

Abstract
The inaccuracy of routine serum 25-hydroxyvitamin D measurements hampers the interpretation of data in patient care and public health research. We developed and validated a candidate reference measurement procedure (RMP) for highly accurate quantitation of two clinically important 25-hydroxyvitamin D metabolites in serum, 25-hydroxyvitamin D2 [25(OH)D2] and 25-hydroxyvitamin D3 [25(OH)D3]. The two compounds of interest together with spiked deuterium-labeled internal standards [d3-25(OH)D2 and d6-25(OH)D3] were extracted from serum via liquid-liquid extraction. The featured isotope-dilution LC-MS/MS method used reversed-phase chromatography and atmospheric pressure chemical ionization in positive ion mode. A pentafluorophenylpropyl-packed UHPLC column together with isocratic elution allowed for complete baseline resolution of 25(OH)D2 and 25(OH)D3 from their structural C-3 isomers within 12 min. We evaluated method trueness, precision, potential interferences, matrix effects, limits of quantitation, and measurement uncertainty. Calibration materials were, or were traceable to, NIST Standard Reference Materials 2972. Within-day and total imprecision (CV) averaged 1.9% and 2.0% for 25(OH)D3, respectively, and 2.4% and 3.5% for 25(OH)D2, respectively. Mean trueness was 100.4% for 25(OH)D3 and 100.3% for 25(OH)D2. The limits of quantitation/limits of detection were 4.61/1.38 nmol/L for 25(OH)D3 and 1.46/0.13 nmol/L for 25(OH)D2. When we compared our RMP results to an established RMP using 40 serum samples, we found a nonsignificant mean bias of 0.2% for total 25(OH)D. This candidate RMP for 25(OH)D metabolites meets predefined method performance specifications (≤5% total CV and ≤1.7% bias) and provides sufficient sample throughput to meet the needs of the Centers for Disease Control and Prevention Vitamin D Standardization Certification Program.
Keywords: Vitamin D, 25-Hydroxyvitamin D3, 25-Hydroxyvitamin D2, reference method, LC-MS/MS
Introduction
Vitamin D is important for bone health [1] and over the years research has focused on additional health benefits such as those relative to improved muscle strength, and reduced risks for cancer and type 2 diabetes [2]. Vitamin D status is routinely assessed in patient care using serum or plasma concentrations of the longer-lived 25-hydroxyvitamin D [25(OH)D] metabolites. 25(OH)D represents the sum of 25(OH)D2, a liver metabolite derived from vitamin D2, which is obtained from dietary sources (plant and fortified foods and dietary supplements) or prescribed medication, and 25(OH)D3, another liver metabolite derived from vitamin D3 formed endogenously in the skin by exposure to sunlight or obtained through dietary sources (animal and fortified foods and dietary supplements).
The most commonly used analytical methods to measure 25(OH)D metabolites are based on immunoassays (e.g., chemiluminescent, enzyme-, or radio-labeled) or chromatographic assays coupled with different detectors [e.g., ultraviolet (HPLC-UV) or tandem mass spectrometry (LC-MS/MS)] [3]. Because competitive immunoassays rely on the interaction of 25(OH)D metabolites with antibodies that may lack specificity, this methodology faces challenges with interfering compounds [4, 5]. The high degree of specificity of chromatographic methods coupled with tandem mass spectrometry makes LC-MS/MS the technique of choice for reference measurement procedures (RMP) intended to assign 25(OH)D3 and 25(OH)D2 target values to materials. Although LC-MS/MS is considered highly specific, the accuracy of mass spectrometric measurements can be affected by isobaric compounds that require chromatographic separation from the analytes of interest. The best known isobaric interference is the C-3 structural analog of 25(OH)D3, 3-epi-25(OH)D3, which may be found circulating in substantial quantities [6] but does not possess the full range of biological activities of 25(OH)D3.
The inadequate accuracy of 25(OH)D measurements [5, 7, 8] hampers the interpretation of data in patient care and public health research. To minimize this problem a reference measurement system is needed that consists of RMPs [9, 10] and higher order standard reference materials (SRM) [11]. Worldwide, vitamin D standardization activities are ongoing [12–14]. As part of these efforts, the Centers for Disease Control and Prevention (CDC) established an international Vitamin D Standardization Certification Program (VDSCP) [15] to improve the accuracy and reliability of 25(OH)D laboratory testing. This program uses approaches and procedures successfully employed in CDC’s Hormone Standardization Program [16, 17]. The program provides serum materials with 25(OH)D values assigned using a RMP. These materials are used to assess and improve assay calibration, and to verify assay accuracy through blinded challenges. Currently, two RMPs for quantitation of 25(OH)D in serum have been reviewed and accepted by the Joint Committee for Tractability in Laboratory Medicine (JCTLM) [9, 10], with one of the methods being primarily used for the certification of SRM materials [9]. To meet the growing demands for RMP services, such as providing 25(OH)D value assignment to a large number of serum materials for assessing measurement accuracy and reliability, we developed and validated a candidate RMP for serum 25(OH)D3 and 25(OH)D2 measurements using isotope dilution LC-MS/MS. This method is designed to assign reference values to serum materials at the highest level of accuracy and metrological traceability [18, 19]. We aimed to develop a metrologically traceable reference method that achieved optimum trueness and precision by meeting the suggested specifications for a RMP for 25(OH)D metabolites [20].
Materials and methods
Chemicals and reagents
The following chemicals were purchased: 25(OH)D3 from U.S. Pharmacopeial Convention (Rockville, MD); 25-hydroxycholesterol, 25(OH)D2, and 1-α-hydroxyvitamin D3 from Sigma Aldrich (St. Louis, MO); 7,19,19-trideuterium-25-hydroxyvitamin D2 (d3-25(OH)D2) and 3-epi-25(OH)D3 from IsoSciences (King of Prussia, PA); 7-α-hydroxy-cholesten-3-one and 4β-hydroxycholesterol from Santa Cruz Biotechnology (Santa Cruz, CA); and 24-hydroxycholesterol from Enzo Life Sciences (Farmingdale, NY). 26, 26, 26, 27, 27, 27-Hexadeuterium-25-hydroxyvitamin D3 (d6-25(OH)D3) and 27-hydroxycholesterol were purchased from Medical Isotopes (Pelham, NH). 3-Epi-25(OH)D2 was a gift from the National Institute of Standards and Technology (NIST, Gaithersburg, MD). ACS grade methanol and n-hexane were obtained from Burdick & Jackson (Muskegan, MI). ASC/USP grade ethanol was purchased from Pharmco-AAPER (Brookfield, CT). Purified water (18 MΩ) was obtained from an Aqua Solutions water purification system (Aqua Solutions, Inc., Falmouth, ME). SRM 2972 (25(OH)D2 and 25(OH)D3, solvent-based) and SRM 972a (serum-based) were purchased, and SRM 972 (2 levels, serum-based) was a gift from NIST.
Calibration preparation and control materials
Volumetric steps were gravimetrically controlled, where indicated, using a Mettler-Toledo XP205 analytical balance (Mettler-Toledo LLC, Columbus, OH) equipped with a built-in antistatic electrode. All solutions of standards were made using absolute ethanol. A master stock solution for 25(OH)D3 was gravimetrically prepared by dissolving solid material in absolute ethanol to achieve a concentration of approximately 30–50 μmol/L. Three working standard 25(OH)D3 solutions (targeted range: 150–250 nmol/L) were gravimetrically prepared from the master stock solution and their exact concentrations were determined by comparison with gravimetrically diluted primary reference material SRM 2972 [21] (see Electronic Supplementary Material for details). We gravimetrically added 25(OH)D2 from SRM 2972 to each 25(OH)D3 working standard solution to achieve a targeted range of 25(OH)D2 of 20–40 nmol/L. Master stock solutions of internal standards, d6-25(OH)D3 and d3-25(OH)D2, were made in absolute ethanol to achieve 150 and 60 μmol/L, respectively, based on UV measurements at 265 nm using molar extinction coefficients of 18,300 and 19,400 L mol−1cm−1, respectively. Working internal standard solutions (ISTD) were made by gravimetrically combining different amounts from the master stock solutions to achieve concentrations ranging between 70–90 and 10–12 nmol/L, for d6-25(OH)D3 and d3-25(OH)D2, respectively. For each measurement series, two sets of four working calibrators were gravimetrically prepared from two independent working standard solutions and mixed with ISTD (0.5 mL) to achieve mass ratios of unlabeled to labeled standards of approximately 0.25, 0.50, 1.25, 2.5, resulting in total of 8 different calibration levels for each anlyte. Calibrators were evaporated under vacuum (SpeedVac Plus, Savant Instrument Co., Farmingdale, NY), reconstituted (73% methanol/water), in some cases filtered (0.45 μm PVDF, Captiva 96-well filter plate, Agilent Tech., Santa Clara, CA), and finally injected directly onto the UHPLC column without additional preparation. For each measurement series, we used 3 of the 4 calibration curve levels (from both sets, i.e., 6 calibration points) that bracketed the expected values for the serum samples (based on pre-screened concentrations determined using a routine LC-MS/MS method [22]). The two most common bracketing calibration ranges (3 calibration levels from two independent working standard solutions, resulting in 6 calibration points) were: approximately 10–50 and 20–100 nmol/L for 25(OH)D3 and approximately 1–5 and 2–10 nmol/L for 25(OH)D2.
We prepared all internal quality control materials (IQC) from pooled human serum purchased from U.S. blood banks. All units were screened for 25(OH)D metabolites and in some cases, they were spiked with either 25(OH)D2 or 25(OH)D3, or both, as needed, to achieve desired concentrations. All specimens were stored at −70 °C when not in use. Two IQC pools had values assigned using an established RMP (Laboratory for Analytical Chemistry, University of Ghent, Belgium).
Sample preparation
Volumetric steps were gravimetrically controlled where indicated. Serum was accurately weighted (0.25 to 1 g) and added to water (1 mL; to avoid protein precipitation from subsequent addition of ethanolic ISTD). To this mixture we gravimetrically added ISTD (0.5 g) and allowed equilibration for 1 h at room temperature. Aqueous Na2CO3 (0.1 g/mL, 0.1 mL; pH ~ 11) was added to allow release of 25(OH)D metabolites from binding proteins and the analytes of interest were removed from serum via liquid-liquid extraction (LLE) with hexane (2.5 mL). To maximize extraction efficiency, the LLE step was repeated, the combined organic layers were dried under vacuum at 45 °C, reconstituted in 73% MeOH/water (0.3 mL), filtered (0.45 μm PVDF), and injected for LC-MS/MS analysis.
LC-MS/MS analysis
We used an Accela UHPLC system coupled to a TSQ Quantum Ultra tandem mass spectrometer (both Thermo Fisher Scientific, Waltham, MA) in positive atmospheric pressure chemical ionization (APCI) mode for analysis. Isocratic elution (73% MeOH/water at 0.45 mL/min) for 12 min on an Ascentis F5 analytical column (2.1 mm × 150 mm, 2.7 μm, Sigma-Aldrich, St. Louis, MO) held at 27 °C, followed by a short (1–3 min) column flush with 100% methanol and short (1–3 min) equilibration to initial conditions, resulted in a < 20 min instrument analysis time per sample. The autosampler tray temperature was maintained at 7 °C. Injection volume, using partial loop mode, was 30 μL. Mass detection was carried out under selected reaction monitoring (SRM) conditions using the following transitions: m/z 383.3 → 365.1 (quantitation) and 383.3 → 105.0 (confirmation) for 25(OH)D3, m/z 395.3 → 377.3 (quantitation) and 395.3 → 209.1 (confirmation) for 25(OH)D2, m/z 389.3 → 371.1 for d6-25(OH)D3 and m/z 398.3 → 380.3 for d3-25(OH)D2. Scan time was 0.25 sec. Mass resolution in Q1 and Q3 was set at 0.7 full width at half maximum. We used Xcalibur 2.0.6 software (Thermo Fisher Scientific) for instrument control and data collection and processing.
Data analysis and typical measurement series set-up
In Xcalibur, we built each calibration curve in each measurement series by plotting peak area ratios of the analyte quantification ion divided by the ISTD, i.e., response ratio on the y-axis and the corresponding mass ratio [i.e., the exact weight (g) of the added working standard solution multiplied by its mass fraction (ng/g) divided by the exact weight (g) of the added ISTD solution multiplied by its mass fraction (ng/g)] on the x-axis. We used linear regression with 1/x weighting as curve fit. For details on the calculation of mass fractions and mass concentrations, see Electronic Supplementary Material. Details on a typical run set up are presented in Electronic Supplementary Material Fig. S1.
Value assignment
For value assignment, we first pre-screened unknown samples using our routine in-house LC-MS/MS method [22] to obtain approximate 25(OH)D metabolite concentrations. Then, each unknown sample was prepared for the RMP in at least two independent measurements series, each consisting of duplicate preparations. It should be noted that all samples (calibrators, QC, or unknowns) were injected twice, and thus each individual result was the average of duplicate injections. The final concentrations for 25(OH)D3 and 25(OH)D2 were assigned from the mean of all measurements.
Method validation
Direct calibration
We assessed whether the calibrators need to be carried through the sample preparation procedure or could be directly injected for chromatographic separation and detection (see Electronic Supplementary Material for experimental design). We evaluated the statistical difference between direct and indirect calibration using Student’s 2-sided t-test. All statistical evaluations were performed using Analyse-it (version 2.20, Analyse-it Software, LTD, Leeds, UK), a statistical plug-in for Microsoft Excel.
Interference
We tested eight structural analogs (with typical mass transitions at or close to the m/z values monitored for 25(OH)D3, 25(OH)D2 or the internal standards or capable of producing product ions similar to those produced by the analytes of interest) for potential interference, among which were the 3-epimer analogs of 25(OH)D3 and 25(OH)D2. All compounds were evaluated for their retention time relative to 25(OH)D3 or 25(OH)D2. In addition, to assess for potential interferences we monitored the confirmation ion ratio (confirmation ion to quantitation ion, CI/QI) for 25(OH)D3 and 25(OH)D2 in 148 serum samples, and compared it to the CI/QI ratio of calibration solutions.
Absolute recovery from serum (extraction efficiency)
The recovery of ISTD from the sample preparation procedure was studied using a single donor human serum. Two sets of six replicates were sampled. To the first set, ISTD was added and the samples were carried through the sample preparation procedure as previously described; to the second set, the same amount of ISTD was added after the LLE step. The differences in the isotope ratios of the two sets were calculated, to assess absolute recovery of the sample preparation procedure.
Limit of quantitation (LOQ) and limit of detection (LOD)
We measured the NIST SRM 972 level 1 material (reference concentration 1.46 ± 0.49 nmol/L) in five independent measurement series to estimate the LOQ and LOD for 25(OH)D2. Similar to the approach used by the Ghent RMP, we defined the LOQ at a total CV <7% [10]. For 25(OH)D3, to estimate the LOQ we used two different serum materials (12.0 and 40 nmol/L).We diluted one of the serum materials (12 nmol/L) with de-ionized water up to 5 times, resulting in a concentration range from 2.1 to 12.0 nmol/L and the second serum material (40 nmol/L) approximately 10 and 15 times with de-ionized water, resulting in a concentration range from 2.5 to 4.0 nmol/L. The analyses were performed in at least five independent measurement series and the SD at the estimated zero concentrations (y-intercept: σ0) from the extrapolation of repeated measurements was used to estimate the LOD as 3σ0 and LOQ as 10σ0 for 25(OH)D3 [23].
Imprecision and trueness
The method imprecision was determined using five serum IQC materials: two preparations per level over five days (n = 10 results). The within-day, between-day, and total CV were calculated for each level using CLSI EP-10-A3 as a guide [24]. We assessed method trueness using five NIST secondary reference materials, measured in singlicate (at least five independent measurement series per material). Trueness was expressed as percent difference from the NIST certified value. We also evaluated method accuracy by analyzing 40 individual serum samples, which were value assigned by an established RMP (Laboratory for Analytical Chemistry, University of Ghent, Belgium) [10]. We analyzed these samples in singlicate in six independent measurement series. The mean percent bias of the candidate RMP to the certified RMP was determined using Bland-Altman agreement and linear regression analyses.
Uncertainty of measurements
The standard uncertainty of a measurement results (uc) is a propagation of many uncertainty components (type “A” (random) and “B” (systematic)), and also called combined uncertainty. We evaluated potential sources of uncertainty and included all factors, which might have a significant contribution, in the calculation of standard uncertainty. We calculated the random component of uncertainty from standard deviation from repeated independent measurements (n, referred to in the literature as repeatability) and used it to calculate the measurement standard deviation of the mean. These standard deviations were from three independent measurement series using IQC serum materials prepared in duplicate, with concentrations of 25(OH)D3 ranging from 16.4 to 72.2 nmol/L and for 25(OH)D2 ranging from 3.01 to 17.1 nmol/L. This protocol matches the measurement protocol for unknowns. The systematic component of uncertainty was calculated from four different factors: uncertainty due to the purity of the primary reference material as indicated by NIST (0.8%, SRM 2972, Certificate of Analysis December 07, 2009), uncertainty from determination of the serum density, determination of the weight of each component, and other unspecific interferences (e.g., sample preparation, instrument, undetected interferences, 1%). The combined standard uncertainty was calculated as square root of combined quadratically type A and B components of uncertainty and then multiplied it by a coverage factor, k (k = 2 for 95% confidence level) to obtain the expanded uncertainty (U).
Results and discussion
We developed and validated a candidate RMP with features as described in ISO 15193 for reference methods [25] for quantitation of 25(OH)D3 and 25(OH)D2 in serum. This new method also meets the suggested performance specifications for a total 25(OH)D RMP of maximum total CV of 5% and maximum bias of 1.7% [20].
Sample preparation
As a first step, to obtain approximate 25(OH)D metabolite concentrations, we screened unknown samples using our routine in-house LC-MS/MS method [22]. The typical amount of sample used with the candidate RMP was 0.5 g, however when the screening concentration was above the highest calibrator used in the method, we adjusted the amount of sample to less than 0.5 g, and for samples with 25(OH)D2 at the LOQ or slightly above we adjusted it to more than 0.5g. The typical amount of serum used by our candidate RMP was comparable to that used by the Ghent RMP (250–500 μL) [10], but lower than that used by the NIST RMP (2 g) [9]. To avoid protein precipitation, we diluted serum samples with water prior to adding the ethanolic ISTD. 25(OH)D metabolites are usually released from the vitamin D binding protein (the main carrier of these lipophilic compounds) via extreme pH change [26] or by mechanical shaking with extraction solvents. We used LLE with hexane at basic pH (> 10). As a potentially faster alternative to LLE, we also evaluated supported liquid extraction (SLE). To assure adequate detection of the less abundant 25(OH)D2 metabolite, the SLE cartridges required at least twice as much sample volume compared to our LLE procedure (data not shown). Two additional drawbacks of SLE cartridges were its higher cost and its need for larger volumes of organic solvents compared to LLE. After LLE, the serum samples were re-dissolved in 73% MeOH/water, which typically produced cloudy samples as a result of reduced solubility of fat soluble compounds extracted with hexane in the LLE step. During method development, we incorporated a filtration step (0.45 μm PVDF) to produce clean extracts for UHPLC. Although this step reduced extraction efficiency by a few percent, it substantially extended column life and is routinely used.
Calibration
NIST provides the highest order commercially available primary calibration reference materials for 25(OH)D3 and 25(OH)D2 [24]. To calibrate, we used primary reference material NIST SRM 2972 [for 25(OH)D2] or solutions traceable to SRM 2972 [for 25(OH)D3]. Most reported RMPs use calibration where the mass ratio of the unlabeled (compound of interest) to labeled (ISTD) analyte is close to 1, at which the best measurement precision is achievable [27]. For example, the NIST 25(OH)D JCTLM-accepted RMP uses a 6-point calibration curve with mass ratios between 0.65 and 1.4 [9] while the Ghent method uses a 1-point calibration curve with a ± 25% deviation from the typical mass ratio of 1 [10]. We used a 6-point bracketing calibration curve with approximate mass ratio ranges from 0.25 to 1.25 [for 25(OH)D3 and 25(OH)D2 concentrations in the unknown samples < 75 nmol/L and < 10 nmol/L, respectively]. When the pre-screened concentrations of 25(OH)D3 and 25(OH)D2 were above 75 nmol/L and 10 nmol/L, respectively, the calibration curves had a mass ratio range of approximately 0.5 to 2.5. To achieve that, we varied the amount of working standard solution, while keeping the amount of ISTD the same. Our approach allowed for simplified sample preparation because it did not require individual ISTD concentration adjustment to match the endogenous 25(OH)D3 and 25(OH)D2 concentration individually for each serum sample. Wider mass calibration ranges with multipoint calibration curves have previously been reported for JCTLM-accepted RMPs, e.g., for cholesterol analysis [28] and also in a recently reported candidate RMP for cyclosporine A [29].
We directly analyzed ethanol-based calibrators mixed with the ISTD without carrying them through the sample preparation procedure. To justify the direct use of calibrators, we assessed whether a directly analyzed calibration set provided different results to a set submitted to sample preparation. The mean relative response ratios from the two sets were not statistically significantly different using a two-sided Student’s t-test [−0.2% difference for 25(OH)D2 (P = 0.84) and 0.6% difference for 25(OH)D3 (P = 0.39)], and thus, we considered the direct use of calibrators without subjecting them to sample preparation as appropriate. We obtained linear calibration curves for both analytes with high coefficients of determination, R2 > 0.995. A typical regression line was yresponse ratio = 0.9218 xmass ratio + 0.00445 (R = 0.9997, standard error = 0.0100; n=12) and yresponse ratio= 1.6847xmass ratio + 0.03346 (R = 0.9990; standard error = 0.0319; n=12) for 25(OH)D3 and 25(OH)D2, respectively. We also obtained good method precision and minimal bias with this calibration approach (shown later).
Interference testing
Another important concern was the assessment of potential interferences from isobaric compounds. The C3-epimer form of 25(OH)D3 can be present in significant quantities in serum, especially in specimens with high circulating concentrations of 25(OH)D3 [6]. To avoid overestimation of circulating 25(OH)D3, it is therefore important that the epimer be chromatographically resolved. Our candidate RMP used a reversed-phase pentafluorophenylpropyl column with isocratic elution, which provided complete baseline resolution of 25(OH)D3 from 3-epi-25(OH)D3 (Fig. 1) and 25(OH)D2 from 3-epi-25(OH)D2 (chromatographic data not shown), with relative retention times for both epimers of greater than 1.10 (Table 1). We tested six additional structural analogs known to be present in serum with similar molecular masses to the analytes of interest. The relative retention times of these potential interferents were greater than 1.5, which indicated that their presence is not likely to affect our method (Table 1).
Fig. 1.
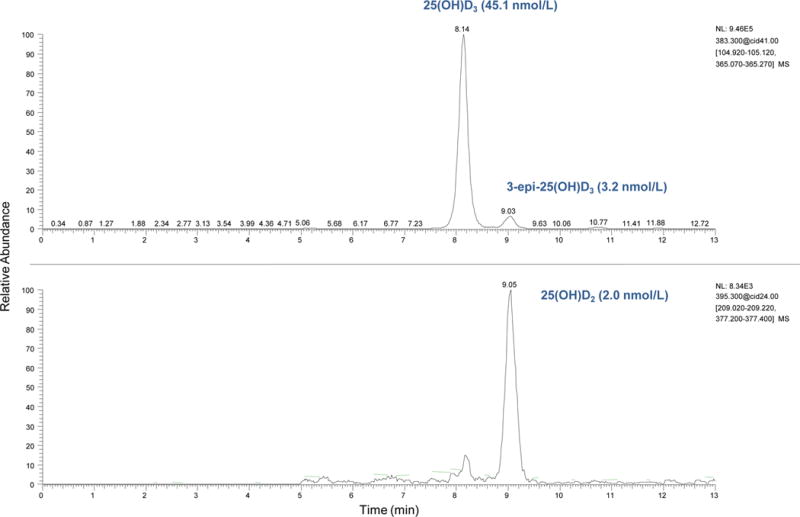
Typical selected reaction monitoring chromatogram of serum NIST SRM 972a level 2 with 25(OH)D3 concentration of 45.1 nmol/L, 3-epi-25(OH)D3 concentration of 3.2 nmol/L and 25(OH)D2 concentration of 2.0 nmol/L.
Table 1.
Vitamin D analogs tested for potential interference
| Vitamin D metabolite or structural analog | Relative retention time a | m/z |
|---|---|---|
| 3-epi-25(OH)D3 | 1.11 | 383→365 |
| 3-epi-25(OH)D2 | 1.12 | 395→377 |
| 7α-hydroxy-4-cholestene-3-one | 2.78 | 383→365 |
| 1α-hydroxyvitamin D3 | 2.25 | 383→365 |
| 4β-hydroxycholesterol | 5.07 | 385→367 |
| 27-hydroxycholesterol | 1.83 | 385→367 |
| 24(S)-hydroxycholesterol | 1.47 | 385→367 |
| 25-hydroxycholesterol | 1.62 | 385→367 |
Relative retention time is calculated relative to 25(OH)D3 (m/z 383→365) or 25(OH)D2 (m/z 395→377)
As compared to the other two JCTLM accepted RMPs for 25(OH)D metabolites, our LC-MS/MS method offers an additional degree of specificity because it can identify potential interferences in serum specimens by monitoring the peak area ratio from two m/z transitions for each analyte. We established acceptability limits for the confirmation ion ratio (peak area ratio of confirmation to quantitation ion) using pure calibration materials (Table 2). We chose variation ranges for the ion ratios of ± 15% for 25(OH)D3 and ± 20% for 25(OH)D2. We accepted a wider range for 25(OH)D2, because of its typical low abundance in serum, resulting in significantly lower confirmation ion peak areas, and thus introducing more variability to the ion ratio calculations. Our choices met or exceeded common “industry standards” for ion ratios. The European Commission uses ± 20–50% depending of the abundance of each ion [30] and the Society of Forensic Toxicologists uses ± 20–30% [31], but we were less stringent than criteria from the U.S. Food and Drug Administration, which uses ± 10% [32]. Our criteria were in line with a recent accepted RMP, which allowed for ± 20% ion ratio variation for measurement of low abundance serum analytes [33]. When we applied the established limits to 25(OH)D3 results from 148 serum samples and to 25(OH)D2 results from 67 serum samples (with concentrations greater than the LOQ), 100% and 98.5% of the samples met the predetermined limits for 25(OH)D3 and 25(OH)D2, respectively (Table 2).
Table 2.
Confirmation ion ratios for a candidate reference measurement procedure for the quantitation of 25(OH)D2 and 25(OH)D3 in calibrators and serum samples
| n | Concentration, nmol/L |
Mean Ion Ratioa (range) |
|
|---|---|---|---|
|
|
|||
| 25(OH)D2 | |||
| Calibrator | 40 | 3.71 | 0.57 (0.45–0.68)b |
| Serum samples | 67 | LOQ-64.9 | 0.56 (0.45–0.69)c |
| 25(OH)D3 | |||
| Calibrators | 80 | 10–100 | 0.57 (0.49–0.66)d |
| Serum samples | 148 | 16–183 | 0.57 (0.51–0.63)c |
Peak area ratios of confirmation (m/z 383.3→105.0) to quantitation (m/z 383.3→365.1) ions
Mean (± 20%) calculated from calibration level 2 in 10 measurement series (2 preparations, 2 injections)
Mean (minimum-maximum) from all serum samples with concentrations >LOQ from 5 measurement series
Mean (± 15%) calculated from all calibration levels in 5 measurement series (4 levels, 2 preparations, 2 injections)
Lastly, we assessed potential interferences due to a carry-over effect from a previous sample, by injecting mobile phase after a high concentration calibrator (140 nmol/L). We did not observe any signal in the monitored mass transitions in the mobile phase blank, thus indicating no evidence of carry-over from a previous injection (data not shown).
Absolute recovery from serum
We assessed the effect of matrix on analyte recovery during the sample preparation procedure. Based on the difference in response ratios from the addition of ISTD prior to and after sample preparation, our extraction method yielded a mean absolute recovery from the sample preparation procedure ± 2-sided confidence interval from replicate analyses of 91% ± 1.4% for 25(OH)D3 and 95% ± 1.2% for 25(OH)D2. This was similar or higher compared to the absolute recovery of the extraction method reported for 25(OH)D3 and 25(OH)D2 by the NIST RMP (97% and 92%, respectively) [9] and the Ghent RMP (71% and 70%, respectively) [10]. Optimizing the sample preparation steps to maximize the recovery from LLE ensures better detection, an aspect that is especially important for 25(OH)D2, typically present in serum at low concentrations.
Limit of quantitation (LOQ) and limit of detection (LOD)
The LOQ for 25(OH)D2 was assessed using SRM 972 level 1 by an approach similar to the one used by the University of Ghent in their JCTLM-accepted RMP [10]. The estimated mean for this material from analysis (n = 5 independent measurement series) with the candidate RMP was 1.25 ± 0.08 nmol/L (2-sided 95% CI) with an absolute difference from the target of −0.21 nmol/L, total CV of 6.1%, and mean signal-to noise (S/N) ratio of 28. The estimated mean was within the NIST uncertainty reported for this level and the CV met the set imprecision criteria. Based on these data, we set the LOQ for 25(OH)D2 to be 1.46 nmol/L, which is the reference value for 25(OH)D2 in this SRM material (1.46 ± 0.49 nmol/L). The LOD for 25(OH)D2 was estimated to be 0.13 nmol/L, at a S/N ratio of 3. For 25(OH)D3, we used a different approach, for which we diluted two serum materials to concentrations close to the expected LOD (based on S/N). The LOQ and LOD for 25(OH)D3 were estimated to be 4.61 nmol/L (CV = 2.9%; S/N ~100) and 1.38 nmol/L (CV ~70%; S/N ~20), respectively [23]. In the end, serum concentrations 25(OH)D3 are rarely less than 10 nmol/L and the absolute LOQ and LOD estimates are inconsequential. Our candidate RMP met the widened imprecision goal of <7% CV at the LOQ for both analytes. The LOQ values of the candidate RMP are sufficiently low to assign target values to unknown serum samples; they are also commensurate with LOQ values reported by the two accepted RMPs [9, 10].
Precision and trueness
Our candidate RMP is intended for target value assignment, not for routine measurement of unknown samples and therefore has to comply with stricter performance criteria. Thus, we assessed method imprecision and bias against performance criteria recommended for a 25-hydroxyvitamin D RMP, namely, total CV ≤ 5% and a systematic bias of ≤ 1.7% [13, 20]. The mean within-day and total imprecision (CV), assessed from five IQC materials were 1.9% and 2.0% for 25(OH)D3 and 2.4% and 3.5% for 25(OH)D2, respectively, (Table 3). We verified that repeated measurements of native serum samples also produced acceptable imprecision: 1.1% for 25(OH)D3 (n = 73) and 2.5% for 25(OH)D2 (n = 26, above the LOQ). The candidate RMP meets current imprecision quality goals and is also in agreement with the imprecision reported by the two JCTLM-accepted RMP for analysis of 25(OH)D metabolites [9,10]. We studied the variability of method performance at different 25(OH)D2 concentrations using nine different serum materials (6 IQC and 3 SRM) analyzed in at least five independent measurement series. The method imprecision becomes more marked as 25(OH)D2 concentrations fall below 2 nmol/L (see Electronic Supplementary Material Fig. S2).
Table 3.
Imprecision of the candidate reference measurement procedure for quantitation of serum concentrations of 25(OH)D2 and 25(OH)D3 in five internal quality control materialsa
| Concentration, nmol/L | CV, %
|
||
|---|---|---|---|
| Within-day | Between-day | Total | |
| 25(OH)D2 | |||
| 3.00 | 1.6 | 2.8 | 3.1 |
| 3.08 | 3.8 | 3.7 | 4.9 |
| 4.66 | 3.4 | 3.0 | 4.1 |
| 9.90 | 1.5 | 2.9 | 3.1 |
| 14.8 | 1.7 | 2.1 | 2.5 |
| 25(OH)D3 | |||
| 23.8 | 2.3 | 1.2 | 2.2 |
| 40.6 | 3.1 | 1.0 | 2.7 |
| 49.2 | 0.6 | 1.5 | 1.6 |
| 65.3 | 1.8 | 0.8 | 1.7 |
| 84.5 | 1.5 | 2.0 | 1.8 |
Duplicate samples of internal quality control serum were tested in five independent measurement series. The within-day, between-day imprecision, and total CV were calculated for each material using CLSI EP-10-A3 document as a guide [24].
We assessed the trueness of our candidate RMP by using five secondary reference materials, NIST SRM 972 and 972a. As shown in Table 4, our mean results were within the uncertainty of the NIST certified values. On average, the systematic deviation from the target for 25(OH)D3 and 25(OH)D2 was 0.4% and 0.3%, respectively. To assess the method performance for 25(OH)D2 at a concentration closer to the patient population, we evaluated a secondary reference material, NIST SRM 972a-level 2, the only material currently available with a low certified 25(OH)D2 value (± U) (2.0 ± 0.2 nmol/L) [34]. We achieved a mean (± U) 25(OH)D2 concentration of 1.97 ± 0.07 nmol/L, with an average deviation from the target of −0.03 nmol/L (−1.5%) in multiple measurement series (n = 10 results). This is an important assessment for 25(OH)D2, which typically circulates at levels around the LOQ or below. We assessed the method performance in a sample with a high 3-epi 25(OH)D3 concentration (SRM 972a-level 4) in three measurement series. We achieved a mean 25(OH)D3 concentration of 28.4 nmol/L with −0.9% deviation from the target. Thus, the candidate RMP meets the set bias quality goal of 1.7%.
Table 4.
Trueness of the candidate reference measurement procedure for quantitation of 25(OH)D2 and 25(OH)D3 using NIST SRM materials with certified values
| SRM
|
|||||
|---|---|---|---|---|---|
| 972a - Level 2 | 972a – Level 3 | 972a – Level 4 | 972 – Level 1 | 972 - Level 3 | |
| 25(OH)D2 | |||||
| na | 10 | 10 | 0 | 0 | 10 |
| Target ± U,b nmol/L | 2.0 ± 0.2 | 32.3 ± 0.8 | 0.54 ± 0.1 | 1.46 ± 0.49 | 64.1 ± 4.8 |
| Meanc (95% CI), nmol/L | 1.97 (1.94–2.00) | 32.7 (32.5–32.9) | __ | __ | 64.2 (63.3–65.1) |
| Truenessd (95% CI), % | 98.5 (97.0–99.9) | 101.4 (100.6–102.0) | __ | __ | 101.1 (101.1–102.1) |
| 25(OH)D3 | |||||
| na | 10 | 10 | 3 | 5 | 10 |
| Target ± U, nmol/L | 45.1 ± 1.1 | 49.4 ± 1.1 | 28.7 ± 0.7 | 59.6 ± 2.1 | 46.2 ± 2.8 |
| Meanb (95% CI), nmol/L | 45.9 (45.4–46.3) | 49.3 (48.5–50.1) | 28.4 (28.4–28.5) | 59.6 (58.0–61.3) | 46.2 (45.5–46.9) |
| Truenessc (95% CI), % | 101.7 (100.7–102.7) | 99.8 (98.2–101.4) | 99.1 (98.9–99.3) | 100.4 (97.9–102.9) | 100.0 (98.6–101.5) |
number of independent analysis measurement series
Singlicate analysis of SRM using candidate RMP in each independent measurement series
Difference between candidate RMP and target concentration as a percentage of target concentration
Method comparison
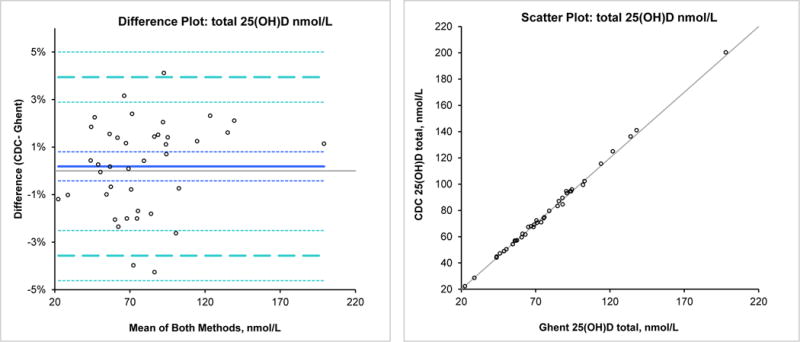
As another way of assessing method trueness, we compared our candidate RMP against a JCTLM-recognized RMP [10] using 40 serum samples. The concentration range was 22.5–198 nmol/L for 25(OH)D3 and 1.7–14.1 nmol/L for 25(OH)D2 (9 samples had 25(OH)D2 values above the LOQ). A Bland-Altman analysis showed excellent agreement between methods with mean bias of −0.9% (95% CI from −1.5 to −0.3 %) for 25(OH)D3, 2.3% (95% CI from −1.4 to 6.0%) for 25(OH)D2, and 0.2% (95% CI from −0.4 to 0.8%) for total 25(OH)D (sum of 25(OH)D2 and 25(OH)D3). The scatter and difference plot are presented in Fig.2. Thus, even with singlicate measurements, the bias goal of <1.7% is met for total 25(OH)D in this reasonably large set of reference samples. The linear regression analysis between the two methods showed excellent agreement (R2=1) with the following equation: CDC total 25(OH)D, nmol/L= −1.118+1.0188* Ghent total 25(OH)D, nmol/L. Neither the slope nor the intercept were significant.
Fig. 2. Bland Altman difference and scatter plots.
 Identity
Identity
 Bias (0.2%)
Bias (0.2%)
 95% CI
95% CI
 95% Limits of agreement (−3.6% to 3.9%)
95% Limits of agreement (−3.6% to 3.9%)
Uncertainty of measurements
The candidate RMP estimated expanded uncertainties, U, ranged from 2.7% to 3.9% with a mean of 3.2% for 25(OH)D3 and from 3.1% to 3.8% with a mean of 3.6% for 25(OH)D2. Detailed information is provided in Electronic Supplemental Material, Table S1.
In summary, we developed a highly accurate, International System of Units (SI) mass-traceable candidate RMP, based on isotope dilution LC-MS/MS, that features good sensitivity despite using a relatively small amount of serum and provides high selectivity for quantitative analysis of serum concentrations of 25(OH)D3 and 25(OH)D2. The performance characteristics of the candidate RMP are comparable to the two currently JCTLM-accepted RMPs. The method meets the needs of the CDC’s Vitamin D Standardization Certification Program.
Supplementary Material
Acknowledgments
The authors thank Dr. Susan Tai from NIST for her guidance and advice during the method development. We would also like to thank Dr. Katleen Van Uytfanghe from the University of Ghent and Dr. Mary Bedner from NIST for their guidance on RMP measurement series acceptance criteria. In addition, we would like to thank all members of the hormone standardization research team at the CDC for providing logistic support.
Footnotes
Conflict of Interest: No specific sources of financial support. The findings and conclusions in this report are those of the authors and do not necessarily represent the official views or positions of the Centers for Disease Control and Prevention/Agency for Toxic Substances and Disease Registry.
References
- 1.Cranney A, Horsley T, O’Donnell S, Weiler H, Puil L, Ooi D, Atkinson S, Ward L, Moher D, Hanley D, Fang M, Yazdi F, Garritty C, Sampson M, Barrowman N, Tsertsvadze A, Mamaladze V. Effectiveness and safety of vitamin D in relation to bone health. Evid Rep Technol Assess (Full Rep) 2007;158:1–235. [PMC free article] [PubMed] [Google Scholar]
- 2.Chung M, Balk EM, Brendel M, Ip S, Lau J, Lee J, Lichtenstein A, Patel K, Raman G, Tatsioni A, Terasawa T, Trikalinos TA. Vitamin D and calcium: a systematic review of health outcomes. Evid Rep Technol Assess (Full Rep) 2009;183:1–420. [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace AM, Gibson S, de la Hunty A, Lamberg-Allardt C, Ashwell M. Measurement of 25-hydroxyvitamin D in clinical laboratory: current procedures, performance characteristics and limitations. Steroids. 2010;75:477–488. doi: 10.1016/j.steroids.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Lind C, Chen J, Byrjalsen I. Enzyme immunoassays for measuring 25-hydroxyvitamin D3 in serum. Clin Chem. 1997;43(6):943–949. [PubMed] [Google Scholar]
- 5.Carter GD, Carter R, Jones J, Berry J. How accurate are assays for 25-hydroxyvitamin D? Data from international Vitamin D External Quality Assessment Scheme. Clin Chem. 2004;50(11):2195–2197. doi: 10.1373/clinchem.2004.040683. [DOI] [PubMed] [Google Scholar]
- 6.Singh RJ, Taylor RL, Reddy S, Grebe S. C-3 epimers can account for a significant proportion of total circulating 25-hydroxyvitamin D in infants, complicating accurate measurement and interpretation of vitamin D status. J Clin Endocrinol Metab. 2006;91(8):3055–3061. doi: 10.1210/jc.2006-0710. [DOI] [PubMed] [Google Scholar]
- 7.Bedner M, Lippa KA, Tai SS. An assessment of 25-hydroxyvitamin D measurements in comparability studies conducted by the Vitamin D Metabolites Quality Assurance Program. Clin Chim Acta. 2013;426:6–11. doi: 10.1016/j.cca.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binkley N, Krueger D, Cowgill CS, Plum L, Lake E, Hansen KE, DeLuca HF, Drezner MK. Assay variation confounds the diagnosis of hypovitaminosis D: a call for standardization. J Clin Endocrinol Metab. 2004;89(7):3152–3157. doi: 10.1210/jc.2003-031979. [DOI] [PubMed] [Google Scholar]
- 9.Tai S, Bedner M, Phinney K. Development of candidate reference measurement procedure for determination of 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal Chem. 2010;82:1942–1948. doi: 10.1021/ac9026862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stepman HCM, Vanderroost A, Van Uytfanghe K, Thienpont LM. Candidate reference measurement procedure for serum 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 by using isotope-dilution liquid chromatography-tandem mass spectrometry. Clin Chem. 2011;53(3):441–448. doi: 10.1373/clinchem.2010.152553. [DOI] [PubMed] [Google Scholar]
- 11.Phinney K, Bedner M, Tai S, Vamathevan V, Sander L, Sharpless K, Wise S, Yen JH, Schleicher RL, Chaudhary-Webb M, Pfeiffer CM, Betz JM, Coates PM, Picciano MF. Development and certification of a standard reference material for Vitamin D metabolites in human serum. Anal Chem. 2012;84(2):956–962. doi: 10.1021/ac202047n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sempos CT, Vesper HW, Phinney KW, Thienpont LM, Coates PM. Vitamin D status as an international issue: national surveys and the problem of standardization. Scand J Clin Lab Invest (Suppl 243) 2012;72:32–40. doi: 10.3109/00365513.2012.681935. [DOI] [PubMed] [Google Scholar]
- 13.Thienpont LM, Stepman HC, Vesper HW. Standardization of measurements of 25-hydroxyvitamin D3 and D2. Scand J Clin Lab Invest (Suppl 243) 2012;72:41–49. doi: 10.3109/00365513.2012.681950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cashman KD, Kiely M, Kinsella M, Durazo-Arvizu RA, Tian L, Zhang Y, Lucey A, Flynn A, Gibney MJ, Vesper HW, Phinney KW, Coates PM, Picciano MF, Sempos CT. Evaluation of Vitamin D Standardization Program protocols for standardizing serum 25-hydrozyvitamin D data: a case study of the program’s potential for national nutrition and health surveys. Am J Clin Nutr. 2013;97(6):1235–1242. doi: 10.3945/ajcn.112.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Centers for Disease Control and Prevention. Laboratory Quality Assurance and Standardization Programs. Atlanta (GA): CDC; http://www.cdc.gov/labstandards/hs.html. Accessed 12 Nov 2014. [Google Scholar]
- 16.Vesper HW, Botelho JC. Standardization of testosterone measurements in humans. J Steroid Biochem Mol Biol. 2010;121(3–5):513–519. doi: 10.1016/j.jsbmb.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 17.Vesper HW, Botelho JC. An overview of CDC’s Standardization Initiative. Clin Lab News. 2012;38(6):10–12. [Google Scholar]
- 18.Bureau International des Poids et Mesures. The international System of Units (SI) http://www.bipm.org/en/about-us/. Accessed 12 Nov 2014.
- 19.Vesper HW, Thienpont LM. Traceability in laboratory medicine. Clin Chem. 2009;55(6):1067–1075. doi: 10.1373/clinchem.2008.107052. [DOI] [PubMed] [Google Scholar]
- 20.Stӧckl D, Sluss PM, Thienpont LM. Specifications for trueness and precision of a reference measurement system for serum/plasma 25-hydroxyvitamin D analysis. Clin Chim Acta. 2009;408:8–13. doi: 10.1016/j.cca.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 21.National Institute of Standards and Technology. Certificate of analysis, standard reference material 2972: 25-hydroxyvitamin D2 and D3 calibration solutions. Gaithersburg (MD): NIST; 2014. [Google Scholar]
- 22.Schleicher RL, Encisco SE, Chaudhary-Webb M, Paliakov EM, McCoy LF, Pfeiffer CM. Isotope dilution ultra performance liquid chromatography-tandem mass spectrometry method for simultaneous measurement of 25-hydroxyvitamin D2, 25-hydroxyvitamin D3 and 3-epi-25-hydroxyvitamin D3 in human serum. Clin Chim Acta. 2011;412:1594–1599. doi: 10.1016/j.cca.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Taylor JK. Quality assurance of chemical measurement. Lewis Publishers; Michigan: 1987. [Google Scholar]
- 24.CLSI. CLSI Document EP10-A3. third. Wayne (PA): CLSI; 2014. Preliminary evaluation of quantitative clinical laboratory measurement procedures; approved guideline. [Google Scholar]
- 25.ISO. ISO document 15193:2009(E) Geneva: ISO; 2009. In vitro diagnostic medical devices-Measurement of quantities in samples of biological origin-Requirements of content and presentation of reference measurement procedures. [Google Scholar]
- 26.Antoni S, Vogl C. Release reagents for Vitamin D compounds. 2013/0078729 A1. US Patent. 2013 Mar 28;
- 27.Thienpont LM, Van Nieuwenhove B, Stöckl D, De Leenheer AP. Calibration for isotope dilution mass spectrometry - description of an alternative to the bracketing procedure. J Mass Spec. 1996;31:1119–1125. [Google Scholar]
- 28.Edwards SH, Kimberly MM, Pyatt SD, Stribling SL, Dobbin KD, Myers GL. Proposed serum cholesterol reference measurement procedure by gas chromatography- isotope dilution mass spectrometry. Clin Chem. 2011;57(4):614–622. doi: 10.1373/clinchem.2010.158766. [DOI] [PubMed] [Google Scholar]
- 29.Grote-Koska D, Czajkowski S, Klauke R, Panten E, Brand K, Schumann G. A candidate reference measurement procedure for cyclosporine A in whole blood. Accred Qual Assur. 2014;19:147–157. [Google Scholar]
- 30.European Union Decision. 2002/657/EC 17.08.2002: commission decision laying down performance criteria for the analytical methods to be used for certain substances and residues thereof in live animals and animal products. Off J Eur Commun. 2002;221:8–32. [Google Scholar]
- 31.Society of Forensic Toxicology/American Academy of Forensic Science. Forensic toxicology laboratory guidelines (2006) Washington (DC): AAFS; 2006. http://www.soft-tox.org/files/Guidelines_2006_Final.pdf. Accessed 12 Nov 2014. [Google Scholar]
- 32.Food and Drug Administration. Guidance for industry: mass spectrometry for confirmation of the identity of animal drug residues. Rockville (MD): FDA; 2003. http://www.fda.gov/downloads/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/UCM052658.pdf. Accessed 12 Nov 2014. [Google Scholar]
- 33.Botelho JC, Shacklady C, Cooper HC, Tai S, Van Uytfanghe K, Thienpont LM, Vesper HW. Isotope-dilution liquid chromatography mass spectrometry candidate reference method for total testosterone in human serum. Clin Chem. 2013;59(2):372–380. doi: 10.1373/clinchem.2012.190934. [DOI] [PubMed] [Google Scholar]
- 34.National Institute of Standards and Technology. Certificate of analysis, standard reference material 972a: Vitamin D metabolites in frozen human serum. Gaithersburg (MD): NIST; 2013. [Google Scholar]
- 35.National Institute of Standards and Technology. Certificate of analysis, standard reference material 972: Vitamin D in human serum. Gaithersburg (MD): NIST; 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.