

The field of abnormal child psychology is facing extraordinary opportunity and challenge in relation to developments emerging in companion fields and in society. In this paper I consider this broader context as enabling an evolving picture of how the etiology of mental disorders might come to be understood in relation to gene × environment interaction and interplay, epigenetic effects, and developmental origins of health and disease. To set the stage for this overview, I consider the fundamental framework under which most nosological, descriptive, and etiological research has been conducted and then consider how this may be changing—and what a promising direction forward might look like.
Changing Context of Child Development and Its Study: Progress and Challenge
Children’s Developmental Context
The contexts in which children are developing are changing rapidly enough that aspects of our literature may be already outdated within a decade of appearing. At the same time, the context for our science is also changing rapidly, commending to us new approaches to solving our perennial questions. First, consider the changing context of child development, which circumscribes any broad framing approach for our field.
Children’s health
In the developed world, obesity and diabetes are exploding among adults and among youth. In the United States, over 30% of adults and 17% of youth age 2–19 are obese (Ogden, Carroll, Kit, & Flegal, 2014) with many more overweight. While the dietary changes that have triggered this epidemic are fairly well understood, the effects on children’s mental health, and the mental health drivers of which children are most vulnerable to these poor health outcomes, are not so clear. However, obesity/overweight and mental health are correlated—ADHD and depression both seem to be related to obesity risk (Cortese et al., 2015; Nigg, Johnstone, Musser, Willoughby, & Shannon, in press). The mind-body implications of these associations are apparent and the potential research avenues are also novel.
Meantime, in the Middle East and Africa, children face extraordinary stressors, including massive dislocation due to war, terrorism, and climate change. Over 40% of the world’s 43 million refugees in 2015 were children (United Nations, 2015). Children continue to be traumatized by being forced into servitude as child soldiers (Betancourt, Newnham, McBain, & Brennan, 2013) and to be victimized by the international human trafficking trade. Their mental health outcomes are widely neglected and yet likely to have substantial consequences for all societies in the coming generation. The results of research conducted in the context of more stable societies are difficult to interpret in relation to these more extreme contexts.
Technology
In the United States, (as reviewed by Livingstone and Smith, 2014) recent data suggest that over 80% of children are online, that the average child sends 30 text messages per day, and that gaming is rapidly supplanting television as the media of choice, with 1–2 hrs. of gaming per day common (Livingstone & Smith, 2014). As those reviewers note, risks of these new developments are still poorly understood. While these statistics pertain primarily to adolescents, younger children are also engaging in these technologies due to the ubiquity of hand-held devices. The effects of immersion in cell phones, internet, and video games on social, emotional, and cognitive development remain poorly known, partly because research has had difficulty keeping up with these rapidly changing contexts. At the same time, the ubiquity of “sensors” (as cell phones are called by some researchers) opens up new opportunities for ambitious data collection efforts that are rapidly coming into play and creating exciting opportunities for rapid, large scale data collection undreamed of just a few years ago.
Neurotoxic environments
While neurotoxins in children’s environment have been a concern for decades, their ubiquity has exploded again in just the past 15 years (Lanphear, 2015). Sufficient study of these risks, particularly in poor countries, remains as needed as it is challenging (Landrigan, 2015). While dozens of chemicals are know to affect neurodevelopment, thousands more are unstudied in regard to their effects on children. Emerging evidence using MRI documents changes in brain development in human children (Peterson et al., 2015) that amplifies prior behavioral and neuropsychological studies. Yet our lack of knowledge of combinations of chemicals, their interaction with stress and nutrition, and genetic moderation of their effects on development is glaring in relation to the topic’s potential importance.
Summary
The environments in which children are developing are changing rapidly and profoundly. What we knew about children’s development in the year 2000 or 2005 may be difficult to generalize to 2015 or 2020. Our researchers face the challenge to imagine both appropriately versatile conceptual models and nimble enough study designs to keep our knowledge base current.
Progress in Psychopathology Research
A second major context for the big picture reflections I am attempting here is that there has been dramatic progress in many aspects of psychopathology research.
Descriptive psychopathology
Here we have seen dramatic advances in the past 20 years, driven in part by better use of advanced statistical models. While progress is notable in integrating the nosology in relation to cross disorder structure via a hierarchical model, we are still far from personalized medicine or understanding the heterogeneity that bedevils the existing nosological categories. Newer longitudinal studies have clarified our knowledge of clinical course and opened the door to trajectory-based understandings of psychopathology. Yet, although we have begun to map major moderators of course, the determinants of variations in clinical course still remain obscure and clinical prediction remains crude. Brain imaging in particular has embarked on work of increased sophistication with consideration of brain topology, organization, and developmental dynamics (Matthews & Fair, 2015). But brain imaging research has lacked clinical impact and it is not clear if MRI measures will get there. The prospects for EEG brain imaging may be more promising in terms of clinical application. In the meantime, phenotype work, while progressing impressively, is far from done.
Treatment
Although it can be fairly argued that major breakthroughs are in short supply, developments have been notable in regard to focused and effective psychosocial treatments for PTSD, anxiety disorders, panic disorders, pain, medical compliance, and addiction. Further, the rapid advance of web based intervention is amplifying the cost-benefit possibilities and has still untapped potential to transform patient access and self-help as well as clinician effectiveness. Computerized training applications and other technical and biologically based interventions are heavily touted and studied; while not yet ready for prime time, their day may yet come and open up further avenues (see Faraone & Antshell (2014)). Presumably, continued refinement of mechanisms, etiology, and nosology will accelerate their relevance.
Psychiatric Genetics
Dramatic progress on genotyping means that single gene diseases are being rapidly explained almost in their entirety. But when it comes to complex disease, like most psychopathology, simple candidate gene studies are largely an historical relic, except when applied to targeted causal designs. The promise of next generation sequencing has yet to really arrive for psychiatry; but in the meantime, GWAS studies are evolving. We now know that most psychopathology is related to accumulation of many genes of small effect, and that some genetic influences are shared across broad swaths of the nosology. Yet many new frontiers remain, including work on rare variants in autism (Iossifov et al., 2014; Krumm et al., 2015; O’Roak et al., 2014), ADHD (Martin, O’Donovan, Thapar, Langley, & Williams, 2015), and child schizophrenia (Ambalavanan et al., 2015); explaining ‘dark heritability” (that is, explaining heritability that is not explained by SNPs), and the complementary application of polygenic scores to a wide range of problems. Now we are entering an age of very large samples; for example, the Psychiatric Genetics Consortium has set a goal of having 40,000 ADHD cases to compare to 90,000 controls in the next 5 years (Faraone, 2015). The same numbers are sought and being attained for other conditions. These sample sizes should enable more refined understanding of subtle genetic effects which in turn can shed light on pathophysiology. Already this work has opened links between psychiatric and physical disease and has broadened the neural targets for developmental disorders.
Environmental effects
From allostatic load (Beauchaine, Neuhaus, Zalewski, Crowell, & Potapova, 2011; Misiak, Frydecka, Zawadzki, Krefft, & Kiejna, 2014) to socialization processes, post-natal environmental mechanisms are now mapped and described with growing methodological and conceptual sophistication. This area is ready for a revival of focus as I note below. Yet, despite impressive progress, further integrative theory remains needed, and still too rare are genetically informed studies of specific environments. G × E effects are likely crucial and I emphasize those below as well.
Changing statistics
Changing statistical norms and methods are overtaking our field. Methodologists have finally run out of patience with inferential hypothesis testing (p<.05). High profile exposes have demonstrated how misleading that practice can be (Ioannidis, 2005). Now the field is under increasing pressure to apply better statistical tools (Collins & Tabak, 2014). These include the development and application of Bayesian models, growing insistence on preregistration of study design, full reporting of null results, replication, use of modern inferential methods (Wilcox, Carlson, Azen, & Clark, 2013), better methods of handling missing data, and greater attention to tools for causal inference in research design, such as sibling designs and Mendelian randomization (Lewis, Relton, Zammit, & Smith, 2013). The students of tomorrow face rather different expectations than the students of yesterday and their statistical applications should be more robust. More fundamentally, however, it remains unclear whether inferential statistics are appropriate to human behavior. One can think of dynamical systems modeling and chaos theory as analogies for the big data, probabilistic and localized predictions that the field likely needs.
Advances in Related Fields
A third critical context for our current work concerns the dramatic advances in neighboring fields that open new avenues for developmental psychopathology research.
Computer and information sciences
The rise of distributed connected computerized sensors, personalized data, web based data collection platforms using proprietary registrants, and big data generally, have opened up previously unimagined opportunities for monitoring and studying large populations in real time. While many of these opportunities have yet to be implemented in psychopathology research, researchers are already beginning to rapidly create study and measure-specific norms with thousands of surveys obtained in a matter of a few days or weeks online, markedly improving validity and generalizability. Related to this, information sciences have entered an era of big data throughput and integration. This work has transformed certain sectors of business and finance, where big data is at hand and the necessary resources are available. With the exception of some work in genetics and neuroscience, for the most part big data throughput has not yet reached child psychopathology. But ongoing, aggressive investments by universities, medical centers, and technology companies in the necessary informatics resources virtually ensure that this opportunity will soon emerge.
Physics
Advances in physics and quantum theory already have profoundly affected our field through the emergence of neuroimaging, gene-chip technology, and molecular sequencing capabilities. The imaging revolution continues as nano-tech, advanced micro-imaging, remote imaging, and field-deployable brain imaging develop further (Chang et al., 2015). Continued rapid advances non-invasive brain stimulation and imaging, may open up opportunities in coming years for novel treatments as well as early detection of risk.
Applied mathematics
Computer power has opened up the opportunity for widespread use of sophisticated tools from applied mathematics. Our field is now using pattern classification, kernel theory and machine learning, graph theory, quantum theory, and statistical simulations. For example, the use of pattern recognition classifiers (e.g., support vector machine, neural network classifiers, linear and logistic discriminant classifiers), to attempt to evaluate biomarkers in psychiatric disorders is now featured in hundreds of studies of brain imaging data alone (Wolfers, Buitelaar, Beckmann, Franke, & Marquand, 2015). While nearly all of those studies are too small to be other than suggestive and results are sometimes oversold without appreciation of the full range of methodological issues, it seems a matter of time until this work scales up to a potentially clinically relevant set of knowledge and potential application.
Epigenetics
The field of epigenetics has been extant for much of the past century. But only recently has it exploded on to the stage of human behavior and development. Now, epigenetic approaches have the potential to enable a substantial sharpening of etiological models and to open new doors to intervention. I develop this point in more detail below.
Systems biology
The explosion of so called ‘OMICS research (i.e., proteomics, metabalomics, along with methylation, RNA expression, and other systems biology approaches) in medicine will eventually wash over abnormal psychology research as well. Despite the inherent difficulty of linking biological markers to complex behavior patterns in humans, the ability, with computer power and new imaging technologies, to begin to search for biomarkers through the full range of molecules in the biome, means that a detailed biological map of how brain and behavior are related to biology is conceivable in a new way, even if its realization is some distance off. In particular, the data acquisition and analytic challenges of handling these opportunities remain to be fully solved, particularly in human research and application. But the opportunity and the context suggest that there will be increasing interest in novel biological applications going forward.
Summary
Progress in our field, and changes in related fields open dramatic new methodologies but also new conceptualizations to our field. In particular, growing recognition of the unitary medicine model (mind-brain-body) that is shaping medicine also increasingly is shaping psychopathology understanding. Conceptualizations of health and illness bring both powerful insights from genetics but also a tempering of the last few decades’ confidence in a simplistic genetic understandings of complex disease, as the potentially decisive importance of several environmental inputs is better specified.
Phenotype Model and Nosology
One of the implications for these developments in both psychology and related fields is a change in our paradigm for developmental psychopathology. (I deliberately use the word paradigm informally).Relevant here are models of etiology (such as G × E), of phenotype (traits versus categories), and of taxonomy (distinct versus related disorders).
Models of etiology
Arguably the dominant, if implicit, model in psychopathology for the past half century was a diathesis stress model (Meehl, 1962; Zubin & Spring, 1977). This model proposed that psychopathology emerged at the interface of a (to-be-specified) susceptibility and (to be specified) life events. The diathesis might be genetic but this is not mandatory. The diathesis-stress logic has been recently invigorated by challenge from a differential susceptibility model (Ellis, Boyce, Belsky, Bakermans-Kranenburg, & van Ijzendoorn, 2011), which postulates differential responsivity to experiences based on genotype and prenatal programming. A related model with considerable influence was the bioecological model (Bronfenbrenner & Ceci, 1994). This model similarly posited that psychopathology emerged from complex interplay of biological vulnerability and context. The more recent developmental psychopathology perspective (Cicchetti, 1984; Cicchetti & Richters, 1997; Sroufe, 2013) is also transactional while adding an emphasis on developmental trajectory, change, and multi-level integration.
At their best, each of these models took the form of sophisticated, well-specified transactional models, although the best has been too rarely in evidence. The obvious but often overlooked implication: we should be studying interactions. However, interactions are difficult to study and difficult to specify, and the specific candidate components for these models were many. To study interactions, considerable methodological care is needed—sampling design (over select extremes?), power (need more for interactions), scaling to enable replicable findings, and careful reflection on the form of the interaction (see (Belsky, Pluess, & Widaman, 2013)). Further, the effect sizes for interactions need scrutiny beyond the simple Cohenian categories—what constitutes an important effect size for a well specified, biologically informative statistical interaction? Perhaps for these reasons, despite some outstanding transactional studies in the past decade and a growing body of work on gene by environment interaction (G ×E; below) and differential susceptibility, much ongoing psychopathology research has defaulted to the study of main effects. Interactions have only in few instances reached solid replication. Few studies are designed to maximize the power to detect interactions, and interactions are too often studied as an afterthought with limited attention to replication.
Short history of nosology and current context
One of the perennial dilemmas in our field is the tension between dimensions and categories in our nosology. Numerous nosologies existed in the 1700’s and 1800’s, but two highlight this issue. First, in his textbook that set the stage for American psychiatry, Rush (1812) emphasized similarity more than difference. For example, he featured a broad mania category that probably would have included today’s mania, psychotic depression, schizophrenia, and perhaps extreme narcissistic personality disorder. In part this seemed to reflect a tendency to see break from reality as a unifying theme that linked diseases, all of which were conceptualized as having multiple environmental causes. While that work was founational, it was also to some extent overturned a century later.
At that time, Kraepelin (1899) and others (notably, Kahlbaum) in Germany concluded that simple symptom observations, like loss of reality testing, are misleading in isolation. Rather, Kraepelin proposed that symptoms be understood in relation to syndromal configuration, time course, and outcome. He and his colleagues differentiated early onset dementia (dementia praecox—later, schizophrenia) from late onset dementia (senile dementia and then, Alzheimer’s disease). Most famous is the Kraepelinian dichotomy: differentiating psychosis accompanied by between-episode negative symptoms (schizophrenia) from psychosis accompanied by between-episode normalization (bipolar illness). While this distinction was noted by Rush (1812), it was not seen then as an important clue to pathophysiology. Kraepelin escalated this type of observation into a decisive indication of a different disease. From there, the differentiations grew as the nosology expanded. Arguably differentiations have expanded far beyond their empirical base, with hundreds of conditions now listed in the DSM while empirical studies suggest only a dozen or so basic psychopathology syndromes.
In the 1970’s the “neo-Kraepelinian” movement sought to overcome what were perceived as excessively inferential clinical syndrome descriptions in DSM-II (which, while accepting many Kraepelinian categories, had poor inter-clinician reliability). The emphasis was on observable, quantifiable or at least countable symptoms. The polythetic symptom list was born and became standard in DSM-III onward. Although reliability duly improved, other well-documented problems with the DSM system increasingly weighed it down. A principal concern is the high co-occurrence of disorders—although important distinctions can be made between major psychopathologies, at the same time they clearly are not entirely discrete diseases in most instances, but rather related and overlapping syndromes that are both partially distinct and share important underlying features. Revised nosology models therefore feature attempts at integration in the form of a hierarchical taxonomy (Markon, Krueger, & Watson, 2005).
The urgency of a better conception of the nosology is underscored by recent studies. For example, similar brain regions and connectivity findings are implicated in many disorders (Goodkind et al., 2015). Genetic influences are likewise shared across disorders that have been studied, albeit not all to the same degree (Hamshere et al., 2013; Psychiatric Genetics Consoritum, 2013). Overall, while there are some important nuances (ADHD is not closely related genetically to bipolar disorder, for example), the picture nonetheless seems to seal the fate of a conception of discrete disorders—the pure or naïve version of a Kraeplinian nosology appears to have failed (Brown, 2015).
While this predicament was well-known to DSM-5 planners, a clear alternative structure was only vaguely articulated in DSM-5. Partly in response, the RDoC initiative (Insel et al., 2010) attempted to increase the emphasis on critical neurobiological dimensions that cut across syndromes. While dimensional research has always featured in psychopathology studies, particularly those in psychology rather than psychiatry, a paradigmatic focus on dimensions of neurobiology (rather than dimensions of symptoms) would be a significant change. The RDoC proposal has led to controversy and efforts at clarification (for one thoughtful perspective, see (Kraemer, 2015)). Now we must ask, how do we do this without returning to the ambiguity of 1812? How do we capitalize on the Kraepelinian insight, and on the progress in diagnostic reliability achieved by the DSM approach, and yet go beyond it to finally make better linkages to neurobiology? I suggest below that a nuanced approach can do so, considering shared underlying spectra in a hierarchical sense while still making critical distinctions that preserve what has already been learned about syndromal surface structure.
Alternative spectra and taxa for neurodevelopmental psychopathology
If child disorders are related in some way, how do we create appropriate spectra, hierarchies, or taxa for child and/or neurodevelopmental disorders? This can be done at the level of phenotypic description and correlation, genetic correlational structure, and/or development. Further, consideration should be given to discrete risks, that is, when we need to shift from dimensional (trait-based) thinking to recognizing discrete injury or insult to the system. When we do that, we in turn must consider adaptive response to insult. Further, this requires that we consider the exposome; a matrix of exposures that may influence development.
In DSM-5, the grouping of neurodevelopmental disorders includes intellectual disability (ID), speech and language disorders, specific learning disorders (LD), motor delays, autism spectrum disorder (ASD), and ADHD. Schizophrenia and psychotic disorders (such as catatonia, but not mania or psychotic depression) form another distinct grouping. Mania and bipolar disorders form yet another distinct grouping (excluding unipolar depression). And, disruptive and impulse control disorders form a separate grouping, and include oppositional defiant and conduct disorder (but not ADHD) along with pyromania and kleptomania. Yet the latter separation from neurodevelopmental disorders may be only an interim picture, if future data can show that early life and subsequent behaviors such as lack of empathy, extreme irritability, and subsequent disruptive behavior reflect early neurodevelopmental changes.
Even so, despite some problems with these groupings, one interesting and provocative aspect of the neurodevelopmental disorder grouping in DSM-5 is moving ADHD away from the DSM-IV “disruptive and impulse control disorders.” The justification is noted momentarily. (Full disclosure, the author was on the DSM-5 workgroup that proposed moving ADHD to the neurodevelopmental category). More provocative would have been to include schizophrenia, which is often thought of as a neurodevelopmental disorder (Debnath, Venkatasubramanian, & Berk, 2015; Howes & Murray, 2014; Pino et al., 2014). Overall, our neurodevelopmental framework still needs further elucidation, rationale, and work. But within this framework, Table 1 lists three alternative, competing spectrum models that might be defensible.
Table 1.
Three competing proposals for a neurodevelopmental or ADHD-related spectrum
| One: Dysregulated grouping (disorders of self control): |
| ADHD, Conduct disorder, oppositional defiant disorder, substance use disorders, antisocial personality disorder, (?) borderline personality disorder. |
| Two: Neurodevelopmental grouping (disorders of early neural development or subtle early neural injury)(as in DSM-5) |
| ADHD, autism, ID, LD, language and communication disorders, motor delays. |
| Three: Neurodevelopmental grouping expanded (disorders of aberrant neurodevelopment): |
| ADHD, ASD, ID, LD, language, motor, and schizophrenia. |
The first grouping model in Table 1 conforms closely to DSM-IV and was one approach advocated for DSM-5. Support for this picture comes from phenotypic and genetic correlations across these conditions (Lahey, Van Hulle, Singh, Waldman, & Rathouz, 2011), (Figure 1). Furthermore, factor analytic studies suggest a broad “externalizing” factor that links these conditions (including substance abuse), and ADHD+conduct disorder provide a powerful route to addiction disorders (S. S. Lee, Humphreys, Flory, Liu, & Glass, 2011). Antisocial personality disorder is included to maintain continuity with conduct disorder. If one eliminated the personality disorder category, however, one can also imagine borderline personality disorder belonging on this list as an adult manifestation. However, arguments against this grouping are equally compelling. As already noted, ADHD seems to have a distinct etiological structure compared to the disruptive behavior disorders. Further, early and stable brain imaging findings as seen in ADHD are not yet reported (though could emerge) for the disruptive behavior disorders in the absence of ADHD. And, cutting the developmental argument the other way, ADHD is may be seen as a neurodevelopmental liability for the other conditions.
Figure 1.
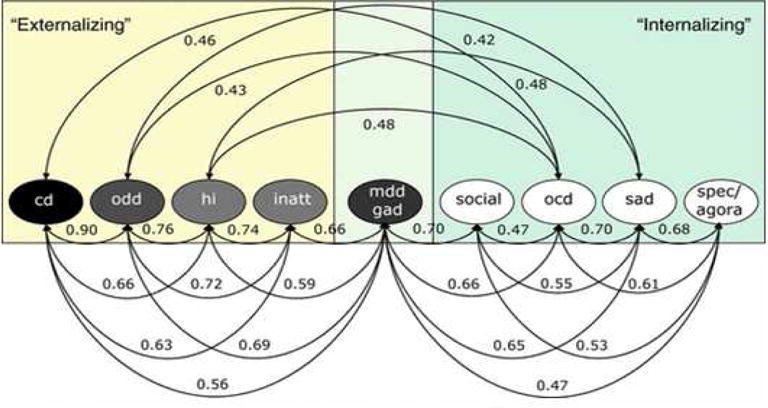
Correlations among latent factors in the best-fitting phenotypic model of caretaker-reported dimensions of common dimensions of child and adolescent psychopathology. Reprinted by permission from Lahey, Rathouz, et al. (2008), figure 5, p. 196
The arguments for the second grouping proposed in Table 1 are largely the converse, as it moves ADHD into a pre-existing neurodevelompental grouping as was ultimately done in DSM-5. The arguments in favor of this include an apparent distinct etiological structure differentiating ADHD and disruptive behavior disorders (Burt, 2009); high comorbidity and shared neural findings in ADHD and ASD (Dougherty, Evans, Myers, Moore, & Michael, 2015; Rommelse, Franke, Geurts, Hartman, & Buitelaar, 2010); shared genetics between ADHD and LD; elevated ASD births from parents with ADHD (Musser (2014);and elevated ADHD in siblings of children with ASD (M. Miller et al., 2015), as well as the well-established overlap of ADHD and ASD with language and learning related problems. However, objections to this grouping are as noted in the arguments in favor of the assignment of ADHD to the dysregulated grouping—there are distinct cognitive findings in ADHD and ASD, as well as distinct brain imaging findings in some instances (Dougherty et al., 2015; Ray et al., 2014). Finally, the surface similarity of ADHD and ASD or ID is arguably quite distinct when set next to the surface similarities of ADHD and other disorders of self regulation as in the dysregulated grouping.
The third grouping model, more provocative, was never seriously considered in the DSM-5 discussions. However, it merits study. It would add schizophrenia to the neurodevelopmental spectrum. Arguments in favor of this include new evidence of shared genetic influences of schizophrenia with ADHD (Hamshere et al., 2013) and ASD (S. H. Lee et al., 2013), elevated rates of schizophrenia in children with ADHD when they are grown up (Shyu et al., 2015), elevated rates of ASD and ADHD in offspring of individuals with schizophrenia (Sanchez-Gistau et al., 2015) and of ADHD in the history of patients with schizophrenia (Dalteg, Zandelin, Tuninger, & Levander, 2014; Rho et al., 2015), and neurodevelopmental changes in offspring of patients with schizophrenia seen on MRI (Francis et al., 2013). Objections include principally the striking dissimilarity of psychotic symptoms to the cognitive and social symptoms of the other neurodevelopmental conditions, the dramatically later typical onset for schizophrenia, and relatively low absolute base rates of elevated rates of schizophrenia in grown up children with ASD or ADHD.
From Table 1 and its discussion, it should be apparent that we can defend rather different spectra concepts. These possibilities open up exciting possibilities for integrating emerging biological findings. Yet, without due care, such approaches could result in one large psychopathological spectrum that is insufficiently differentiated. Thus, to really formalize such spectrum ideas better, we still need accumulating evidence that there really is a shared liability (as seems to be the case for much psychopathology), that the liability is specified (not the case currently), and to be attentive to differential treatment implications that may argue against linking disorders too closely. For example, one of the most important reasons to differentiate ADHD from schizophrenia is that they have rather different treatment implications. Yet the same is also true for ASD and ADHD. In short, to say that they are potentially part of a spectrum is simply a way to form hypotheses about potential etiologies that may cut across disorder (just as early ischemia or key genetic and neural systems may).
Heterogeneity
To this point I have emphasized the problem that disorders in DSM are not really distinct diseases in many instances. From that problem between categories, I now turn to the second and converse problem: the extensive heterogeneity within categories. Balancing our interest in seeing connections among syndromes, we also need the ability to describe accurately and productively the differentiation within a single syndrome. Our lab approaches this in two ways.
In the first approach we can consider an RDoC-like dimension, here related to cognitive control. We utilize differentiated cognitive measures of component abilities like working memory, response inhibition, vigilance/signal detection, temporal information processing, and response variability. Some readers will recognize these as the “usual suspects” when it comes to cognitive “deficit” in ADHD as well as in some other disorders. We can apply clustering algorithms to explore potential ways of organizing the cognitive heterogeneity in the ADHD population. In Fair et al. (2012), we did this and found meaningful subgroups that are theoretically coherent in relation to neurobiology. Figure 2 depicts this. In the figure, high scores are worse performance and the normal comparison group is scaled to zero. One group of children with ADHD has problems in top-down control (working memory, response inhibition). Another has problems with temporal information processing. This picture allows us to hypothesize subgroups that have reached the ADHD profile for different reasons neurobiologically. Some have problems, we can hypothesize, in frontal-parietal networks, while others have problems principally in cerebellar networks. We are following up with MRI studies to test this idea.
Figure 2.
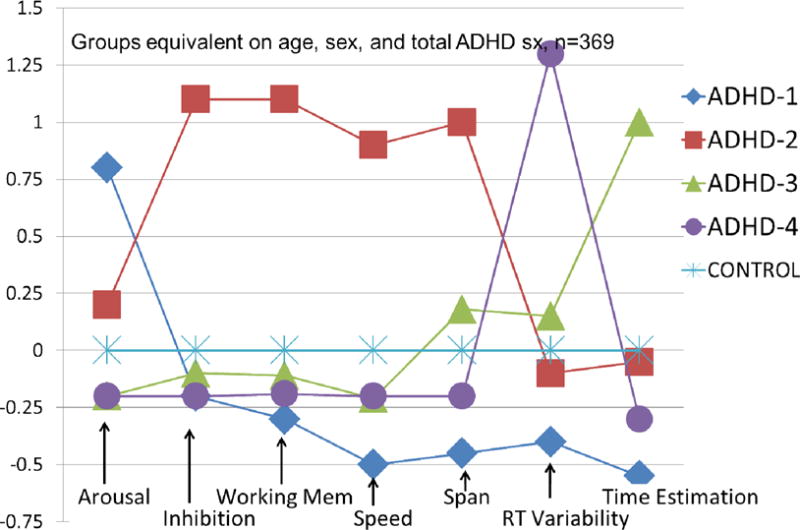
Profiles of four ADHD subgroups identified by mathematical community detection analysis. Profiles were observed in both ADHD and typically developing control children but are scaled here against the control group mean (set at zero) to convey the performance variation in the ADHD group. From Fair et al. (2012) discussed in the text
In Karalunas et al. (2014) we focused on affective dimensions due to their relatively well-characterized neurobiological correlates. Negative affect is closely related to amydgala circuitry. Positive affect is related to frontal-limbic circuitry and includes preferential involvement of the accumbens in reward anticipation (Plichta & Scheres, 2014). Using a simple measure of parent ratings of affective domains among children with ADHD, we again applied clustering and again found neurobiologically meaningful groups. The groups also appear meaningful clinically. They are shown in Figure 3. Again, the control group is scaled to zero. One group fits the literature’s description of “irritable” (Shaw, Stringaris, Nigg, & Leibenluft, 2014). These youth are angry, and have difficulty calming down. The second group we called “exuberant”—they have substantial positive affect. Strikingly, as shown in the circled points on the graph, these groups do not differ in overall impulsivity or in ADHD symptom severity. They get to the same ADHD endpoint by presumably different neurobiological routes. Karalunas et al (2014) also reported that amygdala connectivity differentiated the irritable group, as the clustering result would predict.
Figure 3.
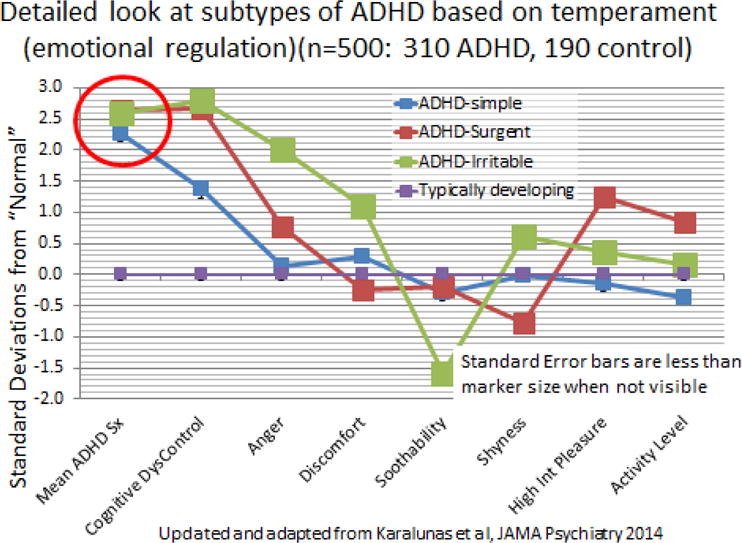
ADHD profiles based on parent-rated emotionality using a temperament scale. The typically developing group did not show distinct subgroups and is scaled to zero on this graph. Squares at the data points approximate the 95% confidence interval. Updated from Karalunas et al., 2014, discussed in the text.
In a second approach, we used brain imaging metrics to create homogenous clusters of children with and without ADHD (Costa Dias et al., 2015). We found that nucleus accumbens-prefrontal connectivity varied in an organized manner within the ADHD and the typically developing populations. When children were nested within their developmental brain profile, only one ADHD subgroup had the marked difficulties with steep reward delay discounting often attributed to ADHD and addiction.
This approach allows us to use the extensive work attempting to validate surface syndromes—not abandon it—but cross it with neurobiologically informative dimensions to add both clinical nuance and etiological signal. Of course, this approach can be extended to other disorders to create a map of disorders and subgroups as they parcel along these neurobiological dimensions and potentially lead to significant re-thinking of nosology. In short, a careful consideration and empirical study of nosology continues to be crucial to our field.
Etiology
Genetics and Epigenetics
Several important goals have opened up promise in the broad genetics domain. One major task is to solve the question of dark heritability (that is, heritability not explained by common gene variants). Psychiatric geneticists have made remarkable progress in a few years with statistical innovations. We now know that polygenic scores show us that common variants explain a substantially greater portion of genetic variance than previously thought—although, crucially, still less than 50% of it (Psychiatric Genetics Consoritum, 2013). Part of the missing heritability is undoubtably from rare variants, de novo mutations, and common variants that are not SnPs, as cited earlier in reviewing changes in the field of genetics. However, perhaps the most important source of missing heritability is G × E interplay, which was introduced earlier. While it is well-known that the heritability estimate in twin data is inflated by interactions of genotype with shared environment (Purcell, 2002), the extent of this effect is largely unknown for psychiatric conditions or behavioral traits.
G × E
The G×E approach caught on conceptually in the past decade and a half. Striking today is the maturation of work in this area via methodological advances, better designs, replication, and improved theory (Petrill, Bartlett, & Blair, 2013). The G × E approach is helpful in that it provides an operational empirical framework for the principle transactional models noted above (Agerbo, Sullivan, Vilhjálmsson, & et al., 2015; South, Hamdi, & Krueger, 2015). Does G ×E deserve to replace diathesis stress as a specific measurement strategy for understanding etiology? It remains daunting to integrate the vast scale of the genome and the vast scale of environments into coherent explanatory models, but the problem is becoming tractable, for example by combining polygenic scores with measured environments in G × E studies (Iyegbe, Campbell, Butler, Ajnakina, & Sham, 2014). Further, studies of specific genes and specific environments are beginning to fill in the details. Although single genes are only a small part of the full transactional story, G × E effects are now essentially established for single genes for both antisocial behavior (Byrd & Manuck, 2014) and depression (van Ijzendoorn, Belsky, & Bakermans-Kranenburg, 2012). These findings should grow stronger with increased use of polygenic scores and large samples. Perhaps the most important implication of G×E however, is that it implies that psychopathology is epigenetic.
Epigenetics
The term epigenetic is used in many ways and so needs a brief explanation. One usage entails conceptual recognition of the broad set of bidirectional processes by which genes and experience interact during development (Gottlieb, 2007). Here, I refer instead to a narrower and more specific usage preferred among contemporary scientists, which refers to specific chemical changes to the chromosome (e.g., DNA methylation, histone modification) that modify gene expression in a manner that endures after cell replication but without altering DNA sequence (Berger, Kouzarides, Shiekhattar, & Shilatifard, 2009). While epigenetic research has been ongoing for decades, its application to human behavior is quite recent.
Not all G×E is epigenetic in this narrower sense. Epigenetic change can be genetic, stochastic, or G × E. G × E can be non-functional or functional, and when it is functional, it may involve epigenetic change. But epigenetic effects are a crucial implication of G×E and vice versa. That is, if we see an epigenetic effect, we are one step closer to mapping the biological mechanism of how a G × E effect influences behavior, brain growth, or other phenotype.
As outlined earlier, our conceptual models must remain transactional. The simple linear model is dead (genes → brain → behavior). Now it is necessarily transactional - G × E → epigenetic change → brain ↔ behavior. The recursive component is able to be instantiated via epigenetic change, such that even as DNA sequence shapes gene expression, brain development and behavior, at the same time behavior and experience influence gene expression and epigenetic change, in turn also shaping neural development and behavior.
Not all epigenetic change is associated with gene expression change; thus epigenetic studies ultimately have to pair with genetic studies and studies of RNA expression. Study of epigenetic effects in human behavior is difficult in that, except in very rare instances, tissue cannot be assayed directly from the living brain. This is important because epigenetic markings are tissue and region specific. In the brain alone, DNA methylation and histone modification may vary widely from one local region to another or one system to another. Generalizing from peripheral tissue to the brain therefore is fraught with difficulty. However, some evidence indicates that despite the predominant tissue-specific variation, there is also sufficient individual-specific patterns of cross-tissue correlation of epigenetic markings that peripheral tissue DNA methylation studies in human psychiatry are generally considered to be informative (Ma et al., 2014). As a result, the potential of finding peripheral epigenetic markers that may inform animal studies, be validated in post-mortem studies, and ultimately inform novel epigenetic intervention to reverse CNS-based psychopathologies is tantalizing (Szyf, 2015).
Crucially, from a conceptual perspective of understanding what psychopathology is, what sort of thing it is, the epigenetic perspective is potentially very important. Why? First, the effects can be very large. Second, effects can carry across generations (they don’t all do so), confounding our understanding of inheritance and inter-generational transmission of psychopathology. Third, these effects are potentially reversible—helping to account for onset, course, and recovery as well as opening new treatment targets. Fourth and most valuable for our purposes, it can provide specific, testable hypotheses for mechanisms that are involved in the effects of G × E on psychopathology.
Developmental Origins
Although environmentally mediated epigenetic changes may occur throughout childhood development, it is widely believed that the biggest effects occur prenatally. Therefore the epigenetic perspective is only completed if we integrate it with a related line of thought, called the developmental origins of health and disease. This approach, emerging in current form through the seminal work of David Barker and colleagues (Barker, 1995), was introduced to child psychopathology in part through efforts of Swanson and colleagues (Wadhwa, Buss, Entringer, & Swanson, 2009). The basic principle here is simple, but the implications are rather striking. The principle is that in early development, the fetus engages in trade-offs to maximize or to attempt to maximize its fitness for its anticipated environment. The anticipation is based on hormonal and other signals from the placenta, transmitting maternal experience—its nutrient level, its stress level, its pollutant level. Alterations in blood flow to the developing fetus, regulated by the placenta, adjust various growth parameters accordingly. This in turn alters gene expression, at least in part via epigenetic mechanisms, changing expression of the immune system, antioxidant defenses, inflammatory responses, and the number and quality of stem cells (Barker & Thornburg, 2013).
Schematically, an adaptive trade off occurs. In the plant world, we might imagine a plant trading off slower rate of growth for better disease resistance or heat tolerance. In the animal world, we might imagine something similar. In humans and non-human animals, we can postulate for example that early life growth might be maximized at the cost of longevity to overcome early adversity. We might hypothesize that fat storage would be more efficient to compensate for anticipated food shortage. These types of effects in fact have been observed.
A classic study in this vein was the Dutch winter famine study. Following up children from the Dutch famine during World War II, investigators compared siblings who were exposed to the famine in utero to those who weren’t. They showed clear differences in methylation of the insulin-like growth factor gene (IGF2), indicating that exposed individuals were prepared to metabolize and handle food very differently than their siblings. This difference was apparent on follow up well into adulthood (Heijmans et al., 2008). The prenatal exposure presumably changed those siblings’ risk for obesity, diabetes, and related ailments as well. Thus, adaptation also changes disease risk—particularly if the actual environment does not match the one “expected” during fetal development. Famine exposure also increased the children’s risk for later psychiatric outcomes. The children with prenatal exposure to famine had elevated adult rates of schizophrenia and CNS abnormalities, such as motor problems (Susser et al., 1996). Numerous such tradeoffs likely occur, can be hypothesized, and are amenable to empirical study, much of it now underway in animal models.
Inflammation as a Candidate Common Mechanistic Pathway
The “Big Three” of prenatal influences on psychopathology risk are (1) maternal and perhaps paternal adiposity and diet (Drake et al., 2012); (2) maternal emotional stress (Sng & Meaney, 2009); and (3) maternal and perhaps paternal toxicant load (Bailey et al., 2013). Each of these has substantial literatures testifying to their prospective association to child neurobehavioral outcomes. These effects are likely mediated by epigenetic changes, as supported by evidence cited elsewhere in this paper. Note that while most work to date has focused on maternal routes of transmission, a growing literature recognizes an important paternal contribution. Paternal aging and paternal obesity, for example, have known associations with epigenetic changes that affect offspring development (McPherson et al., 2015). Further work on the relation of paternal stress and paternal diet and teratogen exposures will be important to complete the picture.
Other known teratogens also remain important, potentially including maternal smoking, alcohol, administration of glucocorticoids in pregnancy, and anti-depressants. Maternal health also remains important, particularly gestational diabetes. Thus, a finite taxonomy of early environments can be imagined that could be paired with key mediating and moderating genes to begin to build a biologically-coherent, etiologically based taxonomy of psychopathology from early in life. These inputs clearly could be moderated by subsequent psychosocial processes.
One important possibility is that all of these three classes of insults exert their effects on neurodevelopment via alterations of inflammatory, oxidative stress, immune response, and related pathways. This is plausible at least as one shared step in the cascade of biological events (Monk, Georgieff, & Osterholm, 2013; Spencer, 2013) and supported at a preliminary level by findings of excess pro-inflammatory markers in most child psychopathologies (Mitchell & Goldstein, 2014) and by initial genetic studies that implicate inflammation-related genes (Psychiatric Genomics Consortium, 2015). An attractive feature of this proposal is that it would also help to account for the shared physical health and mental health effects observed with several of the risk factors noted.
Psychoneuroimmunology is an active field, albeit focused principally on concurrent inflammation and episodes of mental disorder rather than on developmental origins. It underscores the powerful relations between peripheral inflammatory processes in the body and effects in the brain operating via circulating cytokines, microglia, and mediated via vagal nerve pathways. This work highlights the importance of stress response, vagal tone, as well as still-speculative theories about the role of microglia in disorders from ADHD to depression that have recently emerged in our field.
To illustrate, note that the role of inflammation in depression, as well as in schizophrenia, has become a “hot topic” (Chaves, Zuardi, & Hallak, 2015; Dickerson et al., 2015; Khandaker et al., 2015; Pariante, 2015; Yirmiya, Rimmerman, & Reshef, 2015). Annual publications have increased following an exponential curve in the past decade, with PubMed showing over 100 papers per year on schizophrenia and inflammation and over 400 per year on depression and inflammation (Pub Med, November, 2015). Inflammatory chemokines and cytokines appear to influence basic neurodevelopmental processes including neurogenesis and modulation of neurotransmitter synthesis and synaptic development (Borsini, Zunszain, Thuret, & Pariante, 2015; Coiro et al., 2015; Stuart, Singhal, & Baune, 2015). Anti-inflammatory therapies, such as omega-3 supplementation, have been shown to have causally effective benefits on ADHD (Hawkey & Nigg, 2014) and on depression (Su, Matsuoka, & Pae, 2015). Relations of inflammation to depression are fairly straightforward but may relate only to a subset of depressive cases that show sickness like behaviors (e.g., low energy, oversleeping) and a differential treatment response to anti-depressants (Mondelli et al., 2015).
Synthesis: A Testable Paradigm?
Thus, we might be able to arrive at an etiological paradigm that is operational and specific, linking genetic liability, specific prenatal challenges, resulting specific epigenetic change mediated in placenta and then in infancy, with associated inflammatory and other related mechanisms in mother and infant, and consequent alterations in infant brain development. While these brain development alterations would be expected to influence neural development in the entire brain (e.g., via efficiency of axonal growth, pruning, or cell signaling), we would also expect localized effects to be greater on those brain regions that are most resource dependent or longest developing—e.g., prefrontal cortex, certain subcortical structures—and thus most vulnerable to insult. This integrated perspective might help weave together the neuroimaging literature that shows both (a) overlapping findings in psychopathology and (b) both local and distributed brain alterations in many forms of psychopathology. Such a model would also recognize the dynamics of the theory of the developmental origins of disease and developmental programming.
Such a model makes specific, testable predictions. (Other models could be imagined that would also make specific testable predictions, of course). For example, Mendelian designs should demonstrate that diet, toxicants, or stress have causal effects on psychopathology, and should in turn be associated with epigenetic change in biological systems and pathways related to inflammation, oxidative stress, and immune function. Epigenetic change should be observable in placenta, cord blood, and infant blood spots. If effects occur post-natally, this could be discovered by comparing child blood DNA methylation with their cord blood or infant blood spot DNA methylation—samples suitable to such studies already exist and our lab is undertaking a study using this design currently to determine whether epigenetic changes in ADHD occur pre-natally or post-natally. These epigenetic effects should be reproducible and mediating in animal experiments that model the particular stressor.
Preliminary evidence to support this picture is beginning to emerge. For example, in the dietary sphere, Stevenson et al. (2010) found that the effects of food additives on ADHD were moderated by genotype of the histamine degradation gene (HNMT Thr105Ile and HNMT T939C). On this gene, when the T allele is present, the food additive challenge has no effect. When the T allele is absent, the food additives cause more hyperactivity than the placebo.
In the toxicant sphere, Engel et al. (2011) reported that the effect of maternal organophosphate body burden on infant Bayley scores was moderated by maternal genotype on PON1 (Paraoxynase 1 enzyme susceptibility gene; 7q21.3) which moderates the rate of liver metabolism of the pollutant. Nigg et al. (in press) reported that child genotype on the HFE gene (human hemochromatosis protein gene; 6p22.2) moderated the strength of association of children’s blood lead level with their teacher-rated hyperactivity symptoms. This was observed at blood lead levels that are typical in the U.S. population (about 1ug/dL), suggesting these effects may be widespread. Thus, there is evidence of causal associations mediated by biological metabolites in both instances.
Also supporting this picture, epigenetic changes are documented as mediators in animal studies. Pesticide exposure causes substantial epigenetic changes during development and these can be transmitted in the germ line (Skinner et al., 2013). Prenatal stress has well-studied epigenetic effects in animals (Babenko, Kovalchuk, & Metz, 2015; Bale, 2015). Lead similarly, causes hyperactivity in rats and the effect is mediated by epigenetic changes in the brain (Luo et al., 2014). Prenatal variation in omega 3’s intake, total fat intake, and micronutrients all convey epigenetic changes in offspring (Bolton & Bilbo, 2014; Sable, Randhir, Kale, Chavan-Gautam, & Joshi, 2015).
In such an approach to developmental psychopathology, the basic principles would remain transactional, with G × E as a basic axiom proposed to be involved in most psychopathology. DNA structure would be expected in most cases to convey liability or else susceptibility (plasticity). Liability means vulnerability to negative exposures; susceptibility means greater response to exposures for good or for ill. Which of those mechanisms is in play is expected to vary depending on particular syndromes under study. Within this framework, specific genetic constituents can begin to be specified either in relation to general plasticity (a currently in vogue interpretation of the effects of the serotonin transporter gene), or in relation to moderation of particular environmental inputs as noted earlier.
Then, specific mechanistic routes of effect can begin to be mapped in relation to inflammatory systems, epigenetic changes, and routes into brain development, for example via changes in microglia and in neurotransmitter synthesis (A. H. Miller & Raison, 2015; Wang, Yang, Gelernter, & Zhao, 2015). Promising strategies will continue to refine the phenotype (neuroimaging, physiology, cognition, emotion), define the genotype, and define the environment. The critical addition is the measurement of direct mechanisms of transmission from environment to brain and behavior, via epigenetic changes, gene expression, and systemic changes in the inflammatory and glucocorticoid systems.
Designs that are needed to address this include continued genetically informed studies of early and later environment (with improved measurement of the exposome), studies that consider biological and psychosocial environments together (e.g., nutrition, toxicants, and/or psychosocial moderators), differentiated phenotypes (using either common psychopathology factors or differentiated subphenotypes within existing disorders as well as cross-disorder transdiagnostic phenotypes) integrated with differentiated etiologies, to map the true boundaries of etiological effects and their moderators in behavioral expression. Studies of discordant identical twins will be informative in regard to isolating epigenetic effects, but must be paired with case control designs if the quest for clinical biomarkers is to move forward.
A model of this nature has the potential to be able to be extended to extreme environments, to be married to either a categorical taxonomy (DSM) or a dimensional taxonomy (RDoC), and to integrate trauma, allostatic load, and genetic models of key etiological mechanisms. While extremely rich literatures now exist in each of these areas, they tend to remain siloed.
Conclusion
The take home message here is that the many exciting developments in our field and in neighboring fields have launched a new period in conceptualizing psychopathology. Continued refinement of our phenotype nosology, considering both existing psychiatric categories, and refinements in relation to cross-disorder commonalities and within-disorder heterogeneities remain important. Recognition of the changing and powerful environmental influences on children is crucial, and renewed focus on early and even prenatal development is indicated. A fundamentally transactional model of developmental psychopathology remains paramount. However, research going forward increasingly should focus on specific gene or gene pathways, specific environments, and ultimately their combination. Increasingly, attention will be paid to causally informative designs using sibling comparisons, Mendelian randomization, and other opportunities. Important will be the study of mediators, which requires careful animal-human translational work. Here, the targets include epigenetic changes, RNA expression, and physiological implications, with a likely important focus on inflammation as a potential common pathway. Overall, a greater emphasis on environmental contributors to developmental psychopathology is likely even as the genetic advances continue apace.
While the integration of the many factors noted here will take a generation, the potential remains high for breakthroughs in our understanding of developmental psychopathology. The challenge is to move from promise to reproducible proof (Kraemer, 2015). It is incumbent on our field to remain aware of the developments in neighboring fields, to maintain our unique expertise in descriptive psychopathology to avoid the over-simplifications that can bedevil biological studies, and to remain sophisticated and creative in blending the best of biological tools with the best of environmental measures. If we can sustain this perspective, the future for our field is bright.
Acknowledgments
A version of this paper was delivered as the Presidential Address at the Biennial meeting of the International Society for Research on Child and Adolescent Psychopathology, July, 2015. This work was supported by NIMH Grant MH R37-59105.
Footnotes
The author reports no conflicts of interest.
References
- Agerbo E, Sullivan PF, Vilhjálmsson BJ, et al. Polygenic risk score, parental socioeconomic status, family history of psychiatric disorders, and the risk for schizophrenia: A Danish population-based study and meta-analysis. Journal American Medical Association Psychiatry. 2015;72:635–641. doi: 10.1001/jamapsychiatry.2015.0346. [DOI] [PubMed] [Google Scholar]
- Ambalavanan A, Girard SL, Ahn K, Zhou S, Dionne-Laporte A, Spiegelman D, Rouleau GA. De novo variants in sporadic cases of childhood onset schizophrenia. European Journal of Human Genetics. 2015 doi: 10.1038/ejhg.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko O, Kovalchuk I, Metz GA. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neuroscience and Biobehavioral Reviews. 2015;48:70–91. doi: 10.1016/j.neubiorev.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Bailey KA, Wu MC, Ward WO, Smeester L, Rager JE, Garcia-Vargas G, Fry RC. Arsenic and the epigenome: Interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. Journal of Biochemical and Molecular Toxicology. 2013;27:106–115. doi: 10.1002/jbt.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL. Epigenetic and transgenerational reprogramming of brain development. Nature Reviews: Neuroscience. 2015;16:332–344. doi: 10.1038/nrn3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–174. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Thornburg KL. The obstetric origins of health for a lifetime. Clinical Obstetrics and Gynecology. 2013;56:511–519. doi: 10.1097/GRF.0b013e31829cb9ca. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Neuhaus E, Zalewski M, Crowell SE, Potapova N. The effects of allostatic load on neural systems subserving motivation, mood regulation, and social affiliation. Development and Psychopathology. 2011;23:975–999. doi: 10.1017/S0954579411000459. [DOI] [PubMed] [Google Scholar]
- Belsky J, Pluess M, Widaman KF. Confirmatory and competitive evaluation of alternative gene-environment interaction hypotheses. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2013;54:1135–1143. doi: 10.1111/jcpp.12075. [DOI] [PubMed] [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes and Development. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt TS, Newnham EA, McBain R, Brennan RT. Post-traumatic stress symptoms among former child soldiers in sierra leone: Follow-up study. British Journal of Psychiatry. 2013;203:196–202. doi: 10.1192/bjp.bp.112.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Bilbo SD. Developmental programming of brain and behavior by perinatal diet: Focus on inflammatory mechanisms. Dialogues in Clinical Neuroscience. 2014;16:307–320. doi: 10.31887/DCNS.2014.16.3/jbolton. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsini A, Zunszain PA, Thuret S, Pariante CM. The role of inflammatory cytokines as key modulators of neurogenesis. Trends in Neurosciences. 2015;38:145–157. doi: 10.1016/j.tins.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Bronfenbrenner U, Ceci SJ. Nature-nurture reconceptualized in developmental perspective: A bioecological model. Psychological Review. 1994;101:568–586. doi: 10.1037/0033-295x.101.4.568. [DOI] [PubMed] [Google Scholar]
- Brown AS. The kraepelinian dichotomy from the perspective of prenatal infectious and immunologic insults. Schizophrenia Bulletin. 2015;41:786–791. doi: 10.1093/schbul/sbv063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA. Rethinking environmental contributions to child and adolescent psychopathology: A meta-analysis of shared environmental influences. Psychological Bulletin. 2009;135:608–637. doi: 10.1037/a0015702. [DOI] [PubMed] [Google Scholar]
- Byrd AL, Manuck SB. Maoa, childhood maltreatment, and antisocial behavior: Meta-analysis of a gene-environment interaction. Biological Psychiatry. 2014;75:9–17. doi: 10.1016/j.biopsych.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Harford JB, Eaton MA, Boisseau PM, Dube A, Hayeshi R, Lee DS. Nanomedicine: Past, present and future - a global perspective. Biochemical and Biophysical Research Communications. 2015 doi: 10.1016/j.bbrc.2015.10.136. [DOI] [PubMed] [Google Scholar]
- Chaves C, Zuardi AW, Hallak JE. The role of inflammation in schizophrenia: An overview. Trends in Psychiatry Psychotherapy. 2015;37:104–105. doi: 10.1590/2237-6089-2015-0007. [DOI] [PubMed] [Google Scholar]
- Cicchetti D. The emergence of developmental psychopathology. Child Development. 1984;55:1–7. [PubMed] [Google Scholar]
- Cicchetti D, Richters JE. Examining the conceptual and scientific underpinnings of research in developmental psychopathology. Development and Psychopathology. 1997;9:189–191. [PubMed] [Google Scholar]
- Coiro P, Padmashri R, Suresh A, Spartz E, Pendyala G, Chou S, Dunaevsky A. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain, Behavior, and Immunity. 2015 doi: 10.1016/j.bbi.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Tabak LA. Policy: NIH plans to enhance reproducibility. Nature. 2014;505:612–613. doi: 10.1038/505612a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese S, Moreira-Maia CR, St Fleur D, Morcillo-Penalver C, Rohde LA, Faraone SV. Association between ADHD and obesity: A systematic review and meta-analysis. American Journal of Psychiatry. 2015 doi: 10.1176/appi.ajp.2015.15020266. [DOI] [PubMed] [Google Scholar]
- Costa Dias TG, Iyer SP, Carpenter SD, Cary RP, Wilson VB, Mitchell SH, Fair DA. Characterizing heterogeneity in children with and without adhd based on reward system connectivity. Developmental Cognitive Neuroscience. 2015;11:155–174. doi: 10.1016/j.dcn.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalteg A, Zandelin A, Tuninger E, Levander S. Psychosis in adulthood is associated with high rates of adhd and cd problems during childhood. Nord J Psychiatry. 2014;68:560–566. doi: 10.3109/08039488.2014.892151. [DOI] [PubMed] [Google Scholar]
- Debnath M, Venkatasubramanian G, Berk M. Fetal programming of schizophrenia: Select mechanisms. Neuroscience and Biobehavioral Reviews. 2015;49:90–104. doi: 10.1016/j.neubiorev.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson F, Stallings C, Origoni A, Schroeder J, Katsafanas E, Schweinfurth L, Yolken R. Inflammatory markers in recent onset psychosis and chronic schizophrenia. Schizophrenia Bulletin. 2015 doi: 10.1093/schbul/sbv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty CC, Evans DW, Myers SM, Moore GJ, Michael AM. A comparison of structural brain imaging findings in autism spectrum disorder and attention-deficit hyperactivity disorder. Neuropsychology Review. 2015 doi: 10.1007/s11065-015-9300-2. [DOI] [PubMed] [Google Scholar]
- Drake AJ, McPherson RC, Godfrey KM, Cooper C, Lillycrop KA, Hanson MA, Reynolds RM. An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and foetal growth. Clinical Endocrinology. 2012;77:808–815. doi: 10.1111/j.1365-2265.2012.04453.x. [DOI] [PubMed] [Google Scholar]
- Ellis BJ, Boyce WT, Belsky J, Bakermans-Kranenburg MJ, van Ijzendoorn MH. Differential susceptibility to the environment: An evolutionary–neurodevelopmental theory. Development and Psychopathology. 2011;23:7–28. doi: 10.1017/S0954579410000611. [DOI] [PubMed] [Google Scholar]
- Engel SM, Wetmur J, Chen J, Zhu C, Barr DB, Canfield RL, Wolff MS. Prenatal exposure to organophosphates, paraoxonase 1, and cognitive development in childhood. Environmental Health Perspectives. 2011;119:1182–1188. doi: 10.1289/ehp.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair DA, Bathula D, Nikolas MA, Nigg JT. Distinct neuropsychological subgroups in typically developing youth inform heterogeneity in children with adhd. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6769–6774. doi: 10.1073/pnas.1115365109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraone SV. An update on adhd genetics; Paper presented at the World Congress on ADHD; Glasgow, Scotland. 2015. [Google Scholar]
- Faraone SV, Antshel KM. Towards an evidence-based taxonomy of nonpharmacologic treatments for adhd. Child and Adolescent Psychiatric Clinics of North America. 2014;23:965–972. doi: 10.1016/j.chc.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Francis AN, Bhojraj TS, Prasad KM, Montrose D, Eack SM, Rajarethinam R, Keshavan MS. Alterations in the cerebral white matter of genetic high risk offspring of patients with schizophrenia spectrum disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2013;40:187–192. doi: 10.1016/j.pnpbp.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkind M, Eickhoff SB, Oathes DJ, Jiang Y, Chang A, Jones-Hagata LB, Etkin A. Identification of a common neurobiological substrate for mental illness. Journal American Medical Association Psychiatry. 2015;72:305–315. doi: 10.1001/jamapsychiatry.2014.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb G. Probabilistic epigenesis. Dev Sci. 2007;10:1–11. doi: 10.1111/j.1467-7687.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- Hamshere ML, Stergiakouli E, Langley K, Martin J, Holmans P, Kent L, Craddock N. Shared polygenic contribution between childhood attention-deficit hyperactivity disorder and adult schizophrenia. British Journal of Psychiatry. 2013;203:107–111. doi: 10.1192/bjp.bp.112.117432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey E, Nigg JT. Omega-3 fatty acid and adhd: Blood level analysis and meta-analytic extension of supplementation trials. Clinical Psychology Review. 2014;34:496–505. doi: 10.1016/j.cpr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Murray RM. Schizophrenia: An integrated sociodevelopmental-cognitive model. Lancet. 2014;383:1677–1687. doi: 10.1016/s0140-6736(13)62036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, Wang P. Research domain criteria (rdoc): Toward a new classification framework for research on mental disorders. American Journal of Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP. Why most published research findings are false. PLoS Medicine. 2005;2:e124. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Wigler M. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyegbe C, Campbell D, Butler A, Ajnakina O, Sham P. The emerging molecular architecture of schizophrenia, polygenic risk scores and the clinical implications for gxe research. Social Psychiatry and Psychiatric Epidemiology. 2014;49:169–182. doi: 10.1007/s00127-014-0823-2. [DOI] [PubMed] [Google Scholar]
- Karalunas SL, Fair D, Musser ED, Aykes K, Iyer SP, Nigg JT. Subtyping attention-deficit/hyperactivity disorder using temperament dimensions: Toward biologically based nosologic criteria. Journal American Medical Association Psychiatry. 2014;71:1015–1024. doi: 10.1001/jamapsychiatry.2014.763. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry. 2015;2:258–270. doi: 10.1016/s2215-0366(14)00122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer HC. Research domain criteria (rdoc) and the dsm-two methodological approaches to mental health diagnosis. Journal American Medical Association Psychiatry. 2015;72:1163–1164. doi: 10.1001/jamapsychiatry.2015.2134. [DOI] [PubMed] [Google Scholar]
- Kraepelin E. Textbook of psychiatry. (psychiatrie. Ein lehrbuch für studierende und arzte) 6. Leipzig: Earth Publishers; 1899. [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Eichler EE. Excess of rare, inherited truncating mutations in autism. Nature Genetics. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahey BB, Van Hulle CA, Singh AL, Waldman ID, Rathouz PJ. Higher-order genetic and environmental structure of prevalent forms of child and adolescent psychopathology. Archives of General Psychiatry. 2011;68:181–189. doi: 10.1001/archgenpsychiatry.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ. Children’s environmental health: A brief history. Academic Pediatrics. 2015 doi: 10.1016/j.acap.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Lanphear BP. The impact of toxins on the developing brain. Annual Review of Public Health. 2015;36:211–230. doi: 10.1146/annurev-publhealth-031912-114413. [DOI] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Wray NR. Genetic relationship between five psychiatric disorders estimated from genome-wide snps. Nature Genetics. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Humphreys KL, Flory K, Liu R, Glass K. Prospective association of childhood attention-deficit/hyperactivity disorder (adhd) and substance use and abuse/dependence: A meta-analytic review. Clinical Psychology Review. 2011;31:328–341. doi: 10.1016/j.cpr.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SJ, Relton C, Zammit S, Smith GD. Approaches for strengthening causal inference regarding prenatal risk factors for childhood behavioural and psychiatric disorders. Journal of Child Psychology and Psychiatry. 2013;54:1095–1108. doi: 10.1111/jcpp.12127. [DOI] [PubMed] [Google Scholar]
- Livingstone S, Smith PK. Annual research review: Harms experienced by child users of online and mobile technologies: The nature, prevalence and management of sexual and aggressive risks in the digital age. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2014;55:635–654. doi: 10.1111/jcpp.12197. [DOI] [PubMed] [Google Scholar]
- Luo M, Xu Y, Cai R, Tang Y, Ge MM, Liu ZH, Wang HL. Epigenetic histone modification regulates developmental lead exposure induced hyperactivity in rats. Toxicology Letters. 2014;225:78–85. doi: 10.1016/j.toxlet.2013.11.025. [DOI] [PubMed] [Google Scholar]
- Ma B, Wilker EH, Willis-Owen SA, Byun HM, Wong KC, Motta V, Liang L. Predicting DNA methylation level across human tissues. Nucleic Acids Research. 2014;42:3515–3528. doi: 10.1093/nar/gkt1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markon KE, Krueger RF, Watson D. Delineating the structure of normal and abnormal personality: An integrative hierarchical approach. Journal of Personality and Social Psychology. 2005;88:139–157. doi: 10.1037/0022-3514.88.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, O’Donovan MC, Thapar A, Langley K, Williams N. The relative contribution of common and rare genetic variants to adhd. Transl Psychiatry. 2015;5:e506. doi: 10.1038/tp.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews M, Fair DA. Research review: Functional brain connectivity and child psychopathology–overview and methodological considerations for investigators new to the field. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2015;56:400–414. doi: 10.1111/jcpp.12335. [DOI] [PubMed] [Google Scholar]
- McPherson NO, Bell VG, Zander-Fox DL, Fullston T, Wu LL, Robker RL, Lane M. When two obese parents are worse than one! Impacts on embryo and fetal development. American Journal of Physiology: Endocrinology and Metabolism. 2015;309:E568–581. doi: 10.1152/ajpendo.00230.2015. [DOI] [PubMed] [Google Scholar]
- Meehl Paul E. Schizotaxia, schizotypy, schizophrenia. American Psychologist. 1962;17:827. [Google Scholar]
- Miller AH, Raison CL. Are anti-inflammatory therapies viable treatments for psychiatric disorders?: Where the rubber meets the road. Journal American Medical Association Psychiatry. 2015;72:527–528. doi: 10.1001/jamapsychiatry.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Iosif AM, Young GS, Hill M, Phelps Hanzel E, Hutman T, Ozonoff S. School-age outcomes of infants at risk for autism spectrum disorder. Autism Research. 2015 doi: 10.1002/aur.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misiak B, Frydecka D, Zawadzki M, Krefft M, Kiejna A. Refining and integrating schizophrenia pathophysiology - relevance of the allostatic load concept. Neuroscience and Biobehavioral Reviews. 2014;45:183–201. doi: 10.1016/j.neubiorev.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Mitchell RH, Goldstein BI. Inflammation in children and adolescents with neuropsychiatric disorders: A systematic review. Journal of the American Academy of Child and Adolescent Psychiatry. 2014;53:274–296. doi: 10.1016/j.jaac.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Mondelli V, Ciufolini S, Murri MB, Bonaccorso S, Di Forti M, Giordano A, Murray RM. Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophrenia Bulletin. 2015 doi: 10.1093/schbul/sbv028. sbv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk C, Georgieff MK, Osterholm EA. Research review: Maternal prenatal distress and poor nutrition - mutually influencing risk factors affecting infant neurocognitive development. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2013;54:115–130. doi: 10.1111/jcpp.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musser ED, Hawkey E, Kachan-Liu SS, Lees P, Roullet JB, Goddard K, Nigg JT. Shared familial transmission of autism spectrum and attention-deficit/hyperactivity disorders. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2014;55:819–827. doi: 10.1111/jcpp.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg JT, Elmore AL, Natarajan N, Friderici KH, Nikolas MA. Variation in iron metabolism gene moderates the association between low-level blood lead exposure and attention-deficit/hyperactivity disorder. Psychological Science. doi: 10.1177/0956797615618365. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg JT, Johnstone J, Musser ED, Willoughby M, Shannon J. Adhd and risk for obesity: A meta-analysis. Clinical Psychology Review. doi: 10.1016/j.cpr.2015.11.005. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, Eichler EE. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the united states, 2011–2012. JAMA. 2014;311:806–814. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM. Neuroscience, mental health and the immune system: Overcoming the brain-mind-body trichotomy. Epidemiol Psychiatr Sci. 2015:1–5. doi: 10.1017/s204579601500089x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BS, Rauh VA, Bansal R, Hao X, Toth Z, Nati G, Perera F. Effects of prenatal exposure to air pollutants (polycyclic aromatic hydrocarbons) on the development of brain white matter, cognition, and behavior in later childhood. Journal American Medical Association Psychiatry. 2015;72:531–540. doi: 10.1001/jamapsychiatry.2015.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrill SA, Bartlett CW, Blair C. Gene-environment interplay in child psychology and psychiatry–challenges and ways forward. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2013;54:1029. doi: 10.1111/jcpp.12133. [DOI] [PubMed] [Google Scholar]
- Pino O, Guilera G, Gomez-Benito J, Najas-Garcia A, Rufian S, Rojo E. Neurodevelopment or neurodegeneration: Review of theories of schizophrenia. Actas Españolas de Psiquiatría: Acepsi. 2014;42:185–195. [PubMed] [Google Scholar]
- Plichta MM, Scheres A. Ventral-striatal responsiveness during reward anticipation in adhd and its relation to trait impulsivity in the healthy population: A meta-analytic review of the fmri literature. Neuroscience and Biobehavioral Reviews. 2014;38:125–134. doi: 10.1016/j.neubiorev.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric Genetics Consoritum. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet. 2013;381(9875):1371–9. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature Neuroscience. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Research. 2002;5:554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Ray S, Miller M, Karalunas S, Robertson C, Grayson DS, Cary RP, Fair DA. Structural and functional connectivity of the human brain in autism spectrum disorders and attention-deficit/hyperactivity disorder: A rich club-organization study. Human Brain Mapping. 2014;35:6032–6048. doi: 10.1002/hbm.22603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho A, Traicu A, Lepage M, Iyer SN, Malla A, Joober R. Clinical and functional implications of a history of childhood adhd in first-episode psychosis. Schizophrenia Research. 2015;165:128–133. doi: 10.1016/j.schres.2015.03.031. [DOI] [PubMed] [Google Scholar]
- Rommelse NN, Franke B, Geurts HM, Hartman CA, Buitelaar JK. Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. European Child and Adolescent Psychiatry. 2010;19:281–295. doi: 10.1007/s00787-010-0092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush B. Medical inquiries and observations upon the diseases of the mind. Philadelphia, PA: Kimber & Richardson; 1812. [Google Scholar]
- Sable P, Randhir K, Kale A, Chavan-Gautam P, Joshi S. Maternal micronutrients and brain global methylation patterns in the offspring. Nutritional Neuroscience. 2015;18:30–36. doi: 10.1179/1476830513y.0000000097. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gistau V, Romero S, Moreno D, de la Serna E, Baeza I, Sugranyes G, Castro-Fornieles J. Psychiatric disorders in child and adolescent offspring of patients with schizophrenia and bipolar disorder: A controlled study. Schizophrenia Research. 2015;168:197–203. doi: 10.1016/j.schres.2015.08.034. [DOI] [PubMed] [Google Scholar]
- Shaw P, Stringaris A, Nigg J, Leibenluft E. Emotion dysregulation in attention deficit hyperactivity disorder. American Journal of Psychiatry. 2014;171:276–293. doi: 10.1176/appi.ajp.2013.13070966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu YC, Yuan SS, Lee SY, Yang CJ, Yang KC, Lee TL, Wang LJ. Attention-deficit/hyperactivity disorder, methylphenidate use and the risk of developing schizophrenia spectrum disorders: A nationwide population-based study in taiwan. Schizophrenia Research. 2015;168:161–167. doi: 10.1016/j.schres.2015.08.033. [DOI] [PubMed] [Google Scholar]
- Skinner MK, Guerrero-Bosagna C, Haque M, Nilsson E, Bhandari R, McCarrey JR. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. PloS One. 2013;8:e66318. doi: 10.1371/journal.pone.0066318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sng J, Meaney MJ. Environmental regulation of the neural epigenome. Epigenomics. 2009;1:131–151. doi: 10.2217/epi.09.21. [DOI] [PubMed] [Google Scholar]
- South SC, Hamdi N, Krueger RF. Biometric modeling of gene-environment interplay: The intersection of theory and method and applications for social inequality. Journal of Personality. 2015 doi: 10.1111/jopy.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ. Perinatal programming of neuroendocrine mechanisms connecting feeding behavior and stress. Frontiers in Neuroscience. 2013;7:00109. doi: 10.3389/fnins.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroufe LA. The promise of developmental psychopathology: Past and present. Development and Psychopathology. 2013;25:1215–1224. doi: 10.1017/s0954579413000576. [DOI] [PubMed] [Google Scholar]
- Stevenson J, Sonuga-Barke E, McCann D, Grimshaw K, Parker KM, Rose-Zerilli MJ, Warner JO. The role of histamine degradation gene polymorphisms in moderating the effects of food additives on children’s adhd symptoms. American Journal of Psychiatry. 2010;167:1108–1115. doi: 10.1176/appi.ajp.2010.09101529. [DOI] [PubMed] [Google Scholar]
- Stuart MJ, Singhal G, Baune BT. Systematic review of the neurobiological relevance of chemokines to psychiatric disorders. Frontiers in Cellular Neuroscience. 2015;9:357. doi: 10.3389/fncel.2015.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KP, Matsuoka Y, Pae CU. Omega-3 polyunsaturated fatty acids in prevention of mood and anxiety disorders. Clin Psychopharmacol Neurosci. 2015;13:129–137. doi: 10.9758/cpn.2015.13.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, Gorman JM. Schizophrenia after prenatal famine. Further evidence. Archives of General Psychiatry. 1996;53:25–31. doi: 10.1001/archpsyc.1996.01830010027005. [DOI] [PubMed] [Google Scholar]
- Szyf M. Prospects for the development of epigenetic drugs for cns conditions. Nat Rev Drug Discov. 2015;14:461–474. doi: 10.1038/nrd4580. [DOI] [PubMed] [Google Scholar]
- United Nations. Refugees - the numbers. United Nations - Resources for Speakers on Global Issues. 2015 Retrieved from http://www.un.org/en/globalissues/briefingpapers/refugees.
- van Ijzendoorn MH, Belsky J, Bakermans-Kranenburg MJ. Serotonin transporter genotype 5httlpr as a marker of differential susceptibility? A meta-analysis of child and adolescent gene-by-environment studies. Transl Psychiatry. 2012;2:e147. doi: 10.1038/tp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Seminars in Reproductive Medicine. 2009;27:358–368. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yang C, Gelernter J, Zhao H. Pervasive pleiotropy between psychiatric disorders and immune disorders revealed by integrative analysis of multiple gwas. Human Genetics. 2015;134:1195–1209. doi: 10.1007/s00439-015-1596-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox R, Carlson M, Azen S, Clark F. Avoid lost discoveries, because of violations of standard assumptions, by using modern robust statistical methods. Journal of Clinical Epidemiology. 2013;66:319–329. doi: 10.1016/j.jclinepi.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Wolfers T, Buitelaar JK, Beckmann CF, Franke B, Marquand AF. From estimating activation locality to predicting disorder: A review of pattern recognition for neuroimaging-based psychiatric diagnostics. Neuroscience and Biobehavioral Reviews. 2015;57:328–349. doi: 10.1016/j.neubiorev.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends in Neurosciences. 2015;38:637–658. doi: 10.1016/j.tins.2015.08.001.. [DOI] [PubMed] [Google Scholar]
- Zubin J, Spring B. Vulnerability–a new view of schizophrenia. Journal of Abnormal Psychology. 1977;86:103–126. doi: 10.1037//0021-843x.86.2.103. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/858828. [DOI] [PubMed] [Google Scholar]