

Abstract
Objective
Rupture of abdominal aortic aneurysm (AAA), a major cause of death in the aged population, is characterized by vascular inflammation and matrix degradation. Serum amyloid A (SAA), an acute phase reactant linked to inflammation and matrix metalloproteinase induction, correlates with aortic dimensions before aneurysm formation in humans.
We investigated whether SAA deficiency in mice impacts AAA formation during angiotensin II (AngII) infusion.
Approach and Results
Plasma SAA increased ~60-fold in apoE−/− mice 24 hours after i.p. AngII injection (100 μg/kg; n = 4) and ~15-fold after chronic 28-day AngII infusion (1,000 ng/kg/min; n = 9). AAA incidence and severity after 28-day AngII infusion was significantly reduced in apoE−/− mice lacking both acute phase SAA isoforms (SAAKO; n = 20) compared to apoE−/− mice (SAAWT; n = 20) as assessed by in vivo ultrasound and ex vivo morphometric analyses, despite a significant increase in systolic blood pressure in SAAKO mice compared to SAAWT mice after AngII infusion. Atherosclerotic lesion area of the aortic arch was similar in SAAKO and SAAWT mice after 28-day AngII infusion. Immunostaining detected SAA in AAA tissues of AngII-infused SAAWT mice that co-localized with macrophages, elastin breaks, and enhanced matrix metalloproteinase (MMP) activity. MMP-2 activity was significantly lower in aortas of SAAKO mice compared to SAAWT mice after 10-day AngII infusion.
Conclusion
Lack of endogenous acute phase SAA protects against experimental AAA through a mechanism that may involve reduced MMP-2 activity.
Keywords: serum amyloid A, inflammation, acute phase, aneurysm, matrix metalloproteinase
INTRODUCTION
Serum amyloid A (SAA) is a cytokine-inducible acute phase reactant whose plasma concentrations can exceed 1 mg/ml during an acute phase response. Two highly homologous acute phase human isoforms, SAA1 and SAA2, corresponding to mouse SAA1.1 and SAA2.1, are produced mainly in liver during an acute phase response1. Plasma SAA concentrations are also elevated in chronic inflammatory diseases including rheumatoid arthritis2, obesity3, 4, diabetes and insulin resistance3, 5 and with moderate cigarette smoking6. Patients with elevated plasma SAA concentrations are at a greater risk for adverse cardiac events (death and myocardial infarction)7 with some studies reporting that SAA is a better predictor of cardiovascular outcomes than C-reactive protein (CRP)7, 8. In patients with acute myocardial infarction, SAA, but not CRP, is elevated at the site of plaque rupture9.
SAA, but not CRP, has also been positively correlated with abdominal aortic diameter prior to aortic dilation, possibly reflecting early stages of aneurysm formation10. Abdominal aortic aneurysm (AAA), a pathological dilation of the aorta, occurs in ~ 5% of men and ~1% of women 60 years and older. Rupture of AAA accounts for ~15,000 deaths in the US annually11–13. However, this number is likely an underestimate because AAA is typically asymptomatic, and aortic rupture may not be identified without an autopsy. The pathophysiological processes leading to AAA formation are complex, involving medial dissection, luminal dilation and thrombus formation that compromise aortic integrity, ultimately leading to aortic rupture in some individuals. The mechanisms underlying AAA progression are still poorly understood, but inflammation and matrix metalloproteinases (MMPs) are thought to be intimately involved14. There is no proven medical therapy for AAA, with management of patients relying on routine monitoring of AAA size by ultrasound (US) until surgical repair is implemented when the growth rate exceeds 10mm/year or the diameter exceeds 55mm.
Although the physiological function(s) of SAA have not been clearly established, a number of studies link SAA with inflammatory processes implicated in AAA formation and progression, such as leukocyte chemotaxis15, induction of inflammatory cytokines16–18 and upregulation of genes involved in extracellular matrix remodeling, including transforming growth factor-β (TGF-β)19 and MMP expression20–23. To investigate the possibility that SAA plays a role in the initiation and/or progression of AAA, mice lacking both acute phase SAA isoforms24 were bred into a C57BL/6 background and then crossed with apoE−/− mice, which allowed us to take advantage of the widely used AAA model that involves infusing angiotensin II (AngII) into hypercholesterolemic mice. AngII-induced AAAs exhibit many features of human AAA, including medial degeneration, inflammation, thrombus formation and rupture of the abdominal aorta25. Our results demonstrate that SAA augments MMP activity in the abdominal aorta and enhances AAA expansion in AngII-infused apoE−/− mice.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
SAA induces genes implicated in AAA
SAA has been reported to elicit pro-inflammatory effects such as macrophage chemotaxis, inflammatory cytokine and MMP induction in multiple cell types in vitro15, 17, 20, 23, 26, 27. However, conclusions made in many of the published studies have recently come under question, due to the recognition that the commercially available hybrid SAA molecule used in these studies may exert activities not shared by naturally expressed SAA isoforms28, 29. In this study we provide novel data that SAA1.1/SAA2.1 isolated from acute phase mouse plasma stimulates primary mouse aortic SMC (Fig. 1A) and J774 macrophage-like cells (Fig. 1B) to express MMPs and inflammatory cytokines. This result demonstrates that SAA may exert direct effects on vascular cells that impact vessel wall integrity.
Fig 1. Purified mouse SAA induces aortic SMC and J774 macrophage-like cells to express MMPs and inflammatory cytokines.
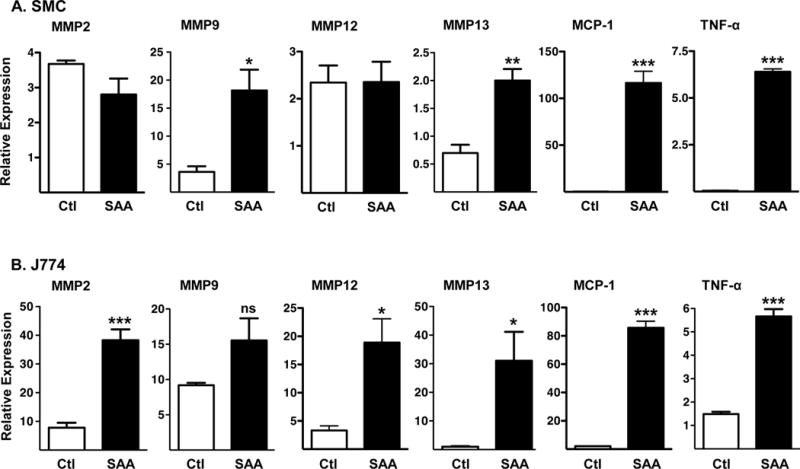
A) SMC prepared from C57BL/6 mouse aortas cultured for 3 days (n = 3) and B) J774 cells (90% confluent; n = 4) were incubated for 8 hours in the absence (control, Ctl) or presence of 50 μg/ml mouse SAA (SAA). qRT-PCR was performed using primers specific for the indicated genes. Values represent the mean ± SEM; *P<0.05; **P<0.01, ***P<0.001
SAA expression is induced in AngII-treated apoE−/− mice
In addition to being a potent vasoactive peptide, AngII is known to elicit proinflammatory effects through activation of NF-κB30. Thus, it was of interest to determine whether AngII administration induces expression of acute phase SAA. Compared to saline-injected apoE−/− mice, hepatic SAA mRNA (Fig 2A) was increased ~26 fold (P < 0.001) and plasma SAA concentrations (Fig 2B) were elevated ~60 fold (P < 0.01) 24 hr after AngII injection (100 μg/kg). Chronic infusion of AngII (1,000 ng kg−1 min−1) by mini-osmotic pump for 28 days resulted in a near significant (P < 0.062) 17-fold increase in hepatic SAA mRNA abundance (Fig 2C) and a significant15-fold increase in plasma SAA concentration (Fig 2D). SAA mRNA expression was also significantly upregulated in abdominal aortas of SAAWT mice after 10 days of AngII infusion (Fig 2E), but not in the thoracic aorta of AngII-infused SAAWT mice (P = 0.083; Fig 2F). Taken together, our data indicate that systemic and vascular expression of acute phase SAA is elevated in AngII-administered apoE−/− mice.
Fig 2. AngII increases hepatic and abdominal aorta SAA mRNA abundance and plasma SAA concentrations in mice.
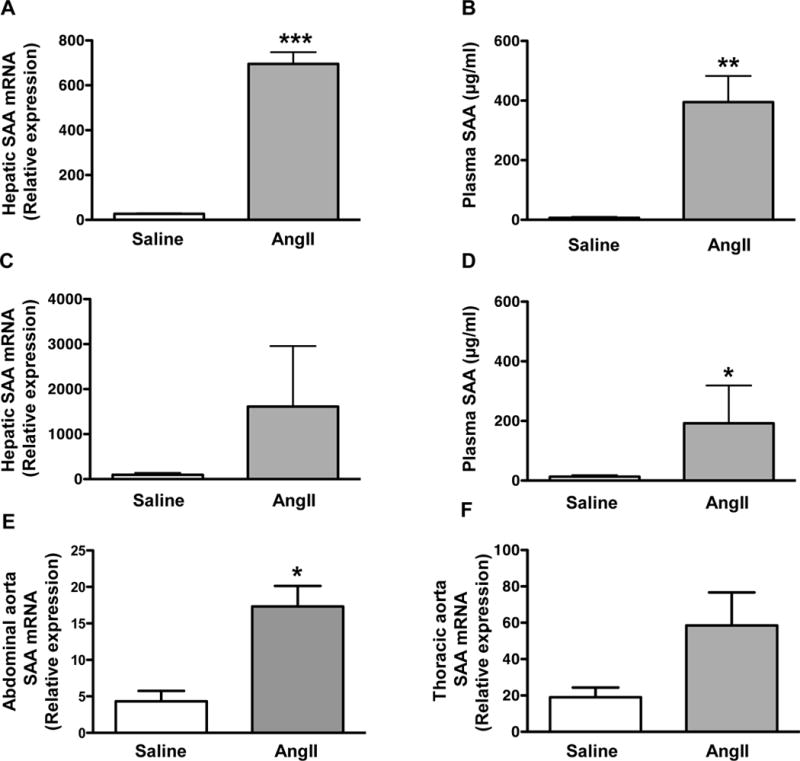
A) SAA mRNA abundance in livers of male apoE−/− mice 24 hours after i.p. injection of saline or 100 μg/kg AngII (n = 4). B) Plasma SAA concentrations in male apoE−/− mice 24 hours after i.p. injection of saline or 100 μg/kg AngII (n = 4). C) SAA mRNA abundance in livers of male apoE−/− mice after 28-day infusion of saline (n = 5) or 1,000 ng kg−1 min−1 AngII (n = 9). D) Plasma SAA concentrations in male apoE−/− mice after 28-day infusion of saline (n = 5) or 1,000 ng kg−1 min−1 AngII (n = 9). E) SAA mRNA abundance in abdominal aortas of male apoE−/− mice after 10-day infusion of saline (n = 4) or 1,000 ng kg−1 min−1 AngII (n = 7). F) SAA mRNA abundance in thoracic aortas of male apoE−/− mice after 10-day infusion of saline (n = 4) or 1,000 ng kg−1 min−1 AngII (n = 6). Values represent the mean ± SEM; *P<0.05, **P<0.01, ***P<0.001 for saline vs AngII.
SAA is present in AngII-induced AAA, co-localized with macrophages, elastin breaks, and matrix metalloproteinase activity
An early response to chronic AngII infusion in hypercholesterolemic mice is macrophage infiltration into the media of the abdominal aorta, particularly in regions of elastin degradation31. To investigate the possibility that SAA may be involved in these events, we performed immunohistochemical analyses throughout the entire length of AAA in apoE−/− mice (SAAWT) infused with AngII for 10 days. This time interval was chosen based on previous studies demonstrating significant tissue remodeling and inflammation after 10-day AngII infusion31, 32. As expected, aneurysmal tissue exhibited considerable heterogeneity along the length of the aorta (Fig 3A), with transmural breaks of elastin fibers evident in some (Fig 3D, asterisk) but not all (Fig 3C) sections. Regions containing elastin breaks were distinguished by intense MMP activity (green fluorescent staining by in situ zymography) and prominent macrophage and SAA immunoreactivity (red staining). In contrast, in regions with intact elastin lamina, MMP activity was primarily localized to acellular regions (i.e., thrombus) and macrophage and SAA immunostaining were relatively less pronounced. The specificity of SAA and macrophage immunoreactivity was confirmed by staining sections in the absence of primary antibodies (Supplemental Fig. I); the specificity of our in situ zymography experiments was confirmed by performing the staining procedure in the presence of an MMP inhibitor (Supplemental Fig. I). In our experience, ~30% of mice do not develop AAA during AngII infusion, consistent with published studies in male apoE−/− mice33. Analysis of the aorta from a non-responsive mouse (Fig 3B) demonstrated the presence of an intact elastin lamina throughout the length of the abdominal aorta, with minimal MMP activity, macrophage infiltration, or SAA immunostaining (Fig 3E). Importantly, SAA can also be detected by immunohistochemistry in human aortic tissue removed during surgical AAA repair (Supplemental Fig II).
Fig 3. SAA is present in AngII-induced AAA and co-localizes with macrophages, elastin breaks and MMP activity.
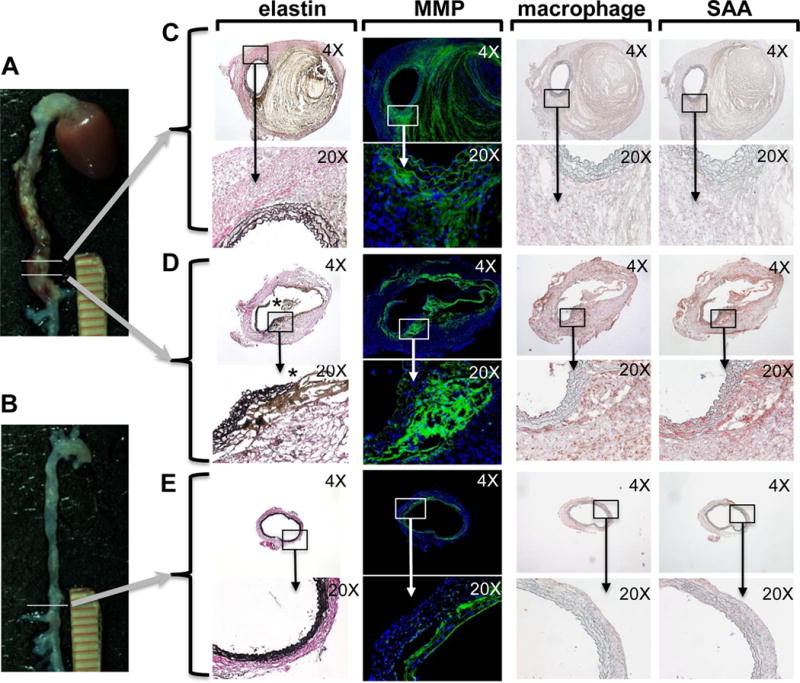
Male apoE−/− mice were infused with 1,000 ng kg−1 min−1 AngII for 10 days. A) Mouse that developed AAA and B) non-responsive mouse. Adjacent sections from two different regions of AAA (denoted by horizontal lines) were analyzed in parallel to allow direct comparisons. C, D) Sections were processed to detect elastin fibers by a modification of Verhoeffs staining (* denotes region with elastin breaks); MMP activity by in situ zymography (green fluorescence); and macrophages and SAA by immunostaining (red chromogen), and then photographed under 4× and 20× objective magnification as indicated. For in situ zymography, nuclei were identified using DAPI (blue fluorescence). E) Sections from a non-responsive mouse processed to detect elastin fibers, MMP activity and immunostaining for macrophages and SAA.
SAA deficiency protects mice from AngII-induced AAA
To investigate the role of SAA in the etiology of AngII-induced AAA, apoE−/− mice were bred with mice lacking both acute phase SAA isoforms, SAA1.1 and SAA2.124, 34. Both apoE−/− and apoE−/− mice lacking SAA (designated “SAAWT“ and “SAAKO”, respectively) were on the C57BL/6 background, bred normally, and appeared to be in good physical health prior to the study. Some mice died during the course of AngII infusion due to aortic rupture, consistent with previous reports31, 32. Although the incidence of rupture for mice infused with AngII for 28 days was similar for SAAWT (4 out 20 mice; 20%) and SAAKO (3 out of 20 mice; 15%), all of the ruptures in SAAKO mice occurred in the thoracic region, whereas 3 out of 4 ruptures were localized to the abdominal region of SAAWT mice (Supplemental Fig III). In subsequent studies whereby mice were infused with AngII for 10 days, we noted a trend for regional differences in aortic rupture in the two strains (see below). AAA progression was assessed in surviving mice by in vivo US and computer-assisted morphometric analyses ex vivo to determine the maximal luminal and external diameters of the abdominal aorta, respectively. Prior to AngII infusion, luminal diameters were not significantly different in SAAWT mice (1.30 ± 0.01 mm; n = 25) compared to SAAKO mice (1.29 ± 0.02 mm; n = 25). As expected, 28-day saline infusion did not significantly alter aortic luminal diameters in either strain (data not shown). Relative to baseline measures, AngII infusion produced a significantly greater increase in maximal diameters of abdominal aorta lumens of SAAWT mice (99.5 ± 12.4% increase) compared to SAAKO mice (44.5 ± 8.5% increase) (P<0.001; Fig 4A; representative US images shown below). Consistently, the maximal external diameter of the abdominal aorta increased significantly with AngII infusion in both genotypes, but was significantly greater for SAAWT mice (2.90 ± 0.30 mm) compared to SAAKO mice (1.53 ± 0.14 mm; P<0.001) after AngII infusion (Fig 4B; representative ex vivo images shown below). The overall incidence, calculated as the percent of total mice that died from aortic rupture plus the percent of total mice that developed AAA, (defined in surviving mice as ≥ 50% dilation of abdominal aorta lumen) was significantly increased in AngII-infused SAAWT mice (80%) compared to SAAKO mice (40%) (P < 0.05; Supplemental Fig III). Thus, both incidence and severity of AngII-induced AAAs were reduced in SAAKO compared to SAAWT mice.
Fig 4. Deficiency of SAA protects mice from AngII-induced AAA.
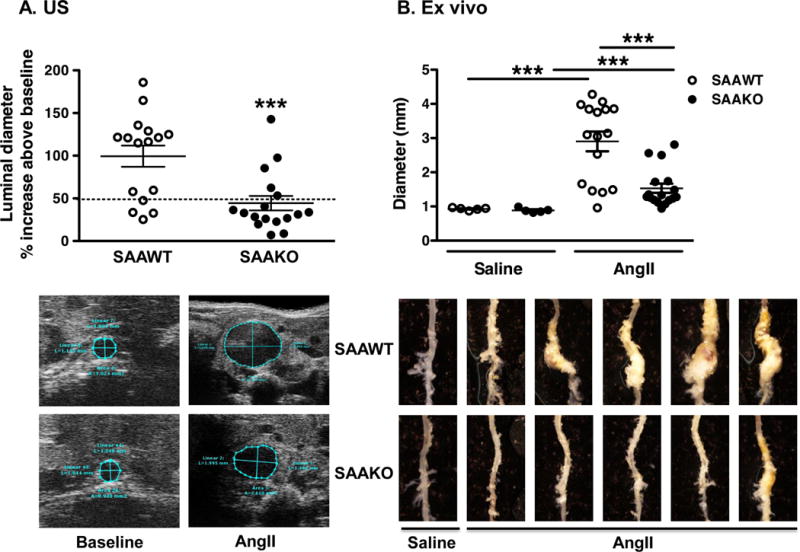
Male SAAWT and SAAKO mice were infused with saline (n = 5) or 1,000 ng kg−1 min−1 AngII (n = 20) for 28 days. A) Abdominal aortas were assessed by in vivo US before and after AngII infusion to determine the percent increase in luminal diameters in individual mice. Values indicating the presence of AAA, defined as a greater than 50% increase in diameter, are indicated above the horizontal line. Representative US images are shown below. B) AAAs were assessed ex vivo by computer-assisted morphometric analysis to determine maximal external diameters of abdominal aortas. Representative ex vivo images are shown below. Each symbol represents one animal; bars represent means ± SEM ***P<0.001
SAA deficiency does not result in a generally suppressed response to AngII infusion
Plasma renin concentrations were similar in SAAWT and SAAKO mice after AngII infusion (0.71 ± 0.06 and 0.73 ± 0.08 pg/ml/30min, respectively; n = 16–17), and significantly lower (P< 0.0001) for both genotypes than in saline-infused mice (3.30 ± 0.66 and 3.38 ± 0.78 pg/ml/30min respectively; n = 5) reflecting an appropriate AT1a receptor-dependent response to AngII in SAAKO mice35. Plasma IL-6 concentrations, a marker of systemic inflammation, were also similar in SAAWT (14.46 ± 4.34 pg/ml) and SAAKO mice (12.25 ± 3.06 pg/ml) after AngII infusion (P=0.682). AngII infusion elicited a hypertensive response in both SAAWT and SAAKO mice (Supplemental Fig IV). However, the mean systolic blood pressure was higher in SAAKO mice compared to SAAWT mice both at baseline (143.5 ± 1.9 mm Hg and 129.9 ± 3.4 mm Hg, respectively; n=24 – 25, P < 0.001) and after AngII infusion (166.4 ± 5.5 mm Hg and 146.9 ± 6.0 mm Hg, respectively; n =16–17, P < 0.001). While the reason for the unexpected differences in systolic blood pressures in the two strains is not known, it is evident that SAA deficiency does not result in a general decrease in responsiveness to AngII.
SAA deficiency does not alter AngII-induced aneurysm formation or atherosclerotic lipid deposition in the aortic arch region of apoE−/− mice
In addition to promoting AAA, subcutaneous infusion of AngII into hypercholesterolemic mice also leads to a significant dilation of the ascending aorta and augments atherosclerotic lipid accumulation36–38. To investigate whether SAA contributes to these AngII-induced vascular pathologies, we quantified the intimal surface area of the aortic arch region as an index of ascending AA36, 39, as well as the atherosclerotic lesion area in the aortic arch region of saline and AngII-infused SAAWT and SAAKO mice. Presence of aneurysms interfered with en face analyses of the thoracic and abdominal regions of AngII-infused mice. The intimal area of the aortic arch was similar in SAAWT and SAAKO mice after saline infusion (Supplemental Fig VA), and AngII infusion significantly increased this area to a similar extent in the two strains (P<0.001 for both genotypes). Atherosclerotic lesions in the aortic arch region were virtually undetectable in saline-infused mice, representing less than 1% of the intimal surface area in both SAAWT and SAAKO mice. AngII infusion resulted in a significant increase in percent lesion areas in both SAAWT and SAAKO mice (P< 0.001). Notably, percent lesion areas were similar for the two strains after AngII infusion (Supplemental Fig VB). Thus, SAA is not required for AngII-induced ascending AA or atherosclerosis in apoE−/− mice.
AngII-induced MMP-2 expression and activity are blunted in abdominal aortas of SAAKO mice
In an earlier study, we reported that 10-day AngII infusion results in significantly increased MMP-2, MMP-13, and MMP-14 mRNA abundance, and enhanced MMP-2 activity, in abdominal aortas of apoE−/− mice32. To investigate whether SAA mediates increased MMP expression in AngII-infused mice, aortas were collected from SAAWT and SAAKO mice after 10-day saline or AngII infusion for qRT-PCR and gel zymography. During the short-term AngII infusion, 5 SAAWT mice (~21%) died from aortic rupture (3 abdominal and 2 thoracic) and 5 SAAKO mice (~21%) died (2 abdominal and 3 thoracic ruptures). A subset of surviving mice was imaged by US 9 days after pump implantation to assess AAAs. Consistent with 28-day infusion studies described above, there were no significant differences in baseline measures of luminal diameters between SAAWT and SAAKO mice, and saline infusion did not cause significant luminal expansion in either strain. Relative to baseline values, maximal luminal diameters were increased to a greater extent in SAAWT (54.7 ± 14.4%, n = 12) compared to SAAKO (28.6 ± 5.1%, n = 8) mice, although the differences in percentage change between the strains was not statistically significant at this early stage of AAA development.
Thoracic and abdominal aortas were analyzed separately to investigate regional effects of SAA deficiency. SAA stimulates VSMC to secrete increased amounts of TGFβ19, a multifunctional cytokine believed to play complex roles in AAA and ascending AA development40. TGF-β mRNA abundance was significantly lower in both the abdominal and thoracic aortas of SAAKO compared to SAAWT mice after saline and AngII infusion (Fig. 5A, 5B). However, aortic TGF-β expression was not altered by 10-day AngII infusion in either strain. Compared to saline-infused mice, MMP-9 mRNA expression was not significantly altered in abdominal aortas of either SAAWT or SAAKO mice after AngII infusion (Fig 5C), whereas AngII infusion evoked a significant increase in MMP-13 and MMP-2 expression in both strains (Fig 5D, 5E). Notably, the induction of MMP-2 mRNA by AngII was significantly blunted in abdominal aortas of SAAKO mice compared to SAAWT mice (Fig 5E), but not thoracic aortas (Fig 5F). Consistent with this finding, results from gel zymography demonstrated significantly reduced abundance of the 70kDa (proMMP-2) and 58kDa (active MMP-2), but not the 64kDa (latent MMP-2) in abdominal aortas of SAAKO mice compared to SAAWT mice (Supplemental Fig VI, A–C), while the levels of these isoforms were similar in thoracic aortas of SAAWT and SAAKO mice (Supplemental Fig VI, D–E). MMP-2 activity was barely detectable in abdominal and thoracic aortas of SAAWT and SAAKO mice infused with saline (data not shown). Taken together, our data indicate that SAA mediates AAA in part by augmenting MMP-2 activity in abdominal aortas of AngII-infused mice.
Fig 5. Abdominal aortas, but not thoracic aortas, of AngII-infused SAAKO mice express less MMP-2.
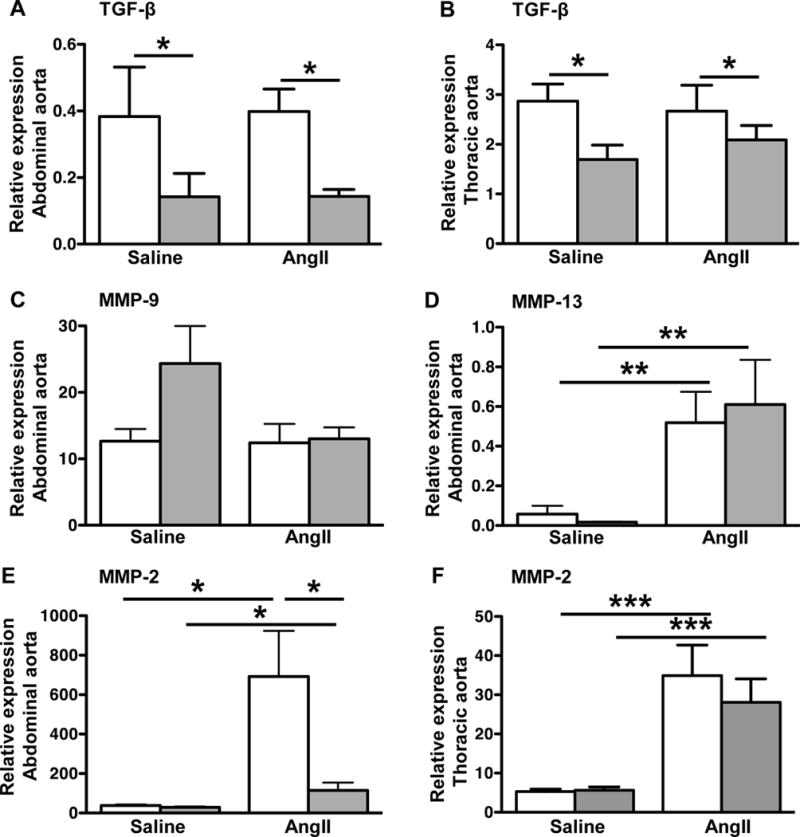
SAAWT and SAAKO mice were infused with saline or 1,000 ng kg−1 min−1 AngII for 10 days. Total RNA was extracted from abdominal and thoracic aortas of SAAWT mice (white bars) and SAAKO mice (grey bars) infused with saline (n = 4) or AngII (n = 7–8) and analyzed by qRT-PCR using primers specific for A and B) TGFβ; C) MMP-9; D) MMP-13; E and F) MMP-2. Values are the mean ± SEM *P<0.05, **P<0.01, ***P<0.001.
DISCUSSION
Understanding the mechanisms underlying AAA formation and expansion is critical for developing non-interventional treatments that inhibit their progression. Obtaining mechanistic insights through the study of human AAA is impeded by the unavailability of aortic tissue from early stages of the disease and the heterogeneous nature of human AAA tissue. Accordingly, researchers have largely relied on animal models to investigate potential factors mediating AAA initiation and progression14, 25. In this study, we utilized the extensively characterized AngII infusion model to investigate whether SAA, a circulating inflammatory mediator previously associated in humans with abdominal aortic diameter prior to aortic dilation10, mediates AAA. AAAs induced by AngII infusion exhibit many features of the human disease, including medial degeneration, inflammation, thrombus formation, and rupture25. Our results clearly show that deficiency of SAA reduces AngII-induced AAA in hypercholesterolemic mice. Both the negative feedback regulation of renin and the hypertensive response elicited by AngII were normal in SAAKO mice, indicating that reduced AAA in these mice was not due to a general loss in their ability to respond to AngII. Our findings have implications with regard to the potential utility of SAA as a biomarker and therapeutic target for AAA.
We demonstrate that one consequence of AngII administration in hyperlipidemic mice is an induction of SAA. Within 24 hours after bolus AngII injection, plasma SAA concentrations increased from almost negligible values at baseline to ~400 μg/ml. We also noted a statistically significant 15-fold increase in plasma SAA concentration in apoE−/− mice chronically infused with AngII. AngII is known to upregulate a number of inflammatory molecules including TNFα and IL-6 through an NF-κB dependent pathway30. Given abundant evidence that these classical inflammatory molecules potently induce SAA41, it is not surprising that AngII leads to increased SAA expression in mice. SAA levels correlated with plasma IL-6 in mice infused with AngII (r2 = 0.5215; data not shown), consistent with a common pathway for the induction of these two acute phase molecules. Moreover, plasma IL-6 concentrations were similar in SAAWT and SAAKO mice after AngII infusion, in accordance with our previous report that SAAKO mice mount a normal inflammatory response42. Notably, the extent of the increase in circulating SAA induced by AngII is similar to what occurs during a robust endotoxemia-induced acute phase response43, 44. The primary source of circulating SAA in mice after acute AngII injection is likely the liver. Hepatic SAA mRNA abundance was increased ~26-fold 24 hours after AngII injection, a time point that may be past the maximal effect of AngII treatment and thus underestimate the extent of gene induction.
In addition to eliciting AAA, AngII infusion also results in the formation of aneurysms in the ascending aortas of mice36. Ascending AAs in AngII-infused mice share many features in common with those that form in mice harboring a mutant allele of fibrillin-1 and are considered a model of Marfan syndrome36, 45. On the other hand, the pathology that develops in the ascending aorta is distinct from AngII-induced AAA, including the absence of thrombi, elastin fragmentation that is associated with intralaminar expansion rather than transmural degradation, macrophage accumulation throughout the medial layers of the ascending aorta rather than localized recruitment to regions of elastin degradation, and SMC hyperplasia rather than hypertrophy36, 46. As noted previously, the marked differences in the pathological characteristics of ascending AA and AAA suggest that distinct pathophysiological pathways may drive their development36, 39, 46. In the current study, SAA deficiency did not significantly impact the development of AngII-induced ascending AA, as assessed by measuring the intimal area of the aortic arch region. It should be noted, however, that the incidence of rupture in the thoracic region in our studies was somewhat more pronounced in SAAKO than SAAWT mice (13.9% vs 6.8% incidence, respectively, for thoracic ruptures; 4.7% and 14% incidence, respectively, for abdominal ruptures). While the incidence of rupture in this animal model is too low for definitive conclusions, it is evident that the effect of SAA deficiency on development of ascending AA versus AAA is distinct. As suggested previously36, 47, regional differences in the embryonic origin of aortic SMC may give rise to functional diversity, and hence the differential effect of SAA deficiency on AngII-induced ascending AA and AAA.
In accordance with previous reports48, 28-day AngII infusion produced a significant increase in atherosclerosis in SAAWT mice; SAA deficiency had no significant effect on atherosclerotic lesion area in either saline or AngII-infused mice. The differential effect of SAA deficiency on AngII-induced AAA versus AngII-induced atherosclerosis is in agreement with previous studies demonstrating that AAA and atherosclerosis represent rather distinct pathologies that are driven by separate mechanisms49–52. We previously reported that deficiency of SAA does not alter atherosclerotic lipid deposition in apoE−/− mice fed either a normal rodent diet for 50 weeks or an atherosclerotic diet for 12 weeks34. Thus, our loss-of-function studies in apoE−/− mice seemingly contradict a large body of epidemiological data associating plasma SAA concentrations with atherosclerotic cardiovascular disease7, 8, 53 as well as reports that increased SAA expression accelerates atherosclerosis in apoE−/− mice16,54. The lack of reduction of atherosclerosis in SAAKO mice does not negate a role for SAA in atherogenesis, but demonstrates it is not absolutely required. In the setting of severe hypercholesterolemia (such as apoE−/− mice), it seems likely there are redundant pathways to promote inflammation/atherogenesis in the absence of SAA. Furthermore, the very low levels of plasma SAA, in addition to the fact that lesions were virtually undetectable in aortas of saline-infused mice, may have precluded our ability to identify a protective effect of SAA deficiency in the current study. We cannot rule out the possibility that SAA deficiency exerts an effect on atherosclerosis during more prolonged AngII infusion. It will be important to determine in future studies whether SAA plays a role in more advanced atherosclerotic lesions, perhaps by modulating plaque stability.
We report positive SAA immunostaining in aneurysmal tissue of humans and AngII-infused mice. Whether positive SAA immunostaining in aneurysmal tissue reflects localized deposition of circulating SAA or expression by vascular cells, or both, requires further investigation. Although SAA is thought to be primarily expressed by the liver during an acute phase response, non-hepatic cells including epithelial cells, fibroblasts, endothelial cells, smooth muscle cells and monocytes/macrophages have all been shown to express SAA in response to inflammatory stimuli41, 55, 56. The cellular source(s) of SAA in our studies remain to be established, but our findings suggest that SAA produced at extra-hepatic sites such as vascular smooth muscle cells56, could play a role in AAA formation through paracrine pathways.
In our studies, SAA was detected in AAA, particularly in regions of elastin breaks and enhanced MMP activity. Our conclusion that SAA mediates AngII-induced AAA in part by promoting elastin degradation is supported by the finding that the induction of MMP-2 expression and activity by AngII is significantly blunted in abdominal aortas, but not thoracic aortas, of SAAKO mice. MMP activity is stringently regulated through multiple mechanisms, including the level of production and secretion of latent enzymes, the extent of activation of latent enzymes, and the relative concentration of specific inhibitors that block the activity of secreted MMPs. We determined that MMPs are transcriptionally regulated by purified mouse SAA1.1/2.1, confirming earlier reports utilizing a recombinant SAA20–22. Substantial data indicate that augmented MMP activity plays an important role in both human and experimental AAA14, 57, although the effectiveness of targeting MMPs for the treatment of human AAA is still under debate58. Doxycycline, a broad-spectrum inhibitor of MMPs, reduces the formation of AngII-induced AAA when given coincident to AngII infusion50, 59. However, according to a more recent report, treatment with doxycycline is not effective in reducing AAA growth or rupture in mice with established AAA60. Thus, the role of MMPs in the etiology of AngII-induced AAA may be particularly important in the early stages of development. The ability of SAA to evoke inflammatory responses, including the induction of chemokines and cytokines implicated in both human and experimental AAA, such as MCP-1 and TNF-α61, 62, may serve to propagate AAA in later stages of the disease.
The possibility that SAA promotes vascular degradation and AAA formation through mechanisms that are independent of MMPs cannot be ruled out. For example, SAA has been shown to form complexes with cystatin C63, an extracellular inhibitor of cysteine proteases, such as cathepsins, that have been implicated in AAA64. Whether SAA mediates AngII-induced pathologies through TGF-β dependent mechanisms merits consideration. TGF-β is a multifunctional cytokine that impacts a number of cellular processes, including cell growth, differentiation and migration, as well as remodeling of the extracellular matrix. Interestingly, TGF-β expression was attenuated in both thoracic and abdominal aortic segments of SAAKO mice compared to SAAWT mice, in accordance with an earlier report that SAA stimulates VSMC to secrete TGF-β19. Reduced TGF-β in SAAKO mice may impact aortic wall integrity, which could be manifested in complex ways under basal conditions and during chronic AngII infusion. For example, alterations in arterial stiffness related to reduced TGF-β expression could underlie differences in mean systolic blood pressure observed in SAAKO mice compared to SAAWT mice both before and after AngII treatment. In the current study, increased blood pressure was associated with reduced AAA in SAAKO mice. Although elevated blood pressure is a common feature in the well-established AAA model, AngII-induced AAA can develop independently of a hypertensive response65. Morphological and biochemical changes in the artery wall occur during AngII infusion66, including a 60% increase in collagen content, a 74% decrease in elastin content and breaks in the elastic lamina, all of which results in arterial stiffening; whether TGF-β modulates these responses is unclear. Accumulating experimental evidence points to a complex interaction between AngII and TGF-β and aortic aneurysm formation (reviewed in 40). Thus, the diverse effects of TGF-β in thoracic and abdominal aneurysms could underlie the increased thoracic, but not abdominal rupture, in AngII-infused SAAKO mice. This potential complex interaction between AngII, TGF-β, and SAA merits further investigation.
In summary, we have established that SAA mediates AAA in a widely used animal model of the disease. A key question that remains to be addressed is whether SAA’s main role is in the initiating events of AAA, the subsequent expansion phase, or all stages of AAA formation. Our data that SAA induces macrophages to express factors implicated in AAA suggests that SAA exacerbates AAA progression, given that macrophage infiltration into the abdominal aorta likely occurs after AAA initiation. Future studies will determine whether SAA serves as a useful biomarker for predicting future AAA expansion and thus identifies individuals who have high risk aneurysms and warrant more vigilant monitoring.
Supplementary Material
SIGNIFICANCE.
Approximately 5–10% of men and 1–2% of women 65–79 years old are living with an abdominal aortic aneurysm (AAA). Although most patients are asymptomatic, the risk of death due to rupture increases greatly as AAAs expand. Current clinical practice is to regularly monitor AAA size by ultrasound with referral for surgery if the growth rate or size exceeds established thresholds. Complicating AAA prognosis is the recognition that some AAAs remain stable, while others undergo rapid increases in diameter or rupture over a short period. Identifying circulating biomarkers that predict expansion would have a major impact on the clinical management of AAA. In this study we determined that purified mouse SAA stimulates VSMCs and macrophages to express MMPs and inflammatory cytokines implicated in AAA. We also demonstrate that apoE−/− mice lacking endogenous SAA are protected from experimental AAA. SAA may serve as a useful biomarker and therapeutic target for human AAA.
Acknowledgments
The authors thank Ms. Vicky English for plasma renin measurements and Drs Ming Gong and Zhongwen Xie for assistance with isolation of VSMC
SOURCES OF FUNDING:
This work was supported by National Institutes of Health Grant P01HL086670. The studies were supported with resources and facilities provided by the Lexington, Kentucky Veterans Affairs Medical Center.
Non-standard Abbreviations and Acronyms
- AngII
angiotensin II
- MMP
matrix metalloproteinase
- RT-PCR
reverse transcription polymerase chain reaction
- SAA
serum amyloid A
- SAAKO mice
apoE−/− mice deficient in SAA1.1 and SAA2.1
- SAAWT mice
apoE−/− mice expressing SAA1.1 and SAA2.1
- US
ultrasound
- SMC
smooth muscle cells
Footnotes
DISCLOSURES:
None.
References
- 1.Sipe J. Revised nomenclature for serum amyloid A (SAA). Nomenclature Committee of the International Society of Amyloidosis. Part 2. Amyloid. 1999;6:67–70. doi: 10.3109/13506129908993291. [DOI] [PubMed] [Google Scholar]
- 2.Wong M, Toh L, Wilson A, Rowley K, Karschimkus C, Prior D, Romas E, Clemens L, Dragicevic G, Harianto H, Wicks I, McColl G, Best J, Jenkins A. Reduced arterial elasticity in rheumatoid arthritis and the relationship to vascular disease risk factors and inflammation. Arthritis Rheum. 2003;48:81–89. doi: 10.1002/art.10748. [DOI] [PubMed] [Google Scholar]
- 3.Leinonen E, Hurt-Camejo E, Wiklund O, Hulten LM, Hiukka A, Taskinen MR. Insulin resistance and adiposity correlate with acute-phase reaction and soluble cell adhesion molecules in type 2 diabetes. Atherosclerosis. 2003;166:387–394. doi: 10.1016/s0021-9150(02)00371-4. [DOI] [PubMed] [Google Scholar]
- 4.Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, Snitker S, Horenstein RB, Hull K, Goldberg NH, Goldberg AP, Shuldiner AR, Fried SK, Gong DW. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006;3:e287. doi: 10.1371/journal.pmed.0030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebeling P, Teppo AM, Koistinen HA, Viikari J, Ronnemaa T, Nissen M, Bergkulla S, Salmela P, Saltevo J, Koivisto VA. Troglitazone reduces hyperglycaemia and selectively acute-phase serum proteins in patients with Type II diabetes. Diabetologia. 1999;42:1433–1438. doi: 10.1007/s001250051315. [DOI] [PubMed] [Google Scholar]
- 6.Bergmann S, Siekmeier R. Influence of smoking and body weight on adipokines in middle aged women. Eur J Med Res. 2009;14(Suppl 4):21–26. doi: 10.1186/2047-783X-14-S4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosuge M, Ebina T, Ishikawa T, Hibi K, Tsukahara K, Okuda J, Iwahashi N, Ozaki H, Yano H, Kusama I, Nakati T, Umemura S, Kimura K. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ J. 2007;71:186–190. doi: 10.1253/circj.71.186. [DOI] [PubMed] [Google Scholar]
- 8.Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, Pepine CJ, Sharaf B, Bairey Merz CN, Sopko G, Olson MB, Reis SE. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE) Circulation. 2004;109:726–732. doi: 10.1161/01.CIR.0000115516.54550.B1. [DOI] [PubMed] [Google Scholar]
- 9.Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, Sutsch G, Roffi M, Neidhart M, Eberli FR, Tanner FC, Gobbi S, von Eckardstein A, Luscher TF. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation. 2005;111:1355–1361. doi: 10.1161/01.CIR.0000158479.58589.0A. [DOI] [PubMed] [Google Scholar]
- 10.Rohde LE, Arroyo LH, Rifai N, Creager MA, Libby P, Ridker PM, Lee RT. Plasma concentrations of interleukin-6 and abdominal aortic diameter among subjects without aortic dilatation. Arterioscler Thromb Vasc Biol. 1999;19:1695–1699. doi: 10.1161/01.atv.19.7.1695. [DOI] [PubMed] [Google Scholar]
- 11.Lederle FA, Johnson GR, Wilson SE. Abdominal aortic aneurysm in women. J Vasc Surg. 2001;34:122–126. doi: 10.1067/mva.2001.115275. [DOI] [PubMed] [Google Scholar]
- 12.Golledge J, Muller J, Daugherty A, Norman P. Abdominal aortic aneurysm: pathogenesis and implications for management. Arterioscler Thromb Vasc Biol. 2006;26:2605–2613. doi: 10.1161/01.ATV.0000245819.32762.cb. [DOI] [PubMed] [Google Scholar]
- 13.Golledge J, Norman PE. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis. 2011;217:57–63. doi: 10.1016/j.atherosclerosis.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Novel mechanisms of abdominal aortic aneurysms. Curr Atheroscler Rep. 2012;14:402–412. doi: 10.1007/s11883-012-0271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HY, Kim SD, Shim JW, Lee SY, Lee H, Cho KH, Yun J, Bae YS. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J Immunol. 2008;181:4332–4339. doi: 10.4049/jimmunol.181.6.4332. [DOI] [PubMed] [Google Scholar]
- 16.Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, Zhang Y, Zhang C, An F. Serum amyloid a directly accelerates the progression of atherosclerosis in apolipoprotein e-deficient mice. Mol Med. 2011;17:1357–1364. doi: 10.2119/molmed.2011.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song C, Hsu K, Yamen E, Yan W, Fock J, Witting PK, Geczy CL, Freedman SB. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis. 2009;207:374–383. doi: 10.1016/j.atherosclerosis.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Furlaneto CJ, Campa A. A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun. 2000;268:405–408. doi: 10.1006/bbrc.2000.2143. [DOI] [PubMed] [Google Scholar]
- 19.Wilson PG, Thompson JC, Webb NR, de Beer FC, King VL, Tannock LR. Serum amyloid A, but not C-reactive protein, stimulates vascular proteoglycan synthesis in a pro-atherogenic manner. Am J Pathol. 2008;173:1902–1910. doi: 10.2353/ajpath.2008.080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Connolly M, Mullan RH, McCormick J, Matthews C, Sullivan O, Kennedy A, FitzGerald O, Poole AR, Bresnihan B, Veale DJ, Fearon U. Acute-phase serum amyloid A regulates tumor necrosis factor alpha and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012;64:1035–1045. doi: 10.1002/art.33455. [DOI] [PubMed] [Google Scholar]
- 21.Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, Eguchi K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest. 1998;78:535–539. [PubMed] [Google Scholar]
- 22.Mullan RH, Bresnihan B, Golden-Mason L, Markham T, O’Hara R, FitzGerald O, Veale DJ, Fearon U. Acute-phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF-kappaB-dependent signal transduction pathway. Arthritis Rheum. 2006;54:105–114. doi: 10.1002/art.21518. [DOI] [PubMed] [Google Scholar]
- 23.Lee HY, Kim MK, Park KS, Bae YH, Yun J, Park JI, Kwak JY, Bae YS. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem Biophys Res Commun. 2005;330:989–998. doi: 10.1016/j.bbrc.2005.03.069. [DOI] [PubMed] [Google Scholar]
- 24.de Beer MC, Webb NR, Wroblewski JM, Noffsinger VP, Rateri DL, Ji A, van der Westhuyzen DR, de Beer FC. Impact of serum amyloid A on high density lipoprotein composition and levels. J Lipid Res. 2010;51:3117–3125. doi: 10.1194/jlr.M005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 26.Badolato R, Johnston JA, Wang JM, McVicar D, Xu LL, Oppenheim JJ, Kelvin DJ. Serum amyloid A induces calcium mobilization and chemotaxis of human monocytes by activating a pertussis toxin-sensitive signaling pathway. J Immunol. 1995;155:4004–4010. [PubMed] [Google Scholar]
- 27.Koga T, Torigoshi T, Motokawa S, Miyashita T, Maeda Y, Nakamura M, Komori A, Aiba Y, Uemura T, Yatsuhashi H, Ishibashi H, Eguchi K, Migita K. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008;582:579–585. doi: 10.1016/j.febslet.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Christenson K, Bjorkman L, Ahlin S, Olsson M, Sjoholm K, Karlsson A, Bylund J. Endogenous Acute Phase Serum Amyloid A Lacks Pro-Inflammatory Activity, Contrasting the Two Recombinant Variants That Activate Human Neutrophils through Different Receptors. Front Immunol. 2013;4:92. doi: 10.3389/fimmu.2013.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MH, de Beer MC, Wroblewski JM, Webb NR, de Beer FC. SAA does not induce cytokine production in physiological conditions. Cytokine. 2013;61:506–512. doi: 10.1016/j.cyto.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Saraff K, Babamusta F, Cassis L, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 32.Zack M, Boyanovsky BB, Shridas P, Bailey W, Forrest K, Howatt DA, Gelb MH, de Beer FC, Daugherty A, Webb NR. Group X secretory phospholipase A(2) augments angiotensin II-induced inflammatory responses and abdominal aortic aneurysm formation in apoE-deficient mice. Atherosclerosis. 2011;214:58–64. doi: 10.1016/j.atherosclerosis.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manning MW, Cassis LA, Huang J, Szilvassy SJ, Daugherty A. Abdominal aortic aneurysms: fresh insights from a novel animal model of the disease. Vasc Med. 2002;7:45–54. doi: 10.1191/1358863x02vm413ra. [DOI] [PubMed] [Google Scholar]
- 34.De Beer MC, Wroblewski JM, Noffsinger VP, Rateri DL, Howatt DA, Balakrishnan A, Ji A, Shridas P, Thompson JC, van der Westhuyzen DR, Tannock LR, Daugherty A, Webb NR, De Beer FC. Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2014;34:255–261. doi: 10.1161/ATVBAHA.113.302247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, Cassis LA. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension. 2012;60:1524–1530. doi: 10.1161/HYPERTENSIONAHA.112.192690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daugherty A, Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor −/− mice. Annals Of The New York Academy Of Sciences. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 38.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E −/− mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP, 3rd, Howatt DA, Subramanian V, Poduri A, Charnigo R, Cassis LA, Daugherty A. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor−/− mice. Circ Res. 2011;108:574–581. doi: 10.1161/CIRCRESAHA.110.222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Lu H, Rateri DL, Cassis LA, Daugherty A. Conundrum of angiotensin II and TGF-beta interactions in aortic aneurysms. Curr Opin Pharmacol. 2013;13:180–185. doi: 10.1016/j.coph.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr Opin Hematol. 2000;7:64–69. doi: 10.1097/00062752-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 42.de Beer MC, Wroblewski JM, Noffsinger VP, Ji A, Meyer JM, van der Westhuyzen DR, de Beer FC, Webb NR. The Impairment of Macrophage-to-Feces Reverse Cholesterol Transport during Inflammation Does Not Depend on Serum Amyloid A. J Lipids. 2013;2013:283486. doi: 10.1155/2013/283486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb NR, de Beer MC, van der Westhuyzen DR, Kindy MS, Banka CL, Tsukamoto K, Rader DL, de Beer FC. Adenoviral vector-mediated overexpression of serum amyloid A in apoA-I-deficient mice. J Lipid Res. 1997;38:1583–1590. [PubMed] [Google Scholar]
- 44.Hosoai H, Webb NR, Glick JM, Tietge UJ, Purdom MS, de Beer FC, Rader DJ. Expression of serum amyloid A protein in the absence of the acute phase response does not reduce HDL cholesterol or apoA-I levels in human apoA-I transgenic mice. J Lipid Res. 1999;40:648–653. [PubMed] [Google Scholar]
- 45.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owens AP, 3rd, Subramanian V, Moorleghen JJ, Guo Z, McNamara CA, Cassis LA, Daugherty A. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differentiation 3. Circ Res. 2010;106:611–619. doi: 10.1161/CIRCRESAHA.109.212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 48.Daugherty A, Cassis L. Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc Med. 2004;14:117–120. doi: 10.1016/j.tcm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 49.Suen RS, Rampersad SN, Stewart DJ, Courtman DW. Differential roles of endothelin-1 in angiotensin II-induced atherosclerosis and aortic aneurysms in apolipoprotein E-null mice. Am J Pathol. 2011;179:1549–1559. doi: 10.1016/j.ajpath.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23:483–488. doi: 10.1161/01.ATV.0000058404.92759.32. [DOI] [PubMed] [Google Scholar]
- 51.Wang S, Subramanian V, Lu H, Howatt DA, Moorleghen JJ, Charnigo R, Cassis LA, Daugherty A. Deficiency of receptor-associated protein attenuates angiotensin II-induced atherosclerosis in hypercholesterolemic mice without influencing abdominal aortic aneurysms. Atherosclerosis. 2012;220:375–380. doi: 10.1016/j.atherosclerosis.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang JA, Chen WA, Wang Y, Zhang S, Bi H, Hong B, Luo Y, Daugherty A, Xie X. Statins exert differential effects on angiotensin II-induced atherosclerosis, but no benefit for abdominal aortic aneurysms. Atherosclerosis. 2011;217:90–96. doi: 10.1016/j.atherosclerosis.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 54.Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, Tannock LR. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J Lipid Res. 2014 doi: 10.1194/jlr.M054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steel DM, Donoghue FC, O’Neill RM, Uhlar CM, Whitehead AS. Expression and regulation of constitutive and acute phase serum amyloid A mRNAs in hepatic and non-hepatic cell lines. Scand J Immunol. 1996;44:493–500. doi: 10.1046/j.1365-3083.1996.d01-341.x. [DOI] [PubMed] [Google Scholar]
- 56.Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci U S A. 1994;91:3186–3190. doi: 10.1073/pnas.91.8.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rizas KD, Ippagunta N, Tilson MD., 3rd Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. 2009;17:201–210. doi: 10.1097/CRD.0b013e3181b04698. [DOI] [PubMed] [Google Scholar]
- 58.Dodd BR, Spence RA. Doxycycline inhibition of abdominal aortic aneurysm growth: a systematic review of the literature. Curr Vasc Pharmacol. 2011;9:471–478. doi: 10.2174/157016111796197288. [DOI] [PubMed] [Google Scholar]
- 59.Turner GH, Olzinski AR, Bernard RE, Aravindhan K, Karr HW, Mirabile RC, Willette RN, Gough PJ, Jucker BM. In vivo serial assessment of aortic aneurysm formation in apolipoprotein E-deficient mice via MRI. Circ Cardiovasc Imaging. 2008;1:220–226. doi: 10.1161/CIRCIMAGING.108.787358. [DOI] [PubMed] [Google Scholar]
- 60.Xie X, Lu H, Moorleghen JJ, Howatt DA, Rateri DL, Cassis LA, Daugherty A. Doxycycline does not influence established abdominal aortic aneurysms in angiotensin II-infused mice. PLoS One. 2012;7:e46411. doi: 10.1371/journal.pone.0046411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Juvonen J, Surcel HM, Satta J, Teppo AM, Bloigu A, Syrjala H, Airaksinen J, Leinonen M, Saikku P, Juvonen T. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 1997;17:2843–2847. doi: 10.1161/01.atv.17.11.2843. [DOI] [PubMed] [Google Scholar]
- 62.Middleton RK, Lloyd GM, Bown MJ, Cooper NJ, London NJ, Sayers RD. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. 2007;45:574–580. doi: 10.1016/j.jvs.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 63.Spodzieja M, Rafalik M, Szymanska A, Kolodziejczyk AS, Czaplewska P. Interaction of serum amyloid A with human cystatin C–assessment of amino acid residues crucial for hCC-SAA formation (part II) J Mol Recognit. 2013;26:415–425. doi: 10.1002/jmr.2283. [DOI] [PubMed] [Google Scholar]
- 64.Qin Y, Cao X, Yang Y, Shi GP. Cysteine protease cathepsins and matrix metalloproteinases in the development of abdominal aortic aneurysms. Future Cardiol. 2013;9:89–103. doi: 10.2217/fca.12.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tham DM, Martin-McNulty B, Wang YX, Da Cunha V, Wilson DW, Athanassious CN, Powers AF, Sullivan ME, Rutledge JC. Angiotensin II injures the arterial wall causing increased aortic stiffening in apolipoprotein E-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1442–1449. doi: 10.1152/ajpregu.00295.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.