

Crystal structures of L. mexicana arginase complexed with two α,α-disubstituted boronic amino-acid inhibitors were determined to resolutions of 1.28 and 1.65 Å, respectively.
Keywords: Leishmania mexicana; arginase; α,α-disubstituted boronic amino-acid inhibitors
Abstract
Leishmania arginase is a potential drug target for the treatment of leishmaniasis because this binuclear manganese metalloenzyme initiates de novo polyamine biosynthesis by catalyzing the hydrolysis of l-arginine to generate l-ornithine and urea. The product l-ornithine subsequently undergoes decarboxylation to yield putrescine, which in turn is utilized for spermidine biosynthesis. Polyamines such as spermidine are essential for the growth and survival of the parasite, so inhibition of enzymes in the polyamine-biosynthetic pathway comprises an effective strategy for treating parasitic infections. To this end, two X-ray crystal structures of L. mexicana arginase complexed with α,α-disubstituted boronic amino-acid inhibitors based on the molecular scaffold of 2-(S)-amino-6-boronohexanoic acid are now reported. Structural comparisons with human and parasitic arginase complexes reveal interesting differences in the binding modes of the additional α-substituents, i.e. the d side chains, of these inhibitors. Subtle differences in the three-dimensional contours of the outer active-site rims among arginases from different species lead to different conformations of the d side chains and thus different inhibitor-affinity trends. The structures suggest that it is possible to maintain affinity while fine-tuning intermolecular interactions of the d side chain of α,α-disubstituted boronic amino-acid inhibitors in the search for isozyme-specific and species-specific arginase inhibitors.
1. Introduction
Leishmaniasis is a neglected tropical disease caused by more than 20 different Leishmania species prevalent in nearly 100 countries, with 1.3 million new cases diagnosed annually (Crompton, 2013 ▸). There are three main forms of the disease: visceral leishmaniasis, which affects the spleen, liver and bone marrow, and is usually fatal if left untreated; cutaneous leishmaniasis, which is characterized by large, disfiguring skin lesions; and mucocutaneous leishmaniasis, which causes painful lesions in nasal and oropharyngeal tissues. It is estimated that 20 000–40 000 people die from visceral leishmaniasis annually (Alvar et al., 2012 ▸). The occurrence of leishmaniasis is on the rise in Western countries owing to travel to and from endemic regions by military and civilian personnel (Antinori et al., 2005 ▸), so this neglected tropical disease comprises an increasingly prominent threat to public health.
Treatment options for patients diagnosed with leishmaniasis vary depending upon the parasite species involved, the organs and tissues affected, and the quality of healthcare available (Desjeux, 1996 ▸, 2001 ▸). For example, many cases of cutaneous leishmaniasis can resolve spontaneously, and those that do not generally respond to treatment with pentavalent antimony-based drugs. Other drugs currently in use for the treatment of cutaneous or visceral leishmaniasis include amphotericin B, pentamidine and miltefosine. Unfortunately, no single leishmaniasis treatment option is ideal owing to financial expense (especially for patients in developing countries), long drug-treatment regimens and side effects. Moreover, the emergence of resistance to currently existing drugs suggests that there is an urgent need for new pharmacological targets for leishmaniasis therapy (Sundar, 2001 ▸; Pérez-Victoria et al., 2002 ▸).
L. mexicana arginase (LmARG) is considered to be an attractive drug target in the exploration of new therapeutic approaches. This binuclear manganese metalloenzyme initiates the first step of de novo polyamine biosynthesis by catalyzing the hydrolysis of l-arginine to generate l-ornithine and urea (Ash et al., 2000 ▸; Christianson, 2005 ▸; Fig. 1 ▸ a); l-ornithine subsequently undergoes decarboxylation to yield putrescine, a polyamine building block (Heby et al., 2003 ▸, 2007 ▸). Studies of arginase-knockout mutants in Leishmania parasites confirm that arginase activity is essential for parasite viability and infectivity (Roberts et al., 2004 ▸; Reguera et al., 2009 ▸; da Silva, Maquiaveli et al., 2012 ▸; da Silva, Zampieri et al., 2012 ▸). Crystal structures of LmARG complexed with first-generation l-amino-acid inhibitors such as 2-(S)-amino-6-boronohexanoic acid (ABH; Fig. 1 ▸ b) revealed the molecular basis of affinity and inhibitory activity (D’Antonio et al., 2013 ▸), thereby setting the stage for the design and evaluation of second-generation inhibitors.
Figure 1.

(a) Arginase utilizes a metal-bridging hydroxide ion to catalyze the hydrolysis of l-arginine to form l-ornithine and urea. (b) The boronic acid analogue of l-arginine, 2-(S)-amino-6-boronohexanoic acid (ABH), undergoes nucleophilic attack by the metal-bridging hydroxide ion to yield a tetrahedral boronate anion that mimics the tetrahedral intermediate and its flanking transition states in the arginase reaction. (c) The arginase inhibitors (R)-2-amino-6-borono-2-[2-(piperidin-1-yl)ethyl]hexanoic acid (ABHPE) and (R)-2-amino-6-borono-2-[1-(3,4-dichlorobenzyl)piperidin-4-yl]hexanoic acid (ABHDP).
To expand the chemical space of arginase-inhibitor design, novel α,α-disubstituted boronic acid inhibitors based on ABH have been developed and evaluated against human arginase I, human arginase II, Plasmodium falciparum arginase and Schistosoma mansoni arginase (Ilies et al., 2011 ▸; Golebiowski et al., 2013 ▸; Van Zandt et al., 2013 ▸; Hai et al., 2014 ▸). While the l-boronic acid side chain of these inhibitors binds in the active-site cleft in an identical fashion to that of the parent compound ABH, the additional d side chain of these inhibitors can make new interactions in the outer rim of the active site.
Here, we report the crystal structures of LmARG with the α,α-disubstituted boronic acid inhibitors (R)-2-amino-6-borono-2-[2-(piperidin-1-yl)ethyl]hexanoic acid (ABHPE) and (R)-2-amino-6-borono-2-[1-(3,4-dichlorobenzyl)piperidin-4-yl]hexanoic acid (ABHDP) (Fig. 1 ▸ c; Golebiowski et al., 2013 ▸; Van Zandt et al., 2013 ▸). Structural comparisons with human and parasitic arginase complexes reveal interesting differences in the binding modes of the d side chains of these inhibitors. Even though the active-site clefts of these enzymes are highly conserved, the outer rims are not. Accordingly, different conformations are observed for the d side chains. As a proof of concept, our structures suggest that it is possible to maintain affinity while fine-tuning the intermolecular interactions of the d side chain of α,α-disubstituted boronic amino-acid inhibitors in the search for isozyme-specific and species-specific arginase inhibitors.
2. Materials and methods
2.1. Crystallization
Recombinant LmARG was expressed and purified as reported previously (D’Antonio et al., 2013 ▸). The LmARG–ABHPE complex was crystallized using the sitting-drop vapor-diffusion method at 21°C by mixing 1 µl LmARG solution (6 mg ml−1 protein pre-incubated with 10 mM ABHPE in 50 mM Bicine pH 8.5, 100 µM MnCl2, 1 mM TCEP, 5% glycerol, 2.5% DMSO) with 1 µl precipitant solution (0.1 M HEPES pH 7.5, 25% PEG 3350). The LmARG–ABHDP complex was crystallized in a similar fashion by mixing 1 µl LmARG solution (6 mg ml−1 protein pre-incubated with 10 mM ABHDP in 50 mM Bicine pH 8.5, 100 µM MnCl2, 1 mM TCEP, 5% glycerol, 2.5% DMSO) with 1 µl precipitant solution (0.1 M HEPES pH 7.2, 22% PEG 3350) at 21°C. Crystals appeared in 3 d and were soaked in a cryoprotectant solution comprised of precipitant solution supplemented with 25–30% glycerol prior to flash-cooling in liquid nitrogen.
2.2. X-ray data collection and processing
X-ray diffraction data were collected on beamline X29 at the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory, New York, USA. Diffraction data were integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▸). Data-collection and reduction statistics are listed in Table 1 ▸. The structure was determined by molecular replacement using Phaser (McCoy et al., 2007 ▸) as implemented in the CCP4 suite of programs, with the atomic coordinates of unliganded LmARG (PDB entry 4ity; D’Antonio et al., 2013 ▸) utilized as a search probe for rotation-function and translation-function calculations. Iterative cycles of refinement and model building were performed using PHENIX (Adams et al., 2010 ▸) and Coot (Emsley et al., 2010 ▸), respectively. The hemihedral twinning operator h, −h − k, −l was included in the refinement strategy as described previously (D’Antonio et al., 2013 ▸). Solvent molecules and inhibitors were added in the final stages of refinement for each structure. The quality of each final model was verified with MolProbity (Chen et al., 2010 ▸). The refinement statistics are reported in Table 1 ▸. Protein structure figures were prepared with PyMOL (http://www.pymol.org) and Adobe Photoshop CS.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| LmARG–ABHPE | LmARG–ABHDP | |
|---|---|---|
| PDB entry | 5hj9 | 5hja |
| Data collection | ||
| Beamline | X29, NSLS | X29, NSLS |
| Wavelength (Å) | 1.075 | 1.075 |
| Temperature (K) | 100 | 100 |
| Detector | ADSC Q315 | ADSC Q315 |
| Space group | R3:H | R3:H |
| Resolution (Å) | 50.0–1.28 | 50.0–1.65 |
| Total reflections measured | 1041671 | 281614 |
| Unique reflections measured | 86060 | 40479 |
| Unit-cell parameters (Å) | a = b = 89.1, c = 113.6 | a = b = 88.9, c = 113.9 |
| Completeness (%) | 99.4 (94.6) | 99.5 (95.3) |
| 〈I/σ(I)〉 | 29.3 (1.2) | 22.4 (2.1) |
| R sym † | 0.091 (1.202) | 0.089 (0.730) |
| R p.i.m. ‡ | 0.029 (0.509) | 0.035 (0.316) |
| Multiplicity | 12.1 (8.5) | 7.0 (5.7) |
| CC1/2 § | 0.956 (0.510) | 0.987 (0.817) |
| Overall B factor from Wilson plot (Å2) | 16 | 24 |
| Twin fraction | 0.15 | 0.08 |
| Refinement | ||
| No. of reflections | ||
| Refinement | 77901 | 36600 |
| Test set | 8159 | 3879 |
| Twin law | h, −h − k, −l | h, −h − k, −l |
| R work ¶ (%) | 14.6 (30.1) | 18.7 (31.8) |
| R free ¶ (%) | 15.9 (31.0) | 21.2 (34.5) |
| No. of non-H atoms per monomer | ||
| Protein | 2425 | 2326 |
| Solvent | 200 | 146 |
| Ligands | 67 | 36 |
| Mn2+ ions | 2 | 2 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.012 | 0.004 |
| Angles (°) | 1.3 | 0.8 |
| Average B factors (Å2) | ||
| Protein | 25 | 35 |
| Solvent | 35 | 40 |
| Mn2+ ions | 14 | 25 |
| Inhibitor | 30 | 55 |
| Other ligands | 32 | 28 |
| Ramachandran | ||
| Favored†† (%) | 97.3 | 96.6 |
| Outliers†† (%) | 0.6 | 0.33 |
R
merge = 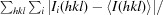
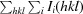 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl, 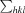 is the sum over all reflections and
is the sum over all reflections and 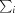 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R
p.i.m. = 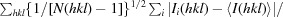 , where N(hkl) is the number of observations (multiplicity) and 〈I(hkl)〉 is the average intensity calculated from replicate data.
, where N(hkl) is the number of observations (multiplicity) and 〈I(hkl)〉 is the average intensity calculated from replicate data.
CC1/2 = στ 2/(στ 2 + σ∊ 2), where στ 2 is the true measurement-error variance and σ∊ 2 is the independent measurement-error variance.
R
work = 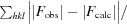
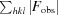 for reflections contained in the working set. R
free =
for reflections contained in the test set held aside during refinement. |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively.
for reflections contained in the working set. R
free =
for reflections contained in the test set held aside during refinement. |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively.
Calculated with MolProbity.
2.3. Inhibitory activity assays
Arginase activity was measured using a colorimetric assay with slight modifications (Archibald, 1945 ▸). Briefly, 0.5–150 mM l-arginine pH 8.0 was added to a solution consisting of 50 mM 4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid (EPPS) pH 8.0, 100 µM MnCl2 and the reaction was initiated by adding 1 µM LmARG in a total volume of 170 µl at 21°C. The reaction was terminated after 20 min using 30 µl of a 3:1(v:v) concentrated acid/dye solution [1:3:1(v:v:v) H2SO4:H3PO4:H2O and 245 mM α-isonitrosopropiophenone in ethanol]. Samples were heated to 90°C for 1 h in a thermocycler to ensure complete reaction of urea with the dye. To quantify urea formation, the absorbance of each sample was measured at λ = 550 nm using a Tecan Infinite M1000 Pro Microplate Reader. Kinetic parameters were determined using GraphPad Prism. For IC50 determinations, 30 mM l-arginine was used and the K i values for ABHPE and ABHDP were calculated based on the Cheng–Prusoff equation [K i = IC50/(1 + [S]/K m)] (Cheng & Prusoff, 1973 ▸).
3. Results and discussion
In comparison with the parent inhibitor ABH, the α,α-disubstituted boronic amino acids ABHPE and ABHDP exhibit modestly improved affinity for S. mansoni arginase (5.0-fold and 2.4-fold, respectively; Hai et al., 2014 ▸), modestly improved inhibitory potency (IC50) against human arginase I (6.5-fold and 7.4-fold, respectively; Van Zandt et al., 2013 ▸) and modestly improved inhibitory potency against human arginase II (3.8-fold and 7.4-fold, respectively; Golebiowski et al., 2013 ▸). However, neither ABHPE nor ABHDP exhibit improved inhibitory potency against LmARG (Table 2 ▸, Fig. 2 ▸). Even so, since the ABHDP sample is a racemic mixture, the effective IC50 of the l-boronic acid side-chain enantiomer should be twofold lower than that measured for the racemic mixture, since only the l-boronic acid side-chain enantiomer binds to the enzyme (see below).
Table 2. Inhibitory activity of ABH derivatives.
| Inhibitor | IC50 (µM) | K i (µM) |
|---|---|---|
| ABH, S-isomer (L) | 1.3 ± 0.2 | 0.9 ± 0.1 |
| ABHPE, S-isomer (L) | 2.1 ± 0.3 | 1.4 ± 0.2 |
| ABHDP, racemic (DL) | 1.7 ± 0.4 | 1.1 ± 0.3 |
Figure 2.

(a) Steady-state kinetics of LmARG yield k cat = 4.1 ± 0.2 s−1 and K m = 61 ± 6 mM. (b) Inhibitory activity assays of LmARG; IC50 values are recorded in Table 2 ▸.
The overall structures of LmARG complexed with ABHPE and ABHDP (Fig. 3 ▸) are essentially identical to the structure of the LmARG–ABH complex (PDB entry 4iu0; D’Antonio et al., 2013 ▸), with r.m.s. deviations of 0.26 Å (ABHPE) and 0.17 Å (ABHDP) for 289 and 286 Cα atoms, respectively. Although racemic ABHDP was used in the crystallization experiment, the electron-density map unambiguously shows that the l stereoisomer binds exclusively in the active site, as also observed for binding to S. mansoni arginase (Hai et al., 2014 ▸). The boronic acid moiety of each inhibitor undergoes nucleophilic attack by the metal-bridging hydroxide ion to form a tetrahedral boronate anion, as observed for the parent compound ABH (Fig. 1 ▸ b; D’Antonio et al., 2013 ▸), a well studied competitive inhibitor (Riley et al., 2011 ▸). This tetrahedral species mimics the tetrahedral intermediate and its flanking transition states in the arginase reaction (Fig. 1 ▸ a). Metal-coordination and hydrogen-bond interactions of the tetrahedral boronate anions in the LmARG complexes with ABH, ABHPE and ABHDP are compared in Table 3 ▸.
Figure 3.

(a) Simulated-annealing OMIT map of ABHPE bound to LmARG contoured at 2.5σ. Mn2+ ions are shown as purple spheres. Ligand atoms are color-coded as follows: C, orange; N, blue; O, red; Cl, dark green; B, green. Metal-coordination and hydrogen-bond interactions are shown as red and black dashed lines, respectively. (b) Simulated-annealing OMIT map of ABHDP bound to LmARG contoured at 2.5σ and color-coded as in (a).
Table 3. Intermolecular interactions of the boronate anion.
| Distance (Å) | ||||
|---|---|---|---|---|
| Interaction | ABH | ABHPE | ABHDP | |
| O1⋯MnA 2+ |
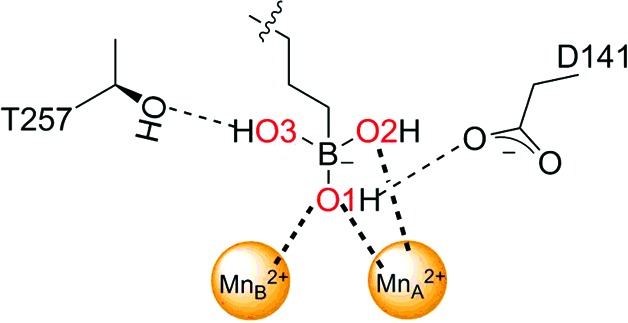
|
2.2 | 2.2 | 1.9 |
| O1⋯MnB 2+ | 2.2 | 2.0 | 2.2 | |
| O1⋯Asp141 | 2.6 | 2.7 | 2.7 | |
| O2⋯MnA 2+ | 2.3 | 2.2 | 2.2 | |
| O3⋯Thr257 | 2.9 | 2.7 | 2.6 | |
The α-carboxylate and α-amino groups are recognized by the conserved l-amino-acid recognition motifs, which saturate the hydrogen-bonding potential and ensure strict molecular recognition of the proper amino-acid stereoisomer. The α-carboxylate group of ABHPE accepts hydrogen bonds from Asn143, Ser150 and two water molecules, and the α-amino group donates hydrogen bonds to Asp194 and two water molecules. It is interesting that the stereoselectivity for l-amino-acid binding is maintained by four water-mediated and three direct hydrogen bonds to the protein. In the LmARG–ABHDP complex, however, the d side chain blocks a water molecule mediating the hydrogen bond between Asp194 and the α-amino group (the direct hydrogen bond between Asp194 and the α-amino group is maintained). Nevertheless, a superposition of the LmARG complexes with ABH, ABHPE and ABHDP shows that the binding modes of the parent ABH backbones are essentially identical (Fig. 4 ▸).
Figure 4.

Superposition of the LmARG–ABH complex (blue C atoms), the LmARG–ABHPE complex (magenta C atoms) and the LmARG–ABHDP complex (orange C atoms) reveals similar overall binding modes of the ABH-based scaffold of each inhibitor but different binding conformations for the d side chains of ABHPE and ABHDP.
Despite similar binding modes for the l-amino-acid portion of each inhibitor, the additional d side chains adopt different conformations (Fig. 4 ▸). In the LmARG–ABHPE complex the piperidine ring is unambiguously assigned as a chair conformation based on high-resolution electron density (Fig. 3 ▸ a). Remarkably, the d side chain of ABHPE makes no direct interaction with LmARG; the protonated tertiary amino group of the piperidine ring is oriented away from Asp194 and forms a hydrogen bond to a water molecule. The binding mode of ABHPE with LmARG contrasts with that observed in S. mansoni arginase (SmARG), where the piperidine amino group donates a hydrogen bond to Asp213 (which corresponds to Asp194 in LmARG; Hai et al., 2014 ▸). The binding mode of ABHPE in SmARG is unique, since only this particular enzyme has a groove that accommodates the piperidine ring of ABHPE (Fig. 5 ▸). Notably, the piperidine ring of ABHPE does not interact directly with the active-site residues in human arginases I and II, although a water-mediated interaction is observed in the human arginase I–ABHPE complex (Van Zandt et al., 2013 ▸).
Figure 5.

Comparison of α,α-disubstituted boronic acid inhibitors bound to arginases from different species. (a) LmARG–ABHPE complex. (b) Human arginase I–ABHPE complex (PDB entry 4hww; Van Zandt et al., 2013 ▸). (c) SmARG–ABHPE complex (PDB entry 4q3s; Hai et al., 2014 ▸). (d) LmARG–ABHDP complex. (e) Human arginase II–ABHCP complex (PDB entry 4ixv; Golebiowski et al., 2013 ▸). (f) SmARG–ABHDP complex (PDB entry 4q3r; Hai et al., 2014 ▸). The conserved aspartate residues (Asp194 in LmARG) potentially interacting with the piperidine group of the d side chain are highlighted as sticks. Ligand atoms are color-coded as follows: C, bright orange; N, blue; O, red; Cl, dark green; B, green. Hydrogen bonds are shown as black dashed lines.
In the LmARG–ABHDP complex, the d side chain of LmARG–ABHDP is characterized by weak and broken electron density, suggesting that it is largely disordered. Regardless, the piperidine ring is best modeled in a chair conformation with the protonated tertiary amino group donating a hydrogen bond to Asp194 (Fig. 3 ▸ b). Interestingly, the dichlorobenzyl group packs against the Ala192 region. This conformation is unique to LmARG, since Ala192 is replaced by a conserved aspartate in human arginase I, human arginase II and SmARG. The bulky, charged aspartate at this position in SmARG causes the d side chain of ABHDP to adopt an alternative conformation (Fig. 5 ▸ f).
It is somewhat surprising that the additional d side-chain substituents of ABHPE and ABHDP do not confer some degree of affinity enhancement relative to ABH. Even though the additional side chains do not make any polar interactions with protein residues, these side chains bind in the so-called ‘d-cleft’ (Ilies et al., 2011 ▸), making van der Waals contacts and displacing the associated water molecules to the bulk solvent. Presumably, the entropic gain provided by the release of water to bulk solvent upon enzyme–inhibitor complexation is offset by the conformational entropic cost of side-chain binding in the d-cleft. Even so, these structures will inform the future design of alternative d substituents for an α,α-disubstituted ABH inhibitor. For example, a bulkier side-chain substituent might thwart the molecular disorder observed for the d side chain of ABHDP. The three-dimensional contour and chemical nature of the d-cleft region can vary in arginases from different species and it is conceivable that interactions of inhibitor substituents in this region could contribute to isozyme or species selectivity.
4. Conclusions
In conclusion, the crystal structures of LmARG in complex with α,α-disubstituted boronic amino-acid inhibitors reported here provide important clues regarding the design of next-generation inhibitors based on the ABH scaffold. While the inhibitory potency of ABH against LmARG is not enhanced by the additional d side-chain substituents present in ABHPE and ABHDP, the inhibitory potency is not compromised either. Thus, ABHPE and ABHDP comprise useful starting points for the design of modified d side-chain substituents that may confer improvements in inhibitory potency and/or species selectivity for arginase inhibition. Future studies in this regard will be reported in due course.
Supplementary Material
PDB reference: L. mexicana arginase, complex with ABHPE, 5hj9
PDB reference: complex with ABHDP, 5hja
Acknowledgments
We thank Dr Michael C. Van Zandt of New England Discovery Partners for the generous gift of samples of ABHPE and ABHDP, and we are grateful to Drs Edward D’Antonio and Buddy Ullman for helpful scientific discussions. We thank the National Synchrotron Light Source at Brookhaven National Laboratory (beamline X29) for access to X-ray crystallographic data-collection facilities. Finally, we thank the NIH for grant GM49758 in support of this research.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Alvar, J., Vélez, I. D., Bern, C., Herrero, M., Desjeux, P., Cano, J., Jannin, J. & den Boer, M. (2012). PLoS One, 7, e35671. [DOI] [PMC free article] [PubMed]
- Antinori, S., Gianelli, E., Calattini, S., Longhi, E., Gramiccia, M. & Corbellino, M. (2005). Clin. Microbiol. Infect. 11, 343–346. [DOI] [PubMed]
- Archibald, R. M. (1945). J. Biol. Chem. 157, 507–518.
- Ash, D. E., Cox, J. D. & Christianson, D. W. (2000). Met. Ions Biol. Syst. 37, 407–428. [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cheng, Y. & Prusoff, W. H. (1973). Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed]
- Christianson, D. W. (2005). Acc. Chem. Res. 38, 191–201. [DOI] [PubMed]
- Crompton, D. W. T. (2013). Editor. Sustaining the Drive to Overcome the Global Impact of Neglected Tropical Diseases: Second WHO Report on Neglected Diseases. Geneva: World Health Organization.
- D’Antonio, E. L., Ullman, B., Roberts, S. C., Dixit, U. G., Wilson, M. E., Hai, Y. & Christianson, D. W. (2013). Arch. Biochem. Biophys. 535, 163–176. [DOI] [PMC free article] [PubMed]
- Desjeux, P. (1996). Clin. Dermatol. 14, 417–423. [DOI] [PubMed]
- Desjeux, P. (2001). Trans. R. Soc. Trop. Med. Hyg. 95, 239–243. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Golebiowski, A. et al. (2013). Bioorg. Med. Chem. Lett. 23, 4837–4841. [DOI] [PubMed]
- Hai, Y., Edwards, J. E., Van Zandt, M. C., Hoffmann, K. F. & Christianson, D. W. (2014). Biochemistry, 53, 4671–4684. [DOI] [PMC free article] [PubMed]
- Heby, O., Persson, L. & Rentala, M. (2007). Amino Acids, 33, 359–366. [DOI] [PubMed]
- Heby, O., Roberts, S. C. & Ullman, B. (2003). Biochem. Soc. Trans. 31, 415–419. [DOI] [PubMed]
- Ilies, M., Di Costanzo, L., Dowling, D. P., Thorn, K. J. & Christianson, D. W. (2011). J. Med. Chem. 54, 5432–5443. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pérez-Victoria, J. M., Di Pietro, A., Barron, D., Ravelo, A. G., Castanys, S. & Gamarro, F. (2002). Curr. Drug Targets, 3, 311–333. [DOI] [PubMed]
- Reguera, R. M., Balaña-Fouce, R., Showalter, M., Hickerson, S. & Beverley, S. M. (2009). Mol. Biochem. Parasitol. 165, 48–56. [DOI] [PMC free article] [PubMed]
- Riley, E., Roberts, S. C. & Ullman, B. (2011). Int. J. Parasitol. 41, 545–552. [DOI] [PMC free article] [PubMed]
- Roberts, S. C., Tancer, M. J., Polinsky, M. R., Gibson, K. M., Heby, O. & Ullman, B. (2004). J. Biol. Chem. 279, 23668–23678. [DOI] [PubMed]
- Silva, E. R. da, Maquiaveli, C. C. & Magalhães, P. P. (2012). Exp. Parasitol. 130, 183–188. [DOI] [PubMed]
- Silva, M. F. da, Zampieri, R. A., Muxel, S. M., Beverley, S. M. & Floeter-Winter, L. M. (2012). PLoS One, 7, e34022. [DOI] [PMC free article] [PubMed]
- Sundar, S. (2001). Trop. Med. Int. Health, 6, 849–854. [DOI] [PubMed]
- Van Zandt, M. C. et al. (2013). J. Med. Chem. 56, 2568–2580.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: L. mexicana arginase, complex with ABHPE, 5hj9
PDB reference: complex with ABHDP, 5hja