

BACKGROUND
Hemophagocytic lymphohistiocytosis (HLH) is a rare but potentially fatal disease that commonly appears in infancy, although it has been reported in adults. Chemoimmunotherapy-based treatments have improved the survival of patients with HLH; however, overall survival is still poor. We retrospectively analyzed the data of 12 HLH patients who were admitted between 2005 and 2014. All patients were Saudi Arabia in origin with a female predominance (75%) and a median age of onset of 9.5 months. The consanguinity rates were significantly high (75%) with a positive family history in 41% of cases. Of the 12 patients, nine were defined as primary HLH patients and three were confirmed to be secondary HLH patients. All patients fulfilled the 2004 diagnostic criteria for HLH and received HLH-2004 treatment. Six of these patients showed a good response to chemotherapy, while the remainder of the patients showed partial or no response to chemotherapy. Five patients in this cohort received stem cell transplant, and these patients are currently in remission. The mortality rate of this cohort is currently 50%. Genetic mutational analysis showed a positive STX11 mutation in five patients and a PRF1 (perforin) mutation in two patients. To the best of our knowledge, this is the first case series of HLH from Saudi Arabia.
Keywords: hemophagocytic lymphohistiocytosis, gene mutations, outcome
Introduction
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a life-threatening condition caused by uncontrolled activation of T lymphocytes and macrophages in addition to an impaired cytotoxic function of natural killer (NK) cells, which results in a massive cytokine release. As a result of failure to downregulate activated macrophages and lymphocytes, a hyper inflammatory phenotype ensues.1 In the absence of treatment with epipodophyllotoxins, immunosuppressive agents, or bone marrow (BM) transplantation, the disease progresses rapidly.2 HLH commonly occurs in infancy, although it has been reported in all age groups. HLH is categorized as primary HLH (familial) or secondary HLH (acquired). Primary HLH is caused by genetic mutations of the FLH loci. Mutations in other genes (PRF1, UNC130, STX11, STBP2, RAB27A, LYST, SH2DIA, and XIAP) usually associated with primary immunodeficiency syndrome have also been implicated in the pathogenesis. Secondary HLH occurs in the setting of bacterial and viral infections, malignancy (including T and B cell lymphomas), autoimmune diseases, and certain metabolic imbalances.3–9
In order to reduce mortality rates, an early diagnosis is crucial. However, due to lack of specificity of current diagnostic criteria, a definitive diagnosis is often difficult. A diagnosis of HLH is made by detecting a specific genetic mutation or by the current HLH-2004 diagnostic criteria (Table 1).10–12
Table 1.
HLH 2004 diagnostic criteria (5/8 must be present in the patient to diagnose HLH).
| 1 | Fever |
| 2 | Splenomegaly |
| 3 | Peripheral cytopenias |
| 4 | Increased triglycerides and hypofibrinogenemia |
| 5 | Hemophagocytosis |
| 6 | Reduced natural killer cell activity |
| 7 | Ferritin >500 |
| 8 | Soluble CD25 >2400 µL/mL |
The difficulties encountered with these criteria is that some patients may have three or four positive markers, which excludes them from a diagnosis of HLH; yet, they have other clinical features of HLH, which responds specifically to HLH treatment protocols.11,12
The characteristic histological finding is accumulation of lymphocytes and non-Langerhans cell histiocytosis BM, spleen, liver, the lymph node, and other organs. The histocytes often shows phagocytosis of blood cells (hemophagocytosis). A lack of demonstration of hemophagocytosis in the BM, spleen, liver, or the lymph node often makes an initial diagnosis difficult.13
Here, we present single-center series of 12 cases of HLH from Saudi Arabia. We retrospectively analyzed the clinical features, molecular profile, diagnostic issues, and treatment outcome in this cohort. To the best of our knowledge, this is the first case series of HLH from Saudi Arabia reported in the literature.
Methods
This study was approved by the Departmental Research and Ethics Committee. Twelve patients were diagnosed with HLH between the period of January 2005 and December 2014 (9-year period) using the 2004 HLH diagnostic criteria (Table 1). All these patients fulfilled at least five fundamental criteria of HLH at the time of diagnosis. We reviewed all medical records of these patients in order to retrieve data such as family history, consanguinity, molecular studies, therapeutic protocols, and overall survival. Patients who were found to have a genetic abnormality and/or early-onset disease (aged 0–2 years) with a family history were considered as having familial HLH. In addition, a recurrence of HLH, in the absence of other causes, such as infection, autoimmune disease, or malignancy, is considered to be good evidence that a patient has primary HLH. Patients whose genetic testing revealed no genetic abnormality and who failed to fulfill the diagnostic criteria were considered as having secondary HLH. Central nervous system involvement was defined by the presence of increased cerebrospinal fluid white blood count and/or neurologic symptoms, or abnormalities in magnetic resonance imaging (MRI).
In our study, 10 patients were tested for three more common gene defects. Genetic testing was not performed in two patients. Commonly occurring genetic mutations analyzed included the following: mutations affecting the STX11 gene, perforin gene (PRF1), and Munc13-4 (UNC13D) gene. Recently, Munc 18–2 or syntax in binding protein 2 (STXBP2 gene), which has been identified as a cause of familial hemophagocytic lymphohistiocytosis type 5 by two separate groups,14,15 has been added to our investigative panel for HLH.
Statistical Methods
Data were recorded systematically on excel spread sheets and analyzed using descriptive statistics for all our calculations, which included age, gender, clinical findings, and biochemical data.
Results
Clinical features, molecular characteristics, and the outcome of the patients are summarized in Table 2.
Table 2.
Patient characteristics.
| # | AGE | SEX | CONSANGUINITY | FAMILY HISTORY | ASSOCIATED INFECTION | HLH CRITERIA | MOLECULAR MUTATION | TREATMENT | HSCT | OUTCOME |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 months | M | + | + | – | 6/8 | PRF1 | HLH 2004 | Haplotype | Alive |
| 2 | 4 months | M | + | + | EBV | 7/8 | STX-11 | HLH 2004 | No | Died |
| 3 | 13 months | F | + | + | Candida | 5/8 | STX-11 | HLH 2004 | No | Died |
| 4 | 3 years | M | – | – | Dengue fever | 5/8 | Negative | HLH 2004 | No | Alive |
| 5 | 8 years | F | – | – | EBV | 7/8 | Not done | HLH 2004 | No | Died (ARDS) |
| 6 | 2 months | F | + | – | – | 6/8 | Negative | HLH 2004 | Haplotype HSCT (Father) | Alive |
| 7 | 11 months | F | + | + | – | 6/8 | STX-11 | HLH 2004 | Cord SCT | Alive |
| 8 | 4 years | F | + | – | Dengue fever | 5/8 | Negative | HLH 2004 | No | Lost follow up |
| 9 | 7 years | F | + | – | – | 5/8 | Not done | Refused treatment | No | Died (relapse) |
| 10 | 3 months | F | + | – | – | 5/8 | PRF1 | HLH 2004 | URD HSCT | Died (relapse) |
| 11 | 6 months | M | + | + | Gram +ve cocci | 6/8 | STX-11 | HLH 2004 | MRD HSCT | Alive |
| 12 | 7 years | F | – | – | HAV | 6/8 | STX-11 | HLH 2004 Anakinra | In process | Alive |
Abbreviations: HSCT, hematopoietic stem cell transplantation; MRD, matched related donor; URD, unrelated donor; PRF1, perforin gene; STX11, syntaxin11; N/A, not applicable; ARDS, acute respiratory distress syndrome.
All patients were Saudi Arabia in origin, between 2 months and 8 years, with a median age of 9.5 months. There was female predominance (75%). The consanguinity rates were significantly high (9/12, 75%). A positive family history was noted in 41% of cases. Of the 12 patients, nine patients were diagnosed with primary HLH and these patients had a median age of six months. The remaining three patients were diagnosed with secondary HLH as a result of concomitant viral infections, including Epstein–Barr virus (EBV; identified in one patient) and dengue virus (identified in two patients). All patients met the criteria of HLH, at least five out of a total of eight criteria (Fig. 1). All patients received the HLH-2004 treatment protocol except one patient who refused complete chemotherapy and was subsequently treated with dexamethasone and cyclophosphamide. A favorable response was noted in 50% of patients. The remainder had partial remission or failed to achieve remission. Five patients received hematopoietic stem cell transplantation (HSCT), of which four patients are currently disease free and stable on follow-up, while the other patient relapsed and subsequently demised.
Figure 1.

Patient clinical and laboratory parameters.
Currently, 6 of the 12 patients are still alive (50%). Two patients demised prior to HSCT, as a result of acute respiratory distress (ARDS) and relapsed disease, respectively. One patient defaulted treatment. The results of genetic mutation analysis were positive for seven patients (five cases for STX-11 and two cases for PRF1 gene mutations) and negative for three patients. Genetic mutational analysis was omitted in two patients for unknown reasons.
In the BM, hemophagocytosis was documented by the presence of mature and immature hematopoietic cells within the macrophages (Fig. 2). Cerebrospinal fluid analysis was performed in all patients and showed no abnormal cells or features suggestive of HLH. However, an MRI scan performed detected structural abnormalities with HLH in three patients (Fig. 3).
Figure 2.
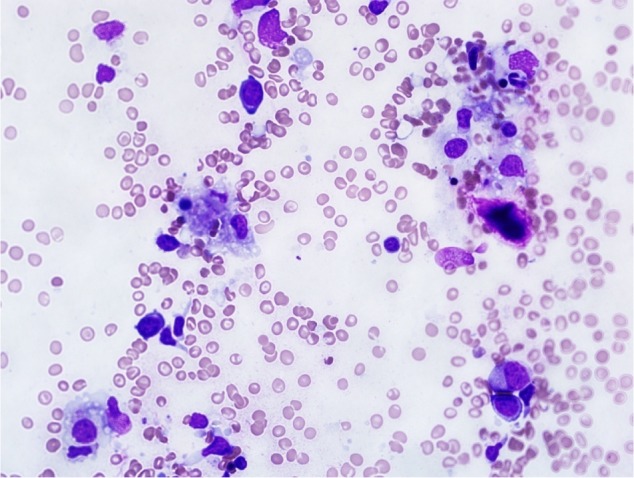
Hemophagocytosis found on BM aspirate (case 12).
Figure 3.

(A) Axial FLAIR and (B) axial T2 MRI showing parieto–occipital lesions of abnormal signal intensity representing brain parenchymal involvement by the disease.
Discussion
The HLH diagnostic criteria, first described in 1994, included five conditions as follows:
Fever,
Splenomegaly,
Bicytopenia (cytopeniasin two cell lines, eg, anemia and thrombocytopenia),
Hypertriglyceridemia and/or hypofibrinogenemia, and
Hemophagocytosis (seen morphologically on BM aspirate specimens).
These guidelines were revised in 2004 with the addition of three new criteria as follows10:
Low or absenceof NK cell activity,
Hyperferritinemia (ferritin levels >500), and
High soluble interleukin 2 receptor levels clusters of differentiation 25 (CD25).
Current practice requires that a minimum of five criteria must be present for a diagnosis of HLH. However, the literature suggests that if a patient has a specific genetic mutation consistent with HLH on molecular testing, then these diagnostic criteria are not essential for diagnosis.10,16,17
In the present study, the results were closely comparable with previously reported cases in the literature with regard to the median age (9.5 months), positive family history (42%), and high rate of consanguinity (75%).18,19 Interestingly, the majority of our patients are females (8/12, 75%). Familial HLH is the most commonly encountered type of syndrome in our institute (9/12), which is due to most probably the high rate of consanguineous marriage in Saudi pedigrees.
In our study, all the patients fulfilled the classical clinical criteria for HLH. Four patients presented with variable neurologic symptoms, ranging from irritability, neck stiffness, and unconsciousness to convulsions. These neurologic symptoms make the diagnosis of HLH difficult and delayed. Other studies have also reported this finding.20
Clinically, the therapy of primary and secondary HLH aims to suppress the exaggerated immune response with the use of immunosuppressive drugs and treatment of the underlying disease, in cases of acquired HLH. Thus, the distinction between primary and secondary HLH is not essential for the initial diagnosis. However, it is important to rule out a genetic mutation, which will be useful for subsequent management. In the HLH-2004 treatment protocol, when a genetic cause of the disease is confirmed by molecular studies, it is recommended to proceed to HSCT.10
In our study, the most common trigger for HLH was viral infections due to EBV and dengue virus, respectively. Fungal infections (Candida albicans) and bacterial infections (Gram-positive cocci) were found in two cases. All these findings concurred with those reported in the literature.4,6,21–23
The overall survival in our cohort was 50%, which is comparable with previously reported results.24–26 The HLH-2004 treatment protocol was the first-line therapy in our study. At present, there are limited data regarding potential second-line therapies. Case reports describing the use of anakinra, infliximab, daclizumab, vincristine, and other agents as salvage therapies for HLH exist.11 Patient 12 was treated with the IL-1 receptor antagonist (IL-1ra) anakinra in a combination as a substitute for etoposide after failing to achieve a response after two weeks. This patient showed improvement in his clinical and laboratory data and is currently stable on follow-up and awaiting HSCT. The promising results shown with the use of IL-1ra for HLH in the present study suggest a potential use of this agent in the treatment of HLH; however, further research and clinical trials are required. Bruck et al reported similar results with the use of anakinra and corticosteroids in systemic juvenile idiopathic arthritis-associated macrophage activation syndrome.27
Peripheral cytopenias was a constant finding in our cohort. Thus, all patients underwent microscopic examination of a BM aspirate and biopsy to detect hemophagocytosis or to detect BM pathology contributing to the cytopenias. Most patients (75%) had hemophagocytosis on BM examination, which concurs with previous studies.28 However, hemophagocytosisis not essential for the diagnosis of HLH. We found that hemophagocytosis failed to be detected initially in 25% of cases, reinforcing that a diagnosis of HLH cannot be excluded solely on the presence or absence of hemophagocytosis in the BM, as this may potentially delay treatment.11,13
We assessed the frequency of specific HLH mutations and found that mutations affecting the STX11 gene (5/10, 50%) and perforin gene (PRF1) (2/10, 20%) occurred commonly in the Saudi population. Only one patient was tested for the STXBP2 mutation and this patient tested negative. A multicenter study is needed to confirm these results due to the relatively small sample size and rarity of HLH.
The differential diagnosis of HLH includes several multisystem illnesses characterized by fever, cytopenia, hepatic failure, and neurologic symptoms. Many of the conditions in the differential diagnosis of HLH can also cause HLH. HLH may simulate a number of common conditions that cause fever, cytopenias, hepatic failure, and neurologic symptoms. In our study, cytopenias, a very high ferritin level, and elevated liver enzymes were prominent findings and assisted in distinguishing HLH from other conditions.
In our study, 83% of patients had abnormal liver function tests, including mildly elevated transaminases and fulminant liver failure. This is a common finding in HLH. Literature suggests that the frequency of hepatitis is high in HLH with some authors hypothesizing that an absence of hepatic abnormalities in suspected HLH should prompt a search for an alternative diagnosis.24,29,30 This suggests a need to modify the current HLH criteria to include abnormal liver function testing. We encountered a delay in diagnosis of HLH in our cohort. This finding is most likely attributable to the complexity of HLH diagnostic criteria, a low index of clinical suspicion, and the rarity of HLH in the general population.
Conclusions
HLH is a rare but rapidly fatal disease with a heterogenous clinical presentation. An early diagnosis is crucial to reduce mortality rates and initiate treatment. Familiarity with the clinical symptoms of the diagnostic criteria of HLH is essential for clinicians, including pediatric hematologists, infectious diseases specialists, gastroenterologists, and intensivists, as early diagnosis and prompt aggressive treatment are vital for patients’ survival and a favorable outcome.
Acknowledgments
The authors thank the Department of Pediatric Hematology/Oncology, King Faisal Specialist Hospital and Research Center for contributions in this work. They also thank Dr. Qanita Sedick in excellent assisting and critical comments on this study.
Footnotes
ACADEMIC EDITOR: Praveen Kumar, Editor in Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1078 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no external funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: GE, AA. Analyzed the data: GE. Wrote the first draft of the manuscript: HE, OA, NO, FA. Contributed to the writing of the manuscript: GE, AA, HE, OA, NO, EA, FA. Agree with manuscript results and conclusions: EA, HE. Jointly developed the structure and arguments for the paper: FA, GE. Made critical revisions and approved final version: GE, AA, HE, OS, NO, EA, FA. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82. doi: 10.1016/j.bbmt.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Blanche S, Caniglia M, Girault D, et al. Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation. Blood. 1991;78:51–4. [PubMed] [Google Scholar]
- 3.Janka G, Imashuku S, Elinder G, et al. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:435–44. doi: 10.1016/s0889-8588(05)70521-9. [DOI] [PubMed] [Google Scholar]
- 4.McClain K, Gehrz R, Grierson H, Purtilo D, Filipovich A. Virus-associated histiocytic proliferations in children. Frequent association with Epstein-Barr virus and congenital or acquired immunodeficiencies. Am J Pediatr Hematol Oncol. 1988;10:196. [PubMed] [Google Scholar]
- 5.Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139(6):713–27. doi: 10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 6.Mou SS, Nakagawa TA, Riemer EC, et al. Hemophagocytic lymphohistiocytosis complicating influenza A infection. Pediatrics. 2006;118:e216. doi: 10.1542/peds.2005-1861. [DOI] [PubMed] [Google Scholar]
- 7.Yuzurihara SS, Ao K, Hara T, et al. Human parechovirus-3 infection in nine neonates and infants presenting symptoms of hemophagocytic lymphohistiocytosis. J Infect Chemother. 2013;19:144. doi: 10.1007/s10156-012-0420-9. [DOI] [PubMed] [Google Scholar]
- 8.Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161(5):609–22. doi: 10.1111/bjh.12293. [DOI] [PubMed] [Google Scholar]
- 9.Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163(3):271–83. doi: 10.1111/j.1365-2249.2010.04302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henter JI, Horne A, Arico M. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 11.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009;1:127–31. doi: 10.1182/asheducation-2009.1.127. [DOI] [PubMed] [Google Scholar]
- 13.Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(3):402–4. doi: 10.1002/pbc.21564. [DOI] [PubMed] [Google Scholar]
- 14.Zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482–92. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brochetta C, Vita F, Tiwari N, et al. Involvement of Munc18 isoforms in the regulation of granule exocytosis in neutrophils. Biochim Biophys Acta. 2008;1783:1781–91. doi: 10.1016/j.bbamcr.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 16.Janka GE, Schenider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2004;124:4–14. doi: 10.1046/j.1365-2141.2003.04726.x. [DOI] [PubMed] [Google Scholar]
- 17.Henter JI, Tondini C, Pritchard J. Histiocytic syndromes. Crit Rev Oncol Hematol. 2004;50:157–74. doi: 10.1016/j.critrevonc.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Horne A, Ramme KG, Rudd E, et al. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol. 2008;143(1):75–83. doi: 10.1111/j.1365-2141.2008.07315.x. [DOI] [PubMed] [Google Scholar]
- 19.Meeths M, Bryceson YT, Rudd E, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. 2010;54(4):563–72. doi: 10.1002/pbc.22357. [DOI] [PubMed] [Google Scholar]
- 20.Henter JI, Nennesmo I. Neuropathologic findings and neurologic symptoms in twenty-three children with hemophagocytic lymphohistiocytosis. J Pediatr. 1997;130(3):358–65. doi: 10.1016/s0022-3476(97)70196-3. [DOI] [PubMed] [Google Scholar]
- 21.Fardet L, Blum L, Kerob D, et al. Human herpesvirus 8-associated hemophagocytic lymphohistiocytosis in human immunodeficiency virus-infected patients. Clin Infect Dis. 2003;37:285. doi: 10.1086/375224. [DOI] [PubMed] [Google Scholar]
- 22.Hegerova LT, Lin Y. Disseminated histoplasmosis: a cause of hemophagocytic syndrome. Mayo Clin Proc. 2013;88:e123. doi: 10.1016/j.mayocp.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 23.Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90:220. doi: 10.1002/ajh.23911. [DOI] [PubMed] [Google Scholar]
- 24.Niece JA, Rogers ZR, Ahmad N, Langevin AM, McClain KL. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer. 2010;54(3):424–8. doi: 10.1002/pbc.22359. [DOI] [PubMed] [Google Scholar]
- 25.Henter JI, Samuelsson-Horne A, Arico M, et al. Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–73. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 26.Trottestam H, Horne A, Aricò M, et al. Histocyte Society. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–84. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol. 2011;17(1):23–7. doi: 10.1097/RHU.0b013e318205092d. [DOI] [PubMed] [Google Scholar]
- 28.Rivière S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127:1118. doi: 10.1016/j.amjmed.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 29.Palazzi DL, McClain KL, Kaplan SL. Hemophagocytic syndrome in children: an important diagnostic consideration in fever of unknown origin. Clin Infect Dis. 2003;36(3):306–12. doi: 10.1086/345903. [DOI] [PubMed] [Google Scholar]
- 30.Arico M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis: report of 122 children from the international registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10(2):197–203. [PubMed] [Google Scholar]
- 31.Pagel J, Beutel K, Lehmberg K, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5) Blood. 2012;119:6016. doi: 10.1182/blood-2011-12-398958. [DOI] [PubMed] [Google Scholar]