
- DAG
diacylglycerol
- DTNB
5,5′-dithio-bis(2-nitrobenzoic acid)
- DTT
dithiothreitol
- EC
endothelial cells
- eNOS
endothelial nitric oxide synthase isoenzyme
- ENOX
disulfide-thiol exchanger
- ETC
electron transport chain
- GAPDH
glyceraldehydes(s) 3-phosphate dehydrogenase
- GPxs
glutathione peroxidases
- GR
glutathione reductase
- GS
glutathione synthetase
- GSH
reduced glutathione
- GSNO
S-nitrosoglutathione
- GSSG
oxidized glutathione
- GSTs
glutathione s-transferases
- GTP
guanosintriphosphat
- H2DCF
dichlorodihydrofluorescein
- H2O2
hydrogen peroxide
- HIF
hypoxia-inducible factor
- HPV
hypoxic pulmonary vasoconstriction
- H2S
hydrogen sulfide
- iNOS
inducible NO synthase isoenzyme
- IP3R
inositol trisphosphate receptor
- K+
potassium
- KATP
ATP-sensitive K+ channel
- KCNE1
potassium voltage-gated channel subfamily E member 1
- KCNH1
potassium voltage-gated channel subfamily H member 1
- KCNQ1
potassium voltage-gated channel, KQT-like subfamily, member 1
- Kir
potassium inwardly rectifying channel
- Kv
voltage-gated potassium channel
- MA
mesenteric artery
- MTSEA
2-aminoethyl methanethiosulfonate hydrobromide
- MTSET
2-(trimethylammonium)ethyl methanethiosulfonate bromide
- NaHS
sodium hydrosulfide
- NCX
Na+/Ca2+ exchanger
- nNOS
neuronal NO synthase isoenzyme
- NO
nitric oxide
- Nox
NADPH oxidase
- NQO1
NAD(P):quinone oxidoreductase 1
- O2•−
superoxide
- Orai
calcium release-activated calcium channel
- PA
pulmonary artery
- PAECs
pulmonary artery endothelial cells
- PAR-2
protease-activated receptor-2
- PDGF
platelet-derived growth factor
- PH
pulmonary hypertension
- PMCA
plasma membrane calcium
- PMET
plasma membrane electron transport
- P-SSG
protein S-glutathionylation
- PTP
protein tyrosine phosphatase
- ROC
receptor-operated Ca2+ channels
- ROS
reactive oxygen species
- RSOH
sulfenic
- RSO2H
sulfinic
- RSO3H
sulfonic
- RTK
receptor tyrosine kinase
- RyR
ryanodine receptor
- RyR2
RyR/Ca2+ release channel
- SERCA
sarcoplasmic/endoplasmic reticulum calcium ATPase
- sGC
soluble guanylate cyclase
- SH
Src homology
- SMC
smooth muscle cell
- SNO
S-nitrosylation
- SOC
store-operated Ca2+ channels
- SOD
superoxide dismutase
- SR
sarcoplasmic reticulum
- src
sarcoma
- STIM
stromal interaction molecule
- TASK-1
TWIK-related acid-sensitive potassium channel 1
- tBOOH
tertiary butyl hydroperoxide
- TGF
transforming growth factor
- t-PMET
trans-plasma membrane electron transport
- TRP
transient-receptor potential
- TRPC
transient-receptor potential cation
- TRPM
transient-receptor potential cation channel, subfamily M
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- VDCC
voltage-dependent Ca2+ channels
- VSMCs
vascular smooth muscle cells
References
- 1.Albarwani S, Robertson BE, Nye PC, and Kozlowski RZ. Biophysical properties of Ca2+- and Mg-ATP-activated K+ channels in pulmonary arterial smooth muscle cells isolated from the rat. Pflügers Arch 428: 446–454, 1994 [DOI] [PubMed] [Google Scholar]
- 2.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garciafigure JG, and Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–H578, 2008 [DOI] [PubMed] [Google Scholar]
- 3.This reference has been deleted.
- 4.Archer SL, Huang J, Henry T, Peterson D, and Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 73: 1100–1112, 1993 [DOI] [PubMed] [Google Scholar]
- 5.Archer SL, Huang JM, Reeve HL, Hampl V, Tolarova S, Michelakis E, and Weir EK. Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res 78: 431–442, 1996 [DOI] [PubMed] [Google Scholar]
- 6.Archer SL, Nelson DP, and Weir EK. Detection of activated O2 species in vitro and in rat lungs by chemiluminescence. J Appl Physiol 67: 1912–1921, 1989 [DOI] [PubMed] [Google Scholar]
- 7.Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, and Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101: 2319–2330, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Archer SL, Tolins JP, Raij L, and Weir EK. Hypoxic pulmonary vasoconstriction is enhanced by inhibition of the synthesis of an endothelium derived relaxing factor. Biochem Biophys Res Commun 164: 1198–1205, 1989 [DOI] [PubMed] [Google Scholar]
- 9.Archer SL, Will JA, and Weir EK. Redox status in the control of pulmonary vascular tone. Herz 11: 127–141, 1986 [PubMed] [Google Scholar]
- 10.Asada K, Kurokawa J, and Furukawa T. Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J Biol Chem 284: 6014–6020, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Audi SH, Bongard RD, Dawson CA, Siegel D, Roerig DL, and Merker MP. Duroquinone reduction during passage through the pulmonary circulation. Am J Physiol Lung Cell Mol Physiol 285: L1116–L1131, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Balzer M, Lintschinger B, and Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res 42: 543–549, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, and Niemeyer BA. Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50: 407–423, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, Takahashi N, Peinelt C, Griesemer D, Bozem M, Mori Y, Hoth M, and Niemeyer BA. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal 3: ra24, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Bongard RD, Krenz GS, Gastonguay AJ, Williams CL, Lindemer BJ, and Merker MP. Characterization of the threshold for NAD(P)H: quinone oxidoreductase activity in intact sulforaphane-treated pulmonary arterial endothelial cells. Free Radic Biol Med 50: 953–962, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bongard RD, Lindemer BJ, Krenz GS, and Merker MP. Preferential utilization of NADPH as the endogenous electron donor for NAD(P)H: quinone oxidoreductase 1 (NQO1) in intact pulmonary arterial endothelial cells. Free Radic Biol Med 46: 25–32, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.This reference has been deleted.
- 18.Bongard RD, Myers CR, Lindemer BJ, Baumgardt S, Gonzalez FJ, and Merker MP. Coenzyme Q(1) as a probe for mitochondrial complex I activity in the intact perfused hyperoxia-exposed wild-type and Nqo1-null mouse lung. Am J Physiol Lung Cell Mol Physiol 302: L949–L958, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, and Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Bradford JR. and Dean HP. The pulmonary circulation. J Physiol 16: 34–96, 1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell DL, Stamler JS, and Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol 108: 277–293, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen B, Nelin VE, Locy ML, Jin Y, and Tipple TE. Thioredoxin-1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 305: L389–L395, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, and Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coggins MP. and Bloch KD. Nitric oxide in the pulmonary vasculature. Arterioscler Thromb Vasc Biol 27: 1877–1885, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Crane FL, Sun IL, Clark MG, Grebing C, and Low H. Transplasma-membrane redox systems in growth and development. Biochim Biophys Acta 811: 233–264, 1985 [DOI] [PubMed] [Google Scholar]
- 26.Day RM, Suzuki YJ, Lum JM, White AC, and Fanburg BL. Bleomycin upregulates expression of gamma-glutamylcysteine synthetase in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 282: L1349–L1357, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Del PD, Avigliano L, Savini I, and Catani MV. Trans-plasma membrane electron transport in mammals: functional significance in health and disease. Antioxid Redox Signal 14: 2289–2318, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Dromparis P, Sutendra G, and Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med (Berl) 88: 1003–1010, 2010 [DOI] [PubMed] [Google Scholar]
- 29.Drose S. and Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 748: 145–169, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Euler v US and Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 12: 301–320, 1946 [Google Scholar]
- 31.Evans AM, Osipenko ON, and Gurney AM. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J Physiol 496: 407–420, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fagan KA, Fouty BW, Tyler RC, Morris KG, Jr., Hepler LK, Sato K, LeCras TD, Abman SH, Weinberger HD, Huang PL, McMurtry IF, and Rodman DM. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest 103: 291–299, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fagan KA, Tyler RC, Sato K, Fouty BW, Morris KG, Jr., Huang PL, McMurtry IF, and Rodman DM. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am J Physiol 277: L472–L478, 1999 [DOI] [PubMed] [Google Scholar]
- 34.Fernandes AP. and Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal 6: 63–74, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Firth AL, Gordienko DV, Yuill KH, and Smirnov SV. Cellular localization of mitochondria contributes to Kv channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am J Physiol Lung Cell Mol Physiol 296: L347–L360, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 459: 923–939, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, and Shapiro MS. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J 25: 4996–5004, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, Golpon H, Toshner M, Grimminger F, and Pascoe S. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med 182: 1171–1177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez R, Nunez L, Vaquero M, Amoros I, Barana A, de PT, Macaya C, Maroto L, Rodriguez E, Caballero R, Lopez-Farre A, Tamargo J, and Delpon E. Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (Ito1). Cardiovasc Res 80: 375–384, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez DR, Beigi F, Treuer AV, and Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A 104: 20612–20617, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham S, Ding M, Ding Y, Sours-Brothers S, Luchowski R, Gryczynski Z, Yorio T, Ma H, and Ma R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J Biol Chem 285: 23466–23476, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grebing C, Crane FL, Low H, and Hall K. A transmembranous NADH-dehydrogenase in human erythrocyte membranes. J Bioenerg Biomembr 16: 517–533, 1984 [DOI] [PubMed] [Google Scholar]
- 43.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med 27: 922–935, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Groschner K, Rosker C, and Lukas M. Role of TRP channels in oxidative stress. Novartis Found Symp 258: 222–230, 2004 [PubMed] [Google Scholar]
- 45.Grupe M, Myers G, Penner R, and Fleig A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell Calcium 48: 1–9, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gurney AM. and Joshi S. The role of twin pore domain and other K+channels in hypoxic pulmonary vasoconstriction. Novartis Found Symp 272: 218–228, 2006 [PubMed] [Google Scholar]
- 47.Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, and Kempsill FE. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res 93: 957–964, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Haldar SM. and Stamler JS. S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest 123: 101–110, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, Kurosaki T, Joseph SK, Gill DL, and Madesh M. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol 190: 391–405, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heiner I, Eisfeld J, Halaszovich CR, Wehage E, Jungling E, Zitt C, and Luckhoff A. Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J 371(Pt 3): 1045–1053, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hidalgo C. and Donoso P. Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal 10: 1275–1312, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Hille B. Potassium Channels and Chloride Channels. Ion Channels of Excitable Membranes. Sunderland: Sinauer Associates, Inc., 2001, pp. 131–167 [Google Scholar]
- 53.Hori M. and Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res 81: 457–464, 2009 [DOI] [PubMed] [Google Scholar]
- 54.Hu HL, Zhang ZX, Chen CS, Cai C, Zhao JP, and Wang X. Effects of mitochondrial potassium channel and membrane potential on hypoxic human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 42: 661–666, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Hu XQ. and Zhang L. Function and regulation of large conductance Ca(2+)-activated K+channel in vascular smooth muscle cells. Drug Discov Today 17: 974–987, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jernigan NL, Broughton BR, Walker BR, and Resta TC. Impaired NO-dependent inhibition of store- and receptor-operated calcium entry in pulmonary vascular smooth muscle after chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 290: L517–L525, 2006 [DOI] [PubMed] [Google Scholar]
- 57.Jernigan NL, Walker BR, and Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57a.Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 7:31, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joshi S, Sedivy V, Hodyc D, Herget J, and Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther 329: 368–376, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kilberg MS. and Christensen HN. Electron-transferring enzymes in the plasma membrane of the Ehrlich ascites tumor cell. Biochemistry 18: 1525–1530, 1979 [DOI] [PubMed] [Google Scholar]
- 60.Kilfoil PJ, Tipparaju SM, Barski OA, and Bhatnagar A. Regulation of ion channels by pyridine nucleotides. Circ Res 112: 721–741, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Killilea DW, Hester R, Balczon R, Babal P, and Gillespie MN. Free radical production in hypoxic pulmonary artery smooth muscle cells. Am J Physiol 279: L408–L412, 2000 [DOI] [PubMed] [Google Scholar]
- 62.Knock GA. and Ward JP. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal 15: 1531–1547, 2011 [DOI] [PubMed] [Google Scholar]
- 63.Kolisek M, Beck A, Fleig A, and Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 18: 61–69, 2005 [DOI] [PubMed] [Google Scholar]
- 64.Kuhr FK, Smith KA, Song MY, Levitan I, and Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca(2)(+) signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kukreja RC, Weaver AB, and Hess ML. Sarcolemmal Na(+)-K(+)-ATPase: inactivation by neutrophil-derived free radicals and oxidants. Am J Physiol 259(Pt 2): H1330–H1336, 1990 [DOI] [PubMed] [Google Scholar]
- 66.Kwapiszewska G, Markart P, Dahal BK, Kojonazarov B, Marsh LM, Schermuly RT, Taube C, Meinhardt A, Ghofrani HA, Steinhoff M, Seeger W, Preissner KT, Olschewski A, Weissmann N, and Wygrecka M. PAR-2 inhibition reverses experimental pulmonary hypertension. Circ Res 110: 1179–1191, 2012 [DOI] [PubMed] [Google Scholar]
- 67.Lassegue B. and Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lassegue B, San MA, and Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le Cras TD. and McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol 280: L575–L582, 2001 [DOI] [PubMed] [Google Scholar]
- 70.Le Cras TD, Tyler RC, Horan MP, Morris KG, Tuder RM, McMurtry IF, Johns RA, and Abman SH. Effects of chronic hypoxia and altered hemodynamics on endothelial nitric oxide synthase expression in the adult rat lung. J Clin Invest 101: 795–801, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee YM, Kim BJ, Chun YS, So I, Choi H, Kim MS, and Park JW. NOX4 as an oxygen sensor to regulate TASK-1 activity. Cell Signal 18: 499–507, 2006 [DOI] [PubMed] [Google Scholar]
- 72.Lemos VS, Poburko D, Liao CH, Cole WC, and van BC. Na+entry via TRPC6 causes Ca2+entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem Biophys Res Commun 352: 130–134, 2007 [DOI] [PubMed] [Google Scholar]
- 73.Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, and Rong W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal 12: 1179–1189, 2010 [DOI] [PubMed] [Google Scholar]
- 74.Lima B, Forrester MT, Hess DT, and Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res 106: 633–646, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin SJ. and Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol 15: 241–246, 2003 [DOI] [PubMed] [Google Scholar]
- 75a.Linley JE, Pettinger L, Huang D, Gamper N. M channel enhancers and physiological M channel block. J Physiol. 590:793–807, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu JQ, Sham JS, Shimoda LA, Kuppusamy P, and Sylvester JT. Hypoxic constriction and reactive oxygen species in porcine distal pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 285: L322–L333, 2003 [DOI] [PubMed] [Google Scholar]
- 77.Liu SQ, Jin H, Zacarias A, Srivastava S, and Bhatnagar A. Binding of pyridine nucleotide coenzymes to the beta-subunit of the voltage-sensitive K+channel. J Biol Chem 276: 11812–11820, 2001 [DOI] [PubMed] [Google Scholar]
- 78.Lock JT, Sinkins WG, and Schilling WP. Protein S-glutathionylation enhances Ca2+-induced Ca2+release via the IP3 receptor in cultured aortic endothelial cells. J Physiol 590(Pt 15): 3431–3447, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.This reference has been deleted.
- 80.Lu W, Wang J, Peng G, Shimoda LA, and Sylvester JT. Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+entry and Ca2+responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 297: L17–L25, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu W, Wang J, Shimoda LA, and Sylvester JT. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L104–L113, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lushington GH, Zaidi A, and Michaelis ML. Theoretically predicted structures of plasma membrane Ca(2+)-ATPase and their susceptibilities to oxidation. J Mol Graph Model 24: 175–185, 2005 [DOI] [PubMed] [Google Scholar]
- 83.Ly JD. and Lawen A. Transplasma membrane electron transport: enzymes involved and biological function. Redox Rep 8: 3–21, 2003 [DOI] [PubMed] [Google Scholar]
- 84.Mandegar M. and Yuan JX. Role of K+channels in pulmonary hypertension. Vascul Pharmacol 38: 25–33, 2002 [DOI] [PubMed] [Google Scholar]
- 85.Marshall C, Mamary AJ, Verhoeven AJ, and Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 15: 633–644, 1996 [DOI] [PubMed] [Google Scholar]
- 86.McMurtry IF, Davidson AB, Reeves JT, and Grover RF. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res 38: 99–104, 1976 [DOI] [PubMed] [Google Scholar]
- 87.Mehdi MZ, Pandey NR, Pandey SK, and Srivastava AK. H2O2-induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src. Antioxid Redox Signal 7: 1014–1020, 2005 [DOI] [PubMed] [Google Scholar]
- 88.Mehta JP, Campian JL, Guardiola J, Cabrera JA, Weir EK, and Eaton JW. Generation of oxidants by hypoxic human pulmonary and coronary smooth-muscle cells. Chest 133: 1410–1414, 2008 [DOI] [PubMed] [Google Scholar]
- 89.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, and Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res 90: 1307–1315, 2002 [DOI] [PubMed] [Google Scholar]
- 90.Michelakis ED, Rebeyka I, Wu XC, Nsair A, Thébaud B, Hashimoto K, Dyck JRB, Haromy A, Harry G, Barr A, and Archer SL. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res 91: 478–486, 2002 [DOI] [PubMed] [Google Scholar]
- 91.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, and Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature 340: 230–233, 1989 [DOI] [PubMed] [Google Scholar]
- 92.Mittal M, Gu XQ, Pak O, Pamenter ME, Haag D, Fuchs DB, Schermuly RT, Ghofrani HA, Brandes RP, Seeger W, Grimminger F, Haddad GG, and Weissmann N. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med 52: 1033–1042, 2012 [DOI] [PubMed] [Google Scholar]
- 93.Mohazzab KM. and Wolin MS. Properties of a superoxide anion-generating microsomal NADH oxidoreductase, a potential pulmonary artery PO2 sensor. Am J Physiol 267: L823–L831, 1994 [DOI] [PubMed] [Google Scholar]
- 94.Morio Y. and McMurtry IF. Ca2+ release from ryanodine-sensitive store contributes to mechanism of hypoxic vasoconstriction in rat lungs. J Appl Physiol 92: 527–534, 2002 [DOI] [PubMed] [Google Scholar]
- 95.Moudgil R, Michelakis ED, and Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol 98: 390–403, 2005 [DOI] [PubMed] [Google Scholar]
- 96.Moudgil R, Michelakis ED, and Archer SL. The role of k+channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13: 615–632, 2006 [DOI] [PubMed] [Google Scholar]
- 97.Murphy AJ. Sulfhydryl group modification of sarcoplasmic reticulum membranes. Biochemistry 15: 4492–4496, 1976 [DOI] [PubMed] [Google Scholar]
- 97a.Nagaraj C, Tang B, Bálint Z, Wygrecka M, Hrzenjak A, Kwapiszewska G, Stacher E, Lindenmann J, Weir EK, Olschewski H, Olschewski A. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J. 41:85–95, 2013 [DOI] [PubMed] [Google Scholar]
- 98.Nakashima I, Kato M, Akhand AA, Suzuki H, Takeda K, Hossain K, and Kawamoto Y. Redox-linked signal transduction pathways for protein tyrosine kinase activation. Antioxid Redox Signal 4: 517–531, 2002 [DOI] [PubMed] [Google Scholar]
- 99.Nelson MT. and Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol 268: C799–C822, 1995 [DOI] [PubMed] [Google Scholar]
- 100.Nunez L, Vaquero M, Gomez R, Caballero R, Mateos-Caceres P, Macaya C, Iriepa I, Galvez E, Lopez-Farre A, Tamargo J, and Delpon E. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc Res 72: 80–89, 2006 [DOI] [PubMed] [Google Scholar]
- 101.Olschewski A, Hong Z, Nelson DP, and Weir EK. Graded response of K+ current, membrane potential, and [Ca2+]i to hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 283: L1143–L1150, 2002 [DOI] [PubMed] [Google Scholar]
- 102.Olschewski A, Hong Z, Peterson DA, Nelson DP, Porter VA, and Weir EK. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am J Physiol Lung Cell Mol Physiol 286: L15–L22, 2004 [DOI] [PubMed] [Google Scholar]
- 103.Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, Kwapiszewska G, Klepetko W, Seeger W, and Olschewski H. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res 98: 1072–1080, 2006 [DOI] [PubMed] [Google Scholar]
- 104.Olschewski A. and Weir EK. Hypoxic pulmonary vasoconstriction and hypertension. In: Pulmonary Circulation: Diseases and Their Treatment, edited by Peacock AJ. and Rubin LJ. London: Arnold, 2004, pp. 33–44 [Google Scholar]
- 105.Ooi L, Gigout S, Pettinger L, and Gamper N. Triple cysteine module within M-type K+channels mediates reciprocal channel modulation by nitric oxide and reactive oxygen species. J Neurosci 33: 6041–6046, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Osipenko ON, Evans AM, and Gurney AM. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold, O2-sensing potassium current. Br J Pharmacol 120: 1461–1470, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paddenberg R, Goldenberg A, Faulhammer P, Braun-Dullaeus RC, and Kummer W. Mitochondrial complex II is essential for hypoxia-induced ROS generation and vasoconstriction in the pulmonary vasculature. Adv Exp Med Biol 536: 163–169, 2003 [DOI] [PubMed] [Google Scholar]
- 108.Paky A, Michael JR, Burke-Wolin TM, Wolin MS, and Gurtner GH. Endogenous production of superoxide by rabbit lungs: effects of hypoxia or metabolic inhibitors. J Appl Physiol 74: 2868–2874, 1993 [DOI] [PubMed] [Google Scholar]
- 109.Palace V, Kumar D, Hill MF, Khaper N, and Singal PK. Regional differences in non-enzymatic antioxidants in the heart under control and oxidative stress conditions. J Mol Cell Cardiol 31: 193–202, 1999 [DOI] [PubMed] [Google Scholar]
- 110.Pan Y, Weng J, Cao Y, Bhosle RC, and Zhou M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvbeta1. J Biol Chem 283: 8634–8642, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pan Y, Weng J, Levin EJ, and Zhou M. Oxidation of NADPH on Kvbeta1 inhibits ball-and-chain type inactivation by restraining the chain. Proc Natl Acad Sci U S A 108: 5885–5890, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Park JW, Chun YS, Kim MS, Park YC, Kwak SJ, and Park SC. Metabolic modulation of cellular redox potential can improve cardiac recovery from ischemia-reperfusion injury. Int J Cardiol 65: 139–147, 1998 [DOI] [PubMed] [Google Scholar]
- 113.Park MK, Lee SH, Lee SJ, Ho WK, and Earm YE. Different modulation of Ca-activated K channels by the intracellular redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflügers Arch 430: 308–314, 1995 [DOI] [PubMed] [Google Scholar]
- 114.Peng W, Hoidal JR, and Farrukh IS. Role of a novel KCa opener in regulating K+ channels of hypoxic human pulmonary vascular cells. Am J Respir Cell Mol Biol 20: 737–745, 1999 [DOI] [PubMed] [Google Scholar]
- 115.Perez-Vizcaino F, Cogolludo A, and Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol Neurobiol 174: 212–220, 2010 [DOI] [PubMed] [Google Scholar]
- 116.Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev 72: S69–S88, 1992 [DOI] [PubMed] [Google Scholar]
- 117.Post JM, Gelband CH, and Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 77: 131–139, 1995 [DOI] [PubMed] [Google Scholar]
- 118.Post JM, Hume JR, Archer SL, and Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 262: C882–C890, 1992 [DOI] [PubMed] [Google Scholar]
- 119.Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, and Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem 281: 13588–13595, 2006 [DOI] [PubMed] [Google Scholar]
- 120.Poteser M, Romanin C, Schreibmayer W, Mayer B, and Groschner K. S-nitrosation controls gating and conductance of the alpha 1 subunit of class C L-type Ca(2+) channels. J Biol Chem 276: 14797–14803, 2001 [DOI] [PubMed] [Google Scholar]
- 121.Quinlan TR, Li D, Laubach VE, Shesely EG, Zhou N, and Johns RA. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 279: L641–L650, 2000 [DOI] [PubMed] [Google Scholar]
- 122.Rathore R, Zheng YM, Li XQ, Wang QS, Liu QH, Ginnan R, Singer HA, Ho YS, and Wang YX. Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem Biophys Res Commun 351: 784–790, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, and Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 45: 1223–1231, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ray PD, Huang BW, and Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24: 981–990, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reeve HL, Tolarova S, Nelson DP, Archer S, and Weir EK. Redox control of oxygen sensing in the rabbit ductus arteriosus. J Physiol 533: 253–261, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reeve HL, Weir EK, Nelson DP, Peterson DA, and Archer SL. Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue. Exp Physiol 80: 825–834, 1995 [DOI] [PubMed] [Google Scholar]
- 127.Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther 90: 1–19, 2001 [DOI] [PubMed] [Google Scholar]
- 128.Robertson TP, Hague D, Aaronson PI, and Ward JP. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol 525: 669–680, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rozanski GJ. and Xu Z. Glutathione and K+ channel remodeling in postinfarction rat heart. Am J Physiol Heart Circ Physiol 282: H2346–H2355, 2002 [DOI] [PubMed] [Google Scholar]
- 130.Sahoo N, Schonherr R, Hoshi T, and Heinemann SH. Cysteines control the N- and C-linker-dependent gating of KCNH1 potassium channels. Biochim Biophys Acta 1818: 1187–1195, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schach C, Xu M, Platoshyn O, Keller SH, and Yuan JX. Thiol oxidation causes pulmonary vasodilation by activating K+channels and inhibiting store-operated Ca2+channels. Am J Physiol Lung Cell Mol Physiol 292: L685–L698, 2007 [DOI] [PubMed] [Google Scholar]
- 132.Schumacker PT. Lung cell hypoxia: role of mitochondrial reactive oxygen species signaling in triggering responses. Proc Am Thorac Soc 8: 477–484, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scragg JL, Dallas ML, Wilkinson JA, Varadi G, and Peers C. Carbon monoxide inhibits L-type Ca2+channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J Biol Chem 283: 24412–24419, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Seddon M, Looi YH, and Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93: 903–907, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, Egemnazarov B, Turowska A, Fuchs B, Nikam S, Roth M, Sydykov A, Medebach T, Klepetko W, Jaksch P, Dumitrascu R, Garn H, Voswinckel R, Kostin S, Seeger W, Schermuly RT, Grimminger F, Ghofrani HA, and Weissmann N. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 147: 293–305, 2011 [DOI] [PubMed] [Google Scholar]
- 136.Shimoda LA. Hypoxic regulation of ion channels and transporters in pulmonary vascular smooth muscle. Adv Exp Med Biol 661: 221–235, 2010 [DOI] [PubMed] [Google Scholar]
- 137.Shimoda LA. and Polak J. Hypoxia. 4. Hypoxia and ion channel function. Am J Physiol Cell Physiol 300: C951–C967, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shimoda LA. and Undem C. Interactions between calcium and reactive oxygen species in pulmonary arterial smooth muscle responses to hypoxia. Respir Physiol Neurobiol 174: 221–229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Simon F, Leiva-Salcedo E, Armisen R, Riveros A, Cerda O, Varela D, Eguiguren AL, Olivero P, and Stutzin A. Hydrogen peroxide removes TRPM4 current desensitization conferring increased vulnerability to necrotic cell death. J Biol Chem 285: 37150–37158, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, Schulz R, Seeger W, Grimminger F, and Weissmann N. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J 32: 1639–1651, 2008 [DOI] [PubMed] [Google Scholar]
- 141.Song MY, Makino A, and Yuan JX. Role of reactive oxygen species and redox in regulating the function of transient receptor potential channels. Antioxid Redox Signal 15: 1549–1565, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Steinbicker AU, Liu H, Jiramongkolchai K, Malhotra R, Choe EY, Busch CJ, Graveline AR, Kao SM, Nagasaka Y, Ichinose F, Buys ES, Brouckaert P, Zapol WM, and Bloch KD. Nitric oxide regulates pulmonary vascular smooth muscle cell expression of the inducible cAMP early repressor gene. Nitric Oxide 25: 294–302, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, and Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res 81: 34–41, 1997 [DOI] [PubMed] [Google Scholar]
- 144.Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, Picard MH, and Zapol WM. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest 101: 2468–2477, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, and Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290: L661–L673, 2006 [DOI] [PubMed] [Google Scholar]
- 146.Sun J, Morgan M, Shen RF, Steenbergen C, and Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res 101: 1155–1163, 2007 [DOI] [PubMed] [Google Scholar]
- 147.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, and Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res 98: 403–411, 2006 [DOI] [PubMed] [Google Scholar]
- 148.Sun J, Yamaguchi N, Xu L, Eu JP, Stamler JS, and Meissner G. Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry 47: 13985–13990, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Svoboda LK, Reddie KG, Zhang L, Vesely ED, Williams ES, Schumacher SM, O'Connell RP, Shaw R, Day SM, Anumonwo JM, Carroll KS, and Martens JR. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ Res 111: 842–853, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sylvester JT, Shimoda LA, Aaronson PI, and Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tabet F, Savoia C, Schiffrin EL, and Touyz RM. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol 44: 200–208, 2004 [DOI] [PubMed] [Google Scholar]
- 152.Tang K, Li X, Zheng MQ, and Rozanski GJ. Role of apoptosis signal-regulating kinase-1-c-Jun NH2-terminal kinase-p38 signaling in voltage-gated K+channel remodeling of the failing heart: regulation by thioredoxin. Antioxid Redox Signal 14: 25–35, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.ten Freyhaus H, Dumitrescu D, Berghausen E, Vantler M, Caglayan E, and Rosenkranz S. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs 21: 119–134, 2012 [DOI] [PubMed] [Google Scholar]
- 154.Thuringer D. and Findlay I. Contrasting effects of intracellular redox couples on the regulation of maxi-K channels in isolated myocytes from rabbit pulmonary artery. J Physiol 500: 583–592, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tipparaju SM, Li XP, Kilfoil PJ, Xue B, Uversky VN, Bhatnagar A, and Barski OA. Interactions between the C-terminus of Kv1.5 and Kvbeta regulate pyridine nucleotide-dependent changes in channel gating. Pflugers Arch 463: 799–818, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tipparaju SM, Liu SQ, Barski OA, and Bhatnagar A. NADPH binding to beta-subunit regulates inactivation of voltage-gated K(+) channels. Biochem Biophys Res Commun 359: 269–276, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tipparaju SM, Saxena N, Liu SQ, Kumar R, and Bhatnagar A. Differential regulation of voltage-gated K+channels by oxidized and reduced pyridine nucleotide coenzymes. Am J Physiol Cell Physiol 288: C366–C376, 2005 [DOI] [PubMed] [Google Scholar]
- 158.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7: 833–846, 2006 [DOI] [PubMed] [Google Scholar]
- 159.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, and Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A 105: 9355–9360, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Viola HM, Arthur PG, and Hool LC. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+channel function in the absence of apoptosis in ventricular myocytes. Circ Res 100: 1036–1044, 2007 [DOI] [PubMed] [Google Scholar]
- 161.Wang QS, Zheng YM, Dong L, Ho YS, Guo Z, and Wang YX. Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic Biol Med 42: 642–653, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang X, Tong M, Chinta S, Raj JU, and Gao Y. Hypoxia-induced reactive oxygen species downregulate ETB receptor-mediated contraction of rat pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 290: L570–L578, 2006 [DOI] [PubMed] [Google Scholar]
- 163.Wang Y, Coe Y, Toyoda O, and Coceani F. Involvement of endothelin-1 in hypoxic pulmonary vasoconstriction in the lamb. J Physiol 482: 421–434, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang YX. and Zheng YM. ROS-dependent signaling mechanisms for hypoxic Ca(2+) responses in pulmonary artery myocytes. Antioxid Redox Signal 12: 611–623, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Waypa GB, Chandel NS, and Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res 88: 1259–1266, 2001 [DOI] [PubMed] [Google Scholar]
- 166.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW, and Schumacker PT. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res 99: 970–978, 2006 [DOI] [PubMed] [Google Scholar]
- 167.Waypa GB. and Schumacker PT. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol 98: 404–414, 2005 [DOI] [PubMed] [Google Scholar]
- 168.Waypa GB. and Schumacker PT. Hypoxia-induced changes in pulmonary and systemic vascular resistance: where is the O2 sensor? Respir Physiol Neurobiol 174: 201–211, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, and Luckhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem 277: 23150–23156, 2002 [DOI] [PubMed] [Google Scholar]
- 170.Weir EK, Hong Z, and Chen Y. Superoxide dismutase: master and commander? Eur Respir J 36: 234–236, 2010 [DOI] [PubMed] [Google Scholar]
- 171.Weir EK. and Olschewski A. Role of ion channels in acute and chronic responses of the pulmonary vasculature to hypoxia. Cardiovasc Res 71: 630–641, 2006 [DOI] [PubMed] [Google Scholar]
- 172.Weir EK, Will JA, Lundquist LJ, Eaton JW, and Chesler E. Diamide inhibits pulmonary vasoconstriction induced by hypoxia or prostaglandin F2 alpha. Proc Soc Exp Biol Med 173: 96–103, 1983 [DOI] [PubMed] [Google Scholar]
- 173.Weissmann N, Kuzkaya N, Fuchs B, Tiyerili V, Schafer RU, Schutte H, Ghofrani HA, Schermuly RT, Schudt C, Sydykov A, Egemnazarow B, Seeger W, and Grimminger F. Detection of reactive oxygen species in isolated, perfused lungs by electron spin resonance spectroscopy. Respir Res 6: 86, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Weissmann N, Vogels H, Schermuly RT, Ghofrani HA, Hanze J, Fink L, Rose F, Seeger W, and Grimminger F. Measurement of exhaled hydrogen peroxide from rabbit lungs. Biol Chem 385: 259–264, 2004 [DOI] [PubMed] [Google Scholar]
- 175.Weng J, Cao Y, Moss N, and Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem 281: 15194–15200, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wu W, Platoshyn O, Firth AL, and Yuan JX. Hypoxia divergently regulates production of reactive oxygen species in human pulmonary and coronary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 293: L952–L959, 2007 [DOI] [PubMed] [Google Scholar]
- 177.Xie ZJ, Wang YH, Askari A, Huang WH, Klaunig JE, and Askari A. Studies on the specificity of the effects of oxygen metabolites on cardiac sodium pump. J Mol Cell Cardiol 22: 911–920, 1990 [DOI] [PubMed] [Google Scholar]
- 178.Xu KY. and Becker LC. Ultrastructural localization of glycolytic enzymes on sarcoplasmic reticulum vesticles. J Histochem Cytochem 46: 419–427, 1998 [DOI] [PubMed] [Google Scholar]
- 179.Xu SZ, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang LH, Emery P, Sivaprasadarao A, and Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature 451: 69–72, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yang Y, Jin X, and Jiang C. S-glutathionylation of ion channels: insights into the regulation of channel functions, thiol modification crosstalk, and mechanosensing. Antioxid Redox Signal 20: 937–951, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yang Y, Shi W, Chen X, Cui N, Konduru AS, Shi Y, Trower TC, Zhang S, and Jiang C. Molecular basis and structural insight of vascular K(ATP) channel gating by S-glutathionylation. J Biol Chem 286: 9298–9307, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, and Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol 2: 596–607, 2006 [DOI] [PubMed] [Google Scholar]
- 183.Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 77: 370–378, 1995 [DOI] [PubMed] [Google Scholar]
- 184.Yuan XJ, Wang J, Juhaszova M, Golovina VA, and Rubin LJ. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol 274: L621–L635, 1998 [DOI] [PubMed] [Google Scholar]
- 185.Zhang H, Tao L, Jiao X, Gao E, Lopez BL, Christopher TA, Koch W, and Ma XL. Nitrative thioredoxin inactivation as a cause of enhanced myocardial ischemia/reperfusion injury in the aging heart. Free Radic Biol Med 43: 39–47, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, and Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A 103: 9357–9362, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zheng MQ, Li X, Tang K, Sharma NM, Wyatt TA, Patel KP, Gao L, Bidasee KR, and Rozanski GJ. Pyruvate restores beta-adrenergic sensitivity of L-type Ca(2+) channels in failing rat heart: role of protein phosphatase. Am J Physiol Heart Circ Physiol 304: H1352–H1360, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]