

Abstract
Retinoids, which are vitamin A derivatives, interact through retinoic acid receptors (RARs) and retinoid X receptors (RXRs) and have profound effects on several physiological and pathological processes in the brain. The presence of retinoic acid signaling is extensively detected in the adult central nervous system, including the amygdala, cortex, hypothalamus, hippocampus, and other brain areas. Retinoids are primarily involved in neural patterning, differentiation, and axon outgrowth. Retinoids also play a key role in the preservation of the differentiated state of adult neurons. Impairment in retinoic acid signaling can result in neurodegeneration and progression of Alzheimer’s disease (AD). Recent studies demonstrated severe deficiencies in spatial learning and memory in mice during retinoic acid (vitamin A) deprivation indicating its significance in preserving memory function. Defective cholinergic neurotransmission plays an important role in cognitive deficits in AD. All-trans retinoic acid is known to enhance the expression and activity of choline acetyltransferase in neuronal cell lines. Activation of RAR and RXR is also known to impede the pathogenesis of AD in mice by inhibiting accumulation of amyloids. In addition, retinoids have been shown to inhibit the expression of chemokines and pro-inflammatory cytokines in microglia and astrocytes, which are activated in AD. In this review article, we have described the chemistry and molecular signaling mechanisms of natural and synthetic retinoids and current understandings of their therapeutic potentials in prevention of AD pathology.
Keywords: Alzheimer’s disease, amyloid-β, neuroprotection, regenerative medicine, retinoids
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia that generally appears in humans after age 65. AD is characterized by the pathological accumulation of cerebral amyloid plaques and prominent neurofibrillary tangles in medial temporal lobe structures and also by the loss of neurons and white matter, inflammation, and oxidative damage. The amyloid plaques and neurofibrillary tangles are composed of aggregated amyloid-β (Aβ) peptide and tau protein, respectively [1–4].
Aβ peptide is a natural metabolic product comprised of 36 to 43 amino acids, although Aβ peptides containing 40 and 42 amino acids (Aβ40 and Aβ42) are produced predominately. Aβ peptides originate from proteolysis of amyloid-β precursor protein (AβPP) by the sequential enzymatic actions of β-site amyloid precursor protein cleaving enzyme 1 (BACE-1), β-secretase and γ-secretase, a protein complex with presenilin 1 (PS1) at its catalytic core [1, 3, 5, 6]. Monomers of Aβ40 are much more ubiquitous than the aggregation-prone and detrimental Aβ42 peptides. Thus, deregulation in synthesis and clearance leading to accumulation of aggregated Aβ42 peptides is thought to be the initiating factor in AD.
Recently, Hample and co-workers have shown that chronic inflammation and deregulated lipid homeostasis can lead to AD pathogenesis[7]. Activated microglia and reactive astrocytes were observed to localize near fibrillar plaques in the brains of AD patients [8]. Chronically activated microglia can release chemokines and also cytokines such as interleukin-1 (IL-1), IL-6, and tumor necrosis factor-alpha (TNF-α) [9] and activate the complement system [10]. A recent investigation indicated that high serum cholesterol levels could play a significant role in AβPP processing and Aβ metabolism [11]. Alterations in expression of the genes responsible for cholesterol homeostasis, including a polipoprotein E (APOE), ATP-binding cassette sub-family A member 1 (ABCA1),low density lipoprotein receptor-related protein 1 (LRP1), 24S-cholesterol hydroxylase (CYP46), acyl-coenzyme A cholesterol acyltransferase (ACAT), and liver X receptor-β (LXRβ) are risk factors in AD [12]. However, a genetic defect in glial-derived cholesterol transporter APOE is the major determinant of the risk for late-onset AD [13]. Current AD therapies, including acetylcholinesterase inhibitors and N-methyl-D-aspartate (NMDA) antagonists, possess limited scope within this multifactorial disease and do not target the Aβ aggregation and deposition that are central to AD pathogenesis. Thus, additional investigations are urgently needed to discover more potent drugs that can target multiple pathophysiological pathways to prevent progression of AD.
Retinoids, which are natural and synthetic derivatives of vitamin A, play significant roles in brain development in adult vertebrates. The majority of these functions are performed by the vitamin A metabolite retinoic acid (RA), which binds to receptors of the nuclear receptor superfamily and regulates gene expression [14]. Retinoids modulate the expression of many genes that codes for enzymes, neurotransmitter transporters and receptors, transcription factors, cell surface receptors, and neuropeptide hormones [15]. Retinoids exert effects on gene transcription through interaction with retinoid receptors such as retinoic acid receptors (RARβ, β, and γ) and retinoid X receptors (RXRα, β, and γ) that are primarily concentrated in amygdala, pre-frontal cortex, and hippocampal areas in the brain [16]. Mutation in RARβ and/or RXRγ genes or deficiency in retinoid can inhibit spatial learning and memory and also can promote depression in animals. An earlier study reported deposition of Aβ peptide in the cerebral vessels due to suppression of RARα expression in vitamin A-deprived rats [17]. Retinoids also play a crucial role in neuroprotection by inhibiting inflammatory responses [18]. Many recent studies demonstrated that retinoids could downregulate the microglial expression of cytokines and inflammatory molecules [19]. In addition, altered central cholinergic neurotransmission is one of the crucial factors in AD pathology. Retinoid receptor agonists could augment cholinergic transmission by increasing choline acetyl transferase (ChAT) expression and vesicular acetylcholine transporter gene expression [20].
In this review article, we have described potential roles of natural and synthetic retinoids for functional neuroprotection in AD; more specifically, a number of areas within the neurodegenerative field are described, including the role of retinoids in modulating Aβ aggregation, different RAR signaling pathways, inflammation, oxidative stress, mitochondrial dysfunction, neurotransmission, and regeneration with stem cells as well as development of potential therapeutic agents for regenerative medicine. Our aim in the current review article is promotion of fresh and novel research ideas that will enable the design of new therapeutic strategies to answer the critical questions associated with AD pathology and discover potentials of retinoids as new therapeutic agents to study the pathophysiology of AD and also treatments for AD patients in the near future.
Aβ AGGREGATION IN AD
Aggregation of Aβ and its subsequent deposition as extracellular amyloid plaques in the brain is a major hallmark of AD [21]. Aggregation of Aβ is a nucleation-dependent self-assembly process that begins with monomeric protein, involves transient intermediates, and concludes with the formation of Aβ fibrils, which deposit as plaques in the brain [1, 3, 4, 22]. Aβ fibrils exhibit a cross β-sheet structure in which the β-strands are oriented perpendicular to and hydrogen bonds oriented parallel to the long axis of the aggregate [23], yet Aβ aggregates that form along the aggregation pathway do display diversity within their biophysical properties [24]. Oligomers and protofibrils are the key intermediate species during the aggregation process and possess higher toxicity than mature fibrils [4, 25]. As a result, revisions to the ‘amyloid cascade hypothesis’ indicate a significant role for these soluble aggregates in the pathogenesis of AD. Due to alternative processing or mutations in AβPP, different variants of Aβ peptides may be produced [4]. Several mutations are associated with familial AD, including mutations that increase production of Aβ or the relative amount of Aβ42, which more readily forms soluble oligomeric species, as well as mutations that alter the aggregation of Aβ to lead to the formation of more soluble aggregates or altered distributions of soluble Aβ oligomers [4, 25, 26].
Studies have described soluble Aβ oligomers, Aβ-derived diffusible ligands, globulomers, large fatty acid-derived oligomers, protofibrils of varying shape, and fibrils that vary in the number and orientation of protofilaments [25, 27–32]. Consistent with in vitro aggregation studies, several distinct Aβ aggregates have been extracted from human AD brains by differential ultracentrifugation using different solvents [4, 29]. Ultracentrifugation of homogenized AD brain tissues in Tris-buffered saline (TBS)-sucrose buffer at 14,000×g cannot separate soluble and dispersible fraction [33]. When the supernatant is further ultracentrifuged at 175,000×g, soluble Aβ remains in the supernatant fraction, and the pellet contains insoluble Aβ aggregates. Membrane-associated Aβ and plaque-associated Aβ remain in the pellet following 14,000×g centrifugation of homogenized AD brain tissues in TBS-sucrose buffer, and can be extracted with 2% sodium dodecyl sulfate (SDS) or Triton X [4, 33, 34]. Membrane-associated Aβ remains in the supernatant and plaque-associated Aβ resides in the pellet that can be extracted by 70% formic acid treatment [4, 29, 33]. Among these forms, Rijal Upadhaya and co-workers used a transgenic model of AD to demonstrate that increases in soluble Aβ and dispersible Aβ correlated with AD progression [4, 33]. Further investigations are needed to confirm the roles of various Aβ aggregates in the human AD brain.
SYNTHESIS, CHEMISTRY, AND ACTIVITY OF RETINOIDS
Vitamin A (all-trans-retinol) is considered to be the most multifunctional vitamin, regulating many biological processes such as embryonic development, cell differentiation, cell growth, and apoptosis as well as brain function [35]. It is generally obtained as pro-vitamin A carotenoids from many colorful fruits and vegetables or animal sources like liver, egg yolks, or dairy products. Carotenoids are then converted into vitamin A within the small intestine. Many photosynthetic plants, bacteria, and some fungi can biosynthesize pro-vitamin A carotenoids, but animals must ingest them through dietary supplements. Epidemiological studies indicate that higher dietary intake of pro-vitamin A carotenoids can decrease the risk of many diseases. Vitamin A also effectively increases visual tuning and prevents macular degeneration. The carotenoid precursors and natural retinoids are composed of relatively long chain conjugated polyene structures (Fig. 1).
Fig. 1.

Synthesis of retinoic acid isomers from natural sources.
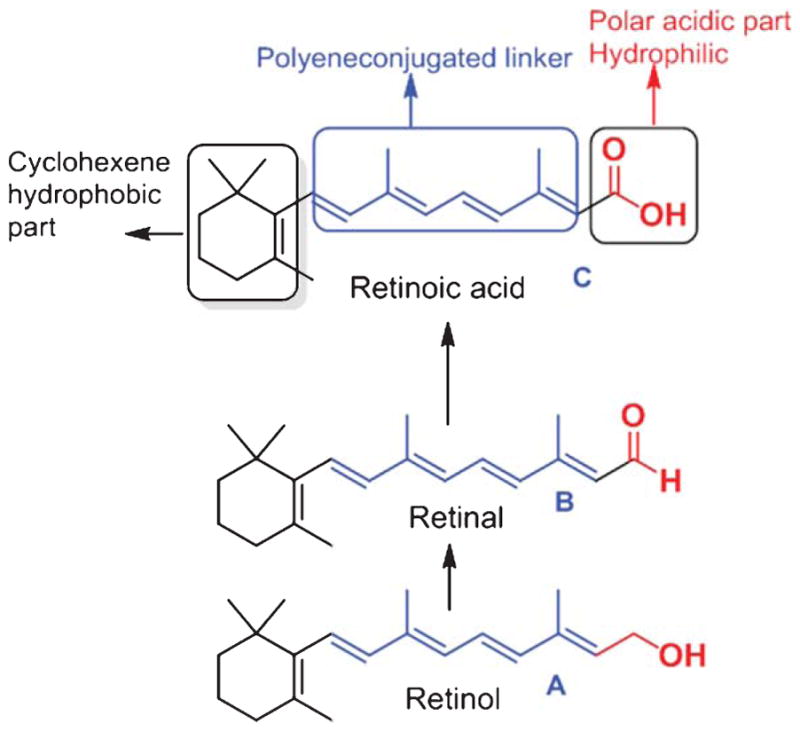
Retinoids contain a group of compounds related to vitamin A, including its natural and synthetic analogs, which have four isoprenoid units joined in a head-to-tail fashion. The basic structure of a retinoid, as shown (Fig. 2), consists of three parts: a trimethylated cyclohexene ring that is a bulky hydrophobic group, a conjugated tetraene side chain that serves as a linker unit, and a polar carbon-oxygen functional group, typically carboxylic acid. Retinoids are unstable due to the presence of conjugated double bonds that easily undergo oxidation and/or isomerization in the presence of oxidants, light or excessive heat. Retinoids that contain alcoholic and carboxylic groups are soluble in methanol and ethanol, whereas the esterified long-chain fatty acid is only slightly soluble in alcohol but highly soluble in hexane.
Fig. 2.
The basic structure of retinoids.
Animals are unable to synthesize vitamin A de novo and therefore they must obtain it through the diets. Dietary intake in the form of retinol, retinyl ester, or β-carotene represents good sources of vitamin A (Fig. 1). Carotenoids, the main precursor of fat-soluble vitamin A, including α/β-carotene are converted into retinal or apocarotenoids and subsequently to retinoids [36]. Unlike most other biological compounds, carotenoids and retinoids have conjugated polyene systems that absorb light in the visible and ultraviolet spectrums around 450 nm and within the range of 325–380 nm, respectively [37]. Colorimetric methods are commonly used for the evaluation of vitamin A and carotenoids.
Many studies have used the Wittig reaction [38] to synthesize retinyl acetate and the ethyl ester of RA. However, the Wittig reaction predominately synthesizes the cis isomer whereas trans olefin linkages are most frequently observed in the natural retinoids. Over the last few decades many modifications and alternatives to the Wittig olefination have been invented. In the Horner Wadsworth Emmons (HWE) modification, replacement of the phosphonium salts with phosphonate esters produced trans or E-olefin. Two groups of investigators [39, 40] employed similar olefination techniques in retinoid syntheses. Recently, researchers have shown that fully functional retinoid receptor agonists can be developed without the classic extended polyene chain. Due to high toxicity profile and off-target binding of ATRA and cis retinoic acid (which are also metabolized by many cytoplasmic enzymes such as Cyp26, isomerases, and others), their full potential as novel pharmacological agents has not been well exploited. To overcome these problems, the synthetic chemistry community has developed many synthetic retinoids [41–43] using SAR (Structure Activity Relationship) analysis and computational modeling.
Thus, the range of potent synthetic retinoid structures from experimental synthesis has expanded dramatically (Fig. 3). A broadened range of structures and chemistry are being used to make analogs that now exist for the synthesis of molecules called retinoids [44]. Recently, many excellent reviews have been published highlighting several natural and synthetic retinoids in the drug discovery process [43, 45]. Several major retinoid-based clinical agents are shown (Fig. 3). Stephens-Jarnagin and co-workers developed a polycyclic structure 4-[(1E)-2-(5, 5, 8, 8-tetramethyl- 5, 6, 7, 8-tetrahydro-2-naphthalenyl)-1-propen-1-yl] benzoic acid (TTNPB) (structure 14, Fig. 3) that is a highly potent retinoid receptor agonist due to its metabolism resistance and high affinity for RARs [46]. The detailed synthetic scheme of TTNPB is also described (Fig. 4). Also, we have described schemes of synthetic retinoids (Figs. 4–10) that can be used as pharmacological agents for the treatment of AD and other diseases.
Fig. 3.

Synthetic retinoids.
Fig. 4.

Synthetic scheme of TTNPB.
Fig. 10.

Synthetic scheme of EC23.
RA SIGNALING PATHWAYS IN AD
Many earlier investigations demonstrated direct correlation of the impaired RA signaling with vitamin A deficiency in vivo. Suppression of RARα and ChAT expression as well as accumulation of Aβ peptide in rat forebrain cortical neurons, all of which are considered to be hallmarks of AD, were observed in vitamin A-deprived conditions [47]. Vitamin A-deprived rats exhibited declined levels of RARβ, AβPP695, BACE, and AβPP-CTF in brain and in the cerebral cortex, whereas RA administration could restore their expression [48].
Many studies involving genetic analysis of AD have confirmed direct correlation between the genes that encode molecules involved in the RA signaling pathway and those that are considered to be involved in the pathogenesis of AD [15]. Altered expression of RARγ, several retinol dehydrogenases, RBP4, and two CYP26 genes are frequently observed in AD. In particular, RBP has been observed at high concentration in amyloid plaques [49]. A few earlier investigations also demonstrated that AβPP could be regulated by retinoids [50, 51]. After alternative splicing of AβPP, three major transcripts (AβPP770, AβPP751, and AβPP695) are generated. Among them, AβPP695 is the most crucial for the pathogenesis of AD. All three products of AβPP undergo proteolytic modification by α-, β-, and γ-secretases. The neuroprotective α-secretase (ADAM10) can be induced by retinoids through interaction with two putative binding sites for RXR resulting in upregulation of long-term potentiation, a model mechanism for synaptic plasticity involved in memory formation [52]. Satoh and Kuroda have demonstrated that the β-secretase BACE-1, which facilitates release of Aβ from AβPP via N-terminal cleavage, is also regulated by retinoids [53]. In contrast, γ-secretase, which facilitates release of Aβ from AβPP via C-terminal cleavage, has been observed to inhibit retinoid-induced neuronal differentiation causing cell death by targeting AβPP-CTF [54]. β-Carotene and retinoids can also exert their neuroprotective effects by suppressing formation of Aβ fibrils and destabilizing pre-formed fibrils in a dose-dependent manner [55].
Down regulation of ChAT expression is frequently observed in the pathogenesis of AD [56]. Retinoids are known to assist in blocking the inhibition of ChAT expression that is caused in AD by Aβ peptides [57]. Many earlier studies confirmed the neuroprotec-tive effects of retinoids through induction of ChAT expression in diverse cell types by modulating RARα [58, 59]. Similarly, suppression of retinaldehyde dehydrogenase 2 (RALDH2) and RARα deficit were observed in vitamin A deficient rats, confirming that disruption of the retinoid signaling pathway could play a crucial role in the onset of AD [60].
NEUROPROTECTIVE POTENTIALS OF RETINOIDS IN AD
Dietary supplementation of carotenoids has been shown to play a crucial role in preventing several neu-rodegenerative diseases, including AD [61]. Retinoids are involved in neuronal patterning, differentiation, and axon outgrowth. Retinoid deprivation leads to impairment of normal brain development and function, resulting in the appearance of symptoms of different neurodegenerative diseases, including AD. Recent investigations indicate that retinoids can induce generation of specific neuronal cell types and also regenerate axons after damage [62]. In addition, retinoids are involved in the maintenance of the differentiated state of adult neurons and neural stem cells as well as altered RA signaling levels. Thus, retinoids appear to be highly effective therapeutic agents for normal maintenance of the nervous system and also for the treatment of different neurodegenerative diseases, including AD.
EFFECTS OF RETINOIDS ON Aβ AGGREGATION
Formation of senile plaques in the cerebral cortex is a major histopathological hallmark of AD [63]. Several in vivo models support the involvement of the altered retinoid signaling in AD pathology. It is suggested that retinoids regulate many biological molecules involved in Aβ aggregate formation in vivo. Many investigations suggest that vitamin A deficiency impairs retinoid signaling in adult rats, causing accumulation of Aβ in the cerebral cortex and cerebral blood vessels [17, 48]. Retinoids can prevent Aβ plaque generation and also disrupt preformed fibrils [55, 64]. Several other recent studies have shown that retinoids regulate the expression of many genes involved in the production of Aβ, including BACE-1, PS1, and PS2 [18, 61]. Jarvis and co-workers reported that induction of the RARβ signaling pathway upregulated production of ADAM10, the α-secretase that could process AβPP into a non-amyloidal pathway, thus reducing formation and accumulation of Aβ [65]. Administration of ATRA to the AβPP/PS1 transgenic mice for 2 months was observed to inhibit Aβ aggregation and tau hyperphosphorylation as well as notably improved performance in the Morris water maze test [66]. Treatment with ATRA can enhance α-secretase activity and also impair AβPP cleavage by delocalizing BACE-1 and PS1 from the target site [67]. Unfortunately, adverse side effects and cytotoxicity at higher concentration of ATRA have restricted its clinical applications. So novel, receptor-specific, less toxic synthetic retinoids are urgently required to overcome these difficulties.
Recently, Fukasawa and co-workers observed that Tamibarotene (Fig. 5), a retinoid receptor agonist, lowered the insoluble Aβ40 and Aβ42 levels in the AβPP23 transgenic mice [68] by upregulating expression of α-secretase. In combination with HX630, an RXR agonist, Tamibarotene drastically enhanced the learning ability of these transgenic mice as observed by the Morris water maze test [68].
Fig. 5.

Synthetic scheme of Tamibarotene (AM80).
Several in vitro and in vivo models of AD indicated the importance of insulin degrading enzyme (IDE) and neprilysin (NEP) in Aβ peptide degradation and clearance in the brain. It has been observed that there is a RA response element (RARE) in the promoter region of IDE, and by interacting through this region retinoids can modulate the transcription of IDE [69]. Accordingly, in AD brain tissues, a direct correlation between lowered IDE mRNA level and elevated Aβ accumulation has been observed. Thus, downregulation of IDE activity could enhance the risk for development of AD. Similarly, NEP-mediated proteolytic cleavage of Aβ may stimulate the RARα signaling pathways both in neurons and microglia [19]. In another study, Jarvis and co-workers observed that AM580 (Fig. 6), a synthetic retinoid, could activate RAR in Tg2576 mice and thus, suppress Aβ formation by increasing ADAM10 and non-amyloidogenic processing of AβPP [65].
Fig. 6.

Synthetic scheme of AM580.
Hence, preclinical investigations strongly suggest that natural and synthetic retinoids can modulate Aβ formation and aggregation, and thus retinoids can be used as potential therapeutic agents against AD pathogenesis. Acitretin (Fig. 7), a synthetic retinoid, is currently undergoing testing in patients suffering from AD in a small Phase II clinical trial [70]; however, there is still no dependable clinical data to support this approach in humans. AD is associated with compromised clearance of Aβ from the brain, and this scavenging process is influenced by APOE. Ligand activated nuclear receptors like liver X receptor (LXR) and peroxisome proliferator-activated receptor gamma (PPARγ) could form heterodimers with RXRs and transcriptionally modulate expression of APOE [71]. LXR:RXR and PPARγ:RXR work in a feed-forward mechanism and promote the expression of APOE and also its lipid transporters ABCA1 and ABCG1 [71]. Prolonged administration of LXR and PPARγ agonists could activate macrophages and microglia resulting in enhancement of phagocytic pathways that bring down Aβ levels with recovery of cognitive function in mouse models of AD [72]. Similarly, RXR agonist could promote Aγ scavenging pathways by inducing APOE expression via activation of LXR and PPARγ in conjunction with RXR [72]. The RXR agonist Bexarotene (Fig. 8), which is an FDA-approved drug, is regularly used for the treatment of breast cancer [73] and non-small cell lung cancer [74]. Although Bexarotene and associated RXR agonists were used for the treatments for cancer, some recent investigations suggested them as potential therapeutic agents against different neurological diseases, including AD [75]. Cramer and co-workers demonstrated rapid clearance of soluble Aγ and Aγ plaques following Bexarotene therapy in transgenic AD mice [76]. But several other investigators could not replicate some aspects of this original report in their studies [77, 78]. Two investigations for assessing memory observed an increase in cognitive function following treatment with Bexarotene [77, 79]. But none of the investigations was able to repeat the reported outcome on fibrillar Aγ and Aγ plaque burden. IRX4204, which is another RXR agonist, shows high selectivity for RXR and does not transactivate RARs, and thus it can avert the adverse side effects associated with RAR agonists [80]. It promotes differentiation of oligodendrocytes and formation of T regulatory cells and suppression of Th17 cells in vitro [80]. Studies are ongoing to observe the efficacy of IRX4204 on AD neuropathology and cognitive impairment. In our laboratories, we are developing novel boron based retinoids or bororetinoids (Fig. 9) for treatment of AD [43]. Obviously, a systematic approach is urgently needed to screen retinoids and RAR agonists for their safe use in humans.
Fig. 7.

Synthestic scheme of Acitretin.
Fig. 8.

Synthetic scheme of Bexarotene.
Fig. 9.

Synthesis of BMS-493 analogs.
INFLAMMATION AND RETINOIDS
Intensification of the inflammatory response is frequently observed in both AD patients and animal models of this disease [81]. Macrophages and microglia act as scavengers to remove fibrillar Aγ aggregates via phagocytosis mediated by the β-1-integrin-dependent pathway in the brain [82]. However, this phagocytosis is blocked in the presence of inflammatory cytokines [82]. During AD pathogenesis, Aγ-stimulated signaling pathways induce synthesis and release of many pro-inflammatory cytokines (IL-1γ, IL-6, TNF-α, etc.), chemokines (CCL2) and acute phase proteins as well as reactive nitrogen species and reactive oxygen species (ROS) that can further promote plaque formation [83].
Several earlier studies reported anti-inflammatory roles of retinoids in neurodegenerative conditions [84]. Since retinoids significantly inhibit generation of IL-6 [85, 86], downregulation of IL-6 by retinoids may be a useful therapeutic strategy against AD. Retinoids have been observed to suppress lipopolysaccharide (LPS)-induced or Aγ-induced TNF-α production and inhibit expression of inducible NO synthase (iNOS) in activated microglia by inhibiting nuclear factor-kappa B nuclear translocation [87–89]. Anti-inflammatory responses of an RAR agonist AM80 (Tamibarotene) have been investigated in the LPS-induced inflammation model in vivo. The results demonstrated that AM80 could promote the production of brain-derived neurotrophic factor providing neuroprotection in pathological conditions [90]. Jarvis and co-workers reported suppression of inflammatory cell death by AM580 in cultured cortical neurons exposed to Aβ [65]. Furthermore, retinoids also affect prostanoid synthesis in cortical astrocytes in culture [91]. Thus, retinoids seem to have significant potential in inhibiting inflammatory responses and promoting amyloid phagocytosis in various neurodegenerative conditions, including AD.
OXIDATIVE STRESS, MITOCHONDRIAL DYSFUNCTION, AND RETINOIDS
Several earlier studies indicated that AD brains have oxidized RNA, nuclear and mtDNA, lipids, and proteins [92, 93]. Oxidative stress due to excessive ROS production is thought to represent an early indication in AD pathogenesis [94]. Mitochondria are the main source of intracellular ROS, and inhibition of ROS clearance leads to mitochondrial dysfunction [95]. Tamagno and co-workers described that an increase in ROS levels could induce Aβ production [96]. Many in vitro studies using PC12, COS, and neuroglioma cells showed that treatment with sodium azide, oligomycin, or carbonyl cyanide m-chlorophenyl hydrazone inhibited normal mitochondrial function with downregulation of release of soluble AβPP derivatives and upregulation of Aβ production [97, 98].
Mitochondrial structural and functional impairments in AD have been investigated in many earlier studies. Several studies indicate that AβPP and Aβ can impair mitochondrial import channels resulting in blockage of electron transfer chain and mitochondrial transport and enhancement of free radical generation causing mitochondrial dysfunction [99, 100]. Mitochondrial dysfunction downregulated expression of peroxisome proliferator-activated receptor gamma co-activator 1-alpha, nuclear respiratory factors 1 and 2, and transcription factor A mitochondrial and induced apoptosis in animal models of AD [101]. Peter-son and co-workers observed significant changes in Ca2+ homeostasis in AD patient fibroblasts leading to defective energy production [102]. Similarly, uncharacteristic mitochondria morphology in degenerating dendrites was observed in brains of AD patients [103]. The activities of many mitochondrial enzymes, such as α-ketoglutarate dehydrogenase complex and pyruvate dehydrogenase complex, are impaired in AD [104, 105]. Thus, it can be hypothesized that AD originates from alteration in brain energy metabolism. Our research group is actively pursuing a program to develop novel imaging and therapeutic agents targeting metabolic pathways for the treatment of AD.
Many in vivo studies reported an increase in oxidative stress in experimental animal models of AD. A similar investigation in Tg2576 AβPP transgenic mice confirmed that oxidative stress was one of the key neuropathological symptoms in AD pathogenesis [106, 107]. The anti-oxidant potential of many retinoids has been investigated both in vitro and in vivo. ATRA can prevent apoptosis in cultured neurons from staurosporine-induced oxidative stress by downregulating superoxide dismutase-1 and manganese superoxide dismutase-2 (MnSOD2) depletion [108, 109]. In addition, ATRA promotes expression of the mitochondrial anti-oxidant enzyme MnSOD2 in human neuroblastoma cells [89]. Hence, therapeutic strategies targeting oxidative stress and mitochondrial dysfunction could be a novel approach for AD treatment, and retinoids appear to be highly potential candidates.
EFFECTS OF RETINOIDS ON NEUROTRANSMISSION IN AD
Perturbations in several neurotransmitter systems, especially the cholinergic and catecholaminergic systems, are prominent in AD [110–112]. A hallmark of AD is degeneration of cholinergic neurons in the basal forebrain that projects to the neocortex, hippocampus, and amygdala. Impairment of cholinergic neurotransmission accompanies the early progression of dementia, and loss of cholinergic neurons results in memory impairments in animal models [113, 114]. In many animal studies, administration of cholinesterase inhibitors (which block the breakdown of acetylcholine and prolong its action in the brain) has been observed to stimulate memory and learning processes and administration of these drugs is currently used to treat the symptoms of AD in humans as well [115]. Retinoids, which are significantly decreased in AD brain [15], can provide an alternative treatment for AD symptoms; since, they have trophic effects on cholinergic neurons. Activation of RARα can upregulate expression of ChAT and vesicular acetylcholine transporter protein, which helps to transport acetylcholine into synaptic vesicles for synaptic release [116]. Retinoids are known to increase levels of acetylcholine and ChAT mRNA [117, 118].
AD is also associated with disruption of monoaminergic systems, including the noradrenergic and dopaminergic systems [112]. Thus, the locus coeruleus, which is one of the main noradrenergic nuclei of the brain and has extensive projection to the cortex and limbic system, exhibits significant degeneration in AD [119]. There are also reduced levels of tyrosine hydroxylase (the rate limiting enzyme for both norepinephrine and dopamine synthesis) and dopamine-beta-hydroxylase (DBH, the enzyme required for the synthesis of norepinephrine) in AD [120]. In addition, AD is associated with reduced levels of: (1) norepinephrine in the cortex [121, 122]; (2) dopamine levels in the cortex, amygdala, and striatum [123]; and (3) dopamine receptors in the striatum [124–126]. Since retinoids regulate the expression of tyrosine hydroxylase and dopamine β-hydroxylase [127] and can also directly modulate the expression of dopamine D2 receptors via interaction with the RARE promoter region [128], their trophic effects on both the noradrenergic and dopaminergic systems may help alleviate AD symptoms.
An interesting study by Shudo and co-workers using a passive avoidance learning test demonstrated the possible efficacy of retinoids in enhancing memory formation [17]. This widely used aversive learning paradigm is known to require activation of receptors in the three neuromodulatory systems, as discussed above, in relation to retinoids and AD: (1) muscarinic cholinergic receptors, (2) β-adrenergic receptors, and (3) D1 and D2 dopamine receptors [129, 130]. It was found that administration of the muscarinic receptor antagonist scopolamine blocked learning as expected, but learning and memory were rescued by administration of RAR and RXR ligands [17]. While the exact mechanism for this effect was not determined, Shudo and co-workers suggested that one possible mechanism was retinoid mediated enhancement of the expression of D2 receptors, since the D2 R gene has an upstream promoter sequence that can be activated by the RAR-RXR family [17].
RETINOIDS, STEM CELL BIOLOGY, AND REGENERATIVE MEDICINE
Several investigations on pattern formation established the significance of RA signaling in CNS development [131]. RA induces differentiation in murine F9 teratocarcinoma stem cell line in vitro [132]. RARβ upregulation could induce neurite progression and enhance recovery of animals with spinal cord injury [133, 134]. Takenaga and co-workers reported a similar therapeutic effect in bodily-injured rats after treatment with the synthetic retinoid Tamibarotene [135]. Thus, retinoids can be potential therapeutic agents for treatment of AD by promoting stem cells that are unspecialized cells, capable of dividing and renewing themselves for long periods and then differentiating into functional phenotypes [136]. These stem cells are generally originated from embryonic, fetal, or adult tissues. Recent investigations confirmed that retinoids induce stem cell differentiation through activation of different transcription factors. Retinoids modulate the transcription of many genes by binding to the nuclear receptors RARα, RARβ, and RARγ. These RARs can form heterodimers with one of the RXRs and can subsequently bind DNA resulting in activation of transcription of RA primary response genes, which are essential for stem cell differentiation. Stem cell differentiation into functional cells within the damaged tissues could be a significant achievement in the field of regenerative medicine. Regenerative therapy, an emerging field of biomedicine, involves the repair, replacement, or regeneration of cells, tissues, or organs. Recently, regenerative therapy has been explored in various diseases including kidney diseases, cardiac diseases, skin diseases, retinal disorders, and also neurodegenerative diseases such as AD and Parkinson’s disease [137–143]. Marder and co-workers observed that synthetic retinoids (Fig. 10) could induce differentiation of embryonal carcinoma stem cells to generate neurons [138].
Serup and co-workers have shown that neural induction in embryonic stem cells requires RA at sub-nanomolar levels to suppress Nodal signaling, suggesting that the mechanism by which Wnt signaling suppresses neural development is through facilitation of Nodal signaling [144]. Thus, there is a growing interest in the potential use of retinoid pathway manipulation as a therapy for neurodegenerative disorders [62].
CONCLUSION
Retinoids could play a crucial role in neural development, nerve regeneration, adult neural plasticity, and different neurodegenerative diseases, including AD (Fig. 11). Application of natural and synthetic retinoids and their receptor agonists are under investigation to regulate the on-going processes of stem cell turnover, cell plasticity, and tissue regeneration. Retinoids are currently used for the treatment of acne vulgaris, neuroblastoma, acute promyelocytic leukemia, and psoriasis [14]. Impaired retinoid signaling promotes AD pathology. Because retinoids are small molecules, they can readily enter the tissues and therefore constitute promising therapeutic candidates. Their application at lower dose or in combination with other neuroprotective drugs could minimize unwanted side toxicity in non-target tissues. Retinoids thus represent a novel therapeutic strategy for AD treatment as they can modulate multiple pathological conditions of this disease, including plaque formation, cholinergic transmission, and inflammatory responses in the brain (Fig. 11). Although many applications of retinoids have been well studied, it will be highly critical to extend the range of applications to neurodegenerative diseases, including AD [70], and also the most important factor is the synthesis of receptor subtype and isotype specific retinoids (to reduce toxicity, off-target binding, and increase specificity). To address these issues, our research group is synthesizing bororetinoids and some of these synthetic compounds are now used in preclinical study [145].
Fig. 11.

Role of RA signaling in AD. Neuronal stem cells (NSC) differentiation, inflammation, and clearance of damaged neuron through the blood-brain barrier (BBB) play a crucial role in AD pathology. RA influences receptors and signaling molecules such as ChAT, dopamine D2 receptor (D2R), brain derived neurotropic factor (BDNF), and tyrosine receptor kinase B (TrkB) that play significant role in memory and learning. RA upregulates of α-secretase activity through ADAM10 resulting in inhibition of Aβ accumulation and enhances Aβ clearance through activation of zona occludens protein 1 (ZO-1). RA suppresses inflammation by inhibiting TNF-α, IL6, IL17, MCP1, and VEGF in astrocytes and microglia. RA inhibits neuronal death by inhibiting Aβ synthesis, augmenting clearance of Aβ, and suppression of inflammation.
Phase II clinical trials (NCT01120002 and NCT01078168) are in progress to determine the potential of retinoids in AD patients. But ATRA is known to be toxic at high dose, which hinders its application to aged-patients. Besides, several RAs generate adverse side effects, like gastrointestinal hemorrhage and abdominal pain in certain patients [65]. Another foremost problem in AD clinical studies is the scarcity of proper animal models that can entirely mimic human AD pathology. Each current animal model can represent one or two main characteristic of AD pathology [146–148]. Another challenge in preclinical studies of AD is the time course of disease in mice models when compared with AD in humans [149]. Thus, AD mice models are usually convenient for investigating some specific features of commencement of AD but not the disease process itself [150]. Besides, dietary habits and environmental factors play significant roles in the disease progression in humans and these are difficult to mimic in AD animal models.
The pathogenesis in AD is frequently described as multifactorial, and such a notion is consistent with the relatively modest benefits from the targeted therapeutic agents [151, 152]. To address this issue, the scientific community may need to design, synthesize, and examine therapeutic agents with multi-target potential rather than single target drugs. The viable options include: (1) a polypharmacy (many dugs with many targets) or (2) a single drug attacking multiple targets. Retinoids are capable of acting upon multiple AD-associated (anti-apoptotic, anti-oxidant, pro-differentiative, Aβ lowering, acetylcholine activation, and autophagy enhancing) targets. Thus, retinoids seem to offer a viable solution as a single drug with multiple targets to treat multifactorial AD.
Acknowledgments
The work was supported in part by University of South Carolina School of Medicine Research Development Fund (USC SOM RDF, Columbia, SC, USA) and South Carolina Spinal Cord Injury Research Fund (SCIRF Columbia, SC, USA) to SKR. Also, BCD is thankful to KUMC for its startup funding.
Footnotes
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0450r2).
References
- 1.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Rafii MS, Aisen PS. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 4.Thal DR, Walter J, Saido TC, Fändrich M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015;129:167–182. doi: 10.1007/s00401-014-1375-y. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer’s disease. Annu Rev Genomics Hum Genet. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- 6.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 7.Hampel H. Current insights into the pathophysiology of Alzheimer’s disease: Selecting targets for early therapeutic intervention. Int Psychogeriatr. 2012;24:S10–S17. doi: 10.1017/S1041610212000579. [DOI] [PubMed] [Google Scholar]
- 8.Wyss-Coray T, Mucke L. Inflammation in neurode-generative disease - a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 9.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer’s disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ledesma MD, Dotti CG. Peripheral cholesterol, metabolic disorders and Alzheimer’s disease. Front Biosci (Elite Ed) 2012;4:181–194. doi: 10.2741/e368. [DOI] [PubMed] [Google Scholar]
- 12.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zlokovic BV. Cerebrovascular effects of apolipoprotein E: Implications for Alzheimer’s disease. JAMA Neurol. 2013;70:440–444. doi: 10.1001/jamaneurol.2013.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerner AJ, Gustaw-Rothenberg K, Smyth S, Casadesus G. Retinoids for treatment of Alzheimer’s disease. Biofactors. 2012;38:84–89. doi: 10.1002/biof.196. [DOI] [PubMed] [Google Scholar]
- 15.Goodman AB. Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer’s disease. J Cell Physiol. 2006;209:598–603. doi: 10.1002/jcp.20784. [DOI] [PubMed] [Google Scholar]
- 16.Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2003;100:2901–2905. doi: 10.1073/pnas.0437937100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shudo K, Fukasawa H, Nakagomi M, Yamagata N. Towards retinoid therapy for Alzheimer’s disease. Curr Alzheimer Res. 2009;6:302–311. doi: 10.2174/156720509788486581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HP, Casadesus G, Zhu X, Lee HG, Perry G, Smith MA, Gustaw-Rothenberg K, Lerner A. All-trans retinoic acid as a novel therapeutic strategy for Alzheimer’s disease. Expert Rev Neurother. 2009;9:1615–1621. doi: 10.1586/ern.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goncalves MB, Clarke E, Hobbs C, Malmqvist T, Deacon R, Jack J, Corcoran JP. Amyloid β inhibits retinoic acid synthesis exacerbating Alzheimer disease pathology which can be attenuated by an retinoic acid receptor α agonist. Eur J Neurosci. 2013;37:1182–1192. doi: 10.1111/ejn.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wetzel R. Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res. 2006;39:671–679. doi: 10.1021/ar050069h. [DOI] [PubMed] [Google Scholar]
- 23.Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol. 2007;17:48–57. doi: 10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Walsh DM, Selkoe DJ. Aβ oligomers - A decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 26.Gessel MM, Bernstein S, Kemper M, Teplow DB, Bowers MT. Familial Alzheimer’s disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem Neurosci. 2012;3:909–918. doi: 10.1021/cn300050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 28.Habicht G, Haupt C, Friedrich RP, Hortschansky P, Sachse C, Meinhardt J, Wieligmann K, Gellermann GP, Brodhun M, Götz J, Halbhuber KJ, Röcken C, Horn U, Fändrich M. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ protofibrils. Proc Natl Acad Sci U S A. 2007;104:19232–19237. doi: 10.1073/pnas.0703793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richard-son PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 31.Krafft GA, Klein WL. ADDLs and the signaling web that leads to Alzheimer’s disease. Neuropharmacol. 2010;59:230–242. doi: 10.1016/j.neuropharm.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 32.Kumar A, Paslay LC, Lyons D, Morgan SE, Correia JJ, Rangachari V. Specific soluble oligomers of amyloid-beta peptide undergo replication and form non-fibrillar aggregates in interfacial environments. J Biol Chem. 2012;287:21253–21264. doi: 10.1074/jbc.M112.355156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rijal Upadhaya A, Capetillo-Zarate E, Kosterin I. Dispersible amyloid β-protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: Their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiol Aging. 2012;33:2641–2660. doi: 10.1016/j.neurobiolaging.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 34.Rijal Upadhaya A, Kosterin I, Kumar S, von Arnim CA, Yamaguchi H, Fändrich M, Walter J, Thal DR. Biochemical stages of amyloid β-peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain. 2014;137:887–903. doi: 10.1093/brain/awt362. [DOI] [PubMed] [Google Scholar]
- 35.Khillan JS. Vitamin A/retinol and maintenance of pluripotency of stem cells. Nutrients. 2014;6:1209–1222. doi: 10.3390/nu6031209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barua AB, Furr HC. Properties of retinoids. Structure, handling, and preparation. Mol Biotechnol. 1998;10:167–182. doi: 10.1007/BF02760863. [DOI] [PubMed] [Google Scholar]
- 37.Furr HC. Analysis of retinoids and carotenoids: Problems resolved and unsolved. J Nutr. 2004;134:281S–285S. doi: 10.1093/jn/134.1.281S. [DOI] [PubMed] [Google Scholar]
- 38.Maercker A. The Wittig reaction. Org React. 1965;14:270–490. [Google Scholar]
- 39.Julia M, Arnould D. Use of sulfones in synthesis. III. Synthesis of vitamin A. Bull Soc Chim Fr. 1973:746–750. [Google Scholar]
- 40.Koch D, Gartner W. Steric hindrance between chro-mophore substituents as the driving force of rhodopsin isomerization: 10-methyl-13-demethyl retinal containing rhodopsin. Photochem Photobiol. 1997;65:181–186. doi: 10.1111/j.1751-1097.1997.tb01896.x. [DOI] [PubMed] [Google Scholar]
- 41.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 42.le Maire A, Alvarez S, Shankaranarayanan P, Lera AR, Bourguet W, Gronemeyer H. Retinoid receptors and therapeutic applications of RAR/RXR modulators. Curr Top Med Chem. 2012;12:505–527. doi: 10.2174/156802612799436687. [DOI] [PubMed] [Google Scholar]
- 43.Das BC, Thapa P, Karki R, Das S, Mahapatra S, Liu TC, Torregroza I, Wallace DP, Kambhampati S, Van Veldhuizen P, Verma A, Ray SK, Evans T. Retinoic acid signaling pathways in development and diseases. Bioorg Med Chem. 2014;22:673–683. doi: 10.1016/j.bmc.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stephens-Jarnagin A, Miller DA, DeLuca HF. The growth supporting activity of a retinoidal benzoic acid derivative and 4,4-difluororetinoic acid. Arch Biochem Biophys. 1985;237:11–16. doi: 10.1016/0003-9861(85)90248-6. [DOI] [PubMed] [Google Scholar]
- 45.Kagechika H, Shudo K. Synthetic retinoids: Recent developments concerning structure and clinical utility. J Med Chem. 2005;48:5875–5883. doi: 10.1021/jm0581821. [DOI] [PubMed] [Google Scholar]
- 46.Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathi-otis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. 2013;9:374–382. doi: 10.1038/nchembio.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corcoran JP, So PL, Maden M. Disruption of the retinoid signalling pathway causes a deposition of amyloid β in the adult rat brain. Eur J Neurosci. 2004;20:896–902. doi: 10.1111/j.1460-9568.2004.03563.x. [DOI] [PubMed] [Google Scholar]
- 48.Husson M, Enderlin V, Delacourte A, Ghenimi N, Alfos S, Pallet V, Higueret P. Retinoic acid normalizes nuclear receptor mediated hypo-expression of proteins involved in beta-amyloid deposits in the cerebral cortex of vitamin A deprived rats. Neurobiol Dis. 2006;23:1–10. doi: 10.1016/j.nbd.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Maury CPJ, Teppo AM. Immunodetection of protein composition in cerebral amyloid extracts in Alzheimer’s disease: Enrichment of retinol-binding protein. J Neurol Sci. 1987;80:221–228. doi: 10.1016/0022-510x(87)90156-0. [DOI] [PubMed] [Google Scholar]
- 50.Lahiri DK, Nall C. Promoter activity of the gene encoding the β-amyloid precursor protein is up-regulated by growth factors, phorbol ester, retinoic acid and interleukin-1. Brain Res Mol Brain Res. 1995;32:233–240. doi: 10.1016/0169-328x(95)00078-7. [DOI] [PubMed] [Google Scholar]
- 51.Yang Y, Quitschke WW, Brewer GJ. Upregulation of amyloid precursor protein gene promoter in rat primary hippocampal neurons by phorbol ester, IL-1 and retinoic acid, but not by reactive oxygen species. Mol Brain Res. 1998;60:40–49. doi: 10.1016/s0169-328x(98)00164-8. [DOI] [PubMed] [Google Scholar]
- 52.Fahrenholz F, Postina R. α-secretase activation - an approach to Alzheimer’s disease therapy. Neurodegener Dis. 2006;3:255–261. doi: 10.1159/000095264. [DOI] [PubMed] [Google Scholar]
- 53.Satoh J, Kuroda Y. Amyloid precursor protein β-secretase (BACE) mRNA expression in human neural cell lines following induction of neuronal differentiation and exposure to cytokines and growth factors. Neuropathology. 2000;20:289–296. doi: 10.1046/j.1440-1789.2000.00349.x. [DOI] [PubMed] [Google Scholar]
- 54.Yoshikawa K, Aizawa T, Hayashi Y. Degeneration in vitro of post-mitotic neurons overexpressing the Alzheimer amyloid protein precursor. Nature. 1992;359:64–67. doi: 10.1038/359064a0. [DOI] [PubMed] [Google Scholar]
- 55.Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Vitamin A exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp Neurol. 2004;189:380–392. doi: 10.1016/j.expneurol.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 56.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 57.Sahin M, Karauzum SB, Perry G, Smith MA, Aliciguzel Y. Retinoic acid isomers protect hippocampal neurons from amyloid-β induced neurodegeneration. Neurotox Res. 2005;7:243–250. doi: 10.1007/BF03036453. [DOI] [PubMed] [Google Scholar]
- 58.Coleman BA, Taylor P. Regulation of acetyl-cholinesterase expression during neuronal differentiation. J Biol Chem. 1996;271:4410–4416. doi: 10.1074/jbc.271.8.4410. [DOI] [PubMed] [Google Scholar]
- 59.Hill DR, Robertson KA. Characterisation of the colinergic neuronal differentiation of the human neuroblastoma cell line LAN-5 after treatment with retinoic acid. Dev Brain Res. 1997;102:53–67. doi: 10.1016/s0165-3806(97)00076-x. [DOI] [PubMed] [Google Scholar]
- 60.Grapin-Botton A, Bonnin MA, Sieweke M, Le Douarin NM. Defined concentrations of a posteriorizing signal are critical for MafB/Kreisler segmental expression in the hind-brain. Development. 1998;125:1173–1181. doi: 10.1242/dev.125.7.1173. [DOI] [PubMed] [Google Scholar]
- 61.Obulesu M, Dowlathabad MR, Bramhachari PV. Carotenoids and Alzheimer’s disease: An insight into therapeutic role of retinoids in animal models. Neurochem Int. 2011;59:535–541. doi: 10.1016/j.neuint.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci. 2007;8:755–765. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]
- 63.Paulson JB, Ramsden M, Forster C, Sherman MA, McGowan E, Ashe KH. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer’s disease. Am J Pathol. 2008;173:762–772. doi: 10.2353/ajpath.2008.080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takasaki J, Ono K, Yoshiike Y, Hirohata M, Ikeda T, Morinaga A, Takashima A, Yamada M. Vitamin A has anti-oligomerization effects on amyloid-β in vitro. J Alzheimers Dis. 2011;27:271–280. doi: 10.3233/JAD-2011-110455. [DOI] [PubMed] [Google Scholar]
- 65.Jarvis CI, Goncalves MB, Clarke E, Dogruel M, Kalindjian SB, Thomas SA, Maden M, Corcoran JP. Retinoic acid receptor-β signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur J Neurosci. 2010;32:1246–1255. doi: 10.1111/j.1460-9568.2010.07426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ding Y, Qiao A, Wang Z, Goodwin JS, Lee ES, Block ML, Allsbrook M, McDonald MP, Fan GH. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J Neurosci. 2008;28:11622–11634. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koryakina A, Aeberhard J, Kiefer S, Hamburger M, Küenzi P. Regulation of secretases by all-transretinoic acid. FEBS J. 2009;276:2645–2655. doi: 10.1111/j.1742-4658.2009.06992.x. [DOI] [PubMed] [Google Scholar]
- 68.Fukasawa H, Nakagomi M, Yamagata N, Katsuki H, Kawahara K, Kitaoka K, Miki T, Shudo K. Tamibarotene: A candidate retinoid drug for Alzheimer’s disease. Biol Pharm Bull. 2012;35:1206–1212. doi: 10.1248/bpb.b12-00314. [DOI] [PubMed] [Google Scholar]
- 69.Melino G, Draoui M, Bernardini S, Bellincampi L, Reichert U, Cohen P. Regulation by retinoic acid of insulin-degrading enzyme and of a related endoprotease in human neuroblastoma cell lines. Cell Growth Differ. 1996;7:787–796. [PubMed] [Google Scholar]
- 70.Corbett A, Pickett J, Burns A, Corcoran J, Dunnett SB, Edison P, Hagan JJ, Holmes C, Jones E, Katona C, Kearns I, Kehoe P, Mudher A, Passmore A, Shepherd N, Walsh F, Ballard C. Drug repositioning for Alzheimer’s disease. Nat Rev Drug Discov. 2012;11:833–846. doi: 10.1038/nrd3869. [DOI] [PubMed] [Google Scholar]
- 71.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 72.Mandrekar-Colucci S, Landreth GE. Nuclear receptors as therapeutic targets for Alzheimer’s disease. Expert Opin Ther Targets. 2011;15:1085–1097. doi: 10.1517/14728222.2011.594043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Esteva FJ, Glaspy J, Baidas S, Laufman L, Hutchins L, Dickler M, Tripathy D, Cohen R, DeMichele A, Yocum RC, Osborne CK, Hayes DF, Hortobagyi GN, Winer E, Demetri GD. Multicenter phase II study of oral bexarotene for patients with metastatic breast cancer. J Clin Oncol. 2003;21:999–1006. doi: 10.1200/JCO.2003.05.068. [DOI] [PubMed] [Google Scholar]
- 74.Dragnev KH, Petty WJ, Shah SJ, Lewis LD, Black CC, Memoli V, Nugent WC, Hermann T, Negro-Vilar A, Rigas JR, Dmitrovsky E. A proof-of-principle clinical trial of bexarotene in patients with non-small cell lung cancer. Clin Cancer Res. 2007;13:1794–1800. doi: 10.1158/1078-0432.CCR-06-1836. [DOI] [PubMed] [Google Scholar]
- 75.Tousi B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: Current evidence. Neuropsychiatr Dis Treat. 2015;11:311–315. doi: 10.2147/NDT.S61309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fitz NF, Cronican AA, Lefterov I, Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–c. doi: 10.1126/science.1235809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Price AR, Xu G, Siemienski ZB, Smithson LA, Borchelt DR, Golde TE, Felsenstein KM. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–d. doi: 10.1126/science.1234089. [DOI] [PubMed] [Google Scholar]
- 79.Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, Jucker M, Holtzman DM, Tanzi RE, Vassar R, Sisodia SS. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–f. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]
- 80.Collins, Thomas R. News from the ANN annual meeting: RXR agonist shows potential for myelin repair in vitro, new study suggests. Neurology Today. 2014;14:22–23. [Google Scholar]
- 81.Johnston H, Boutin H, Allan SM. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem Soc Trans. 2011;39:886–890. doi: 10.1042/BST0390886. [DOI] [PubMed] [Google Scholar]
- 82.Weitz TM, Town T. Microglia in Alzheimer’s disease: It’s all about context. Int J Alzheimers Dis. 2012;2012:314185. doi: 10.1155/2012/314185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2005;7:221–232. doi: 10.3233/jad-2005-7304. [DOI] [PubMed] [Google Scholar]
- 84.Kuenzli S, Tran C, Saurat JH. Retinoid receptors in inflammatory responses: A potential target for pharmacology. Curr Drug Targets Inflamm Allergy. 2004;3:355–360. doi: 10.2174/1568010042634587. [DOI] [PubMed] [Google Scholar]
- 85.Zitnik RJ, Kotloff RM, Latifpour J, Zheng T, Whiting NL, Schwalb J, Elias JA. Retinoic acid inhibition of IL-1-induced IL-6 production by human lung fibroblasts. J Immunol. 1994;152:1419–1427. [PubMed] [Google Scholar]
- 86.Kagechika H, Kawachi E, Fukasawa H, Saito G, Iwanami N, Umemiya H, Hashimoto Y, Shudo K. Inhibition of IL-1-induced IL-6 production by synthetic retinoids. Biochem Biophys Res Commun. 1997;231:243–248. doi: 10.1006/bbrc.1997.6087. [DOI] [PubMed] [Google Scholar]
- 87.Dheen ST, Jun Y, Yan Z, Tay SS, Ling EA. Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia. 2005;50:21–31. doi: 10.1002/glia.20153. [DOI] [PubMed] [Google Scholar]
- 88.Kaur C, Sivakumar V, Dheen ST, Ling EA. Insulin-like growth factor I and II expression and modulation in amoeboid microglial cells by lipopolysaccharide and retinoic acid. Neuroscience. 2006;138:1233–1244. doi: 10.1016/j.neuroscience.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 89.Carratú MR, Marasco C, Signorile A, Scuderi C, Steardo L. Are retinoids a promise for Alzheimer’s disease management? Curr Med Chem. 2012;19:6119–6125. doi: 10.2174/092986712804485700. [DOI] [PubMed] [Google Scholar]
- 90.Katsuki H, Kurimoto E, Takemori S, Kurauchi Y, Hisatsune A, Isohama Y, Izumi Y, Kume T, Shudo K, Akaike A. Retinoic acid receptor stimulation protects midbrain dopaminergic neurons from inflammatory degeneration via BDNF-mediated signaling. J Neurochem. 2009;110:707–718. doi: 10.1111/j.1471-4159.2009.06171.x. [DOI] [PubMed] [Google Scholar]
- 91.Kampmann E, Johann S, van Neerven S, Beyer C, Mey J. Anti-inflammatory effect of retinoic acid on prostaglandin synthesis in cultured cortical astrocytes. J Neurochem. 2008;106:320–332. doi: 10.1111/j.1471-4159.2008.05395.x. [DOI] [PubMed] [Google Scholar]
- 92.Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 93.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nunomura A, Perry G, Aliev G. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 95.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 97.Gasparini L, Racchi M, Benussi L, Curti D, Binetti G, Bianchetti A, Trabucchi M, Govoni S. Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett. 1997;231:113–117. doi: 10.1016/s0304-3940(97)00536-3. [DOI] [PubMed] [Google Scholar]
- 98.Webster MT, Pearce BR, Bowen DM, Francis PT. The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm. 1998;105:839–853. doi: 10.1007/s007020050098. [DOI] [PubMed] [Google Scholar]
- 99.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 100.Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120:419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peterson C, Goldman JE. Alterations in calcium content and biochemical processes in cultured skin fibroblasts from aged and Alzheimer donors. Proc Natl Acad Sci U S A. 1986;83:2758–2762. doi: 10.1073/pnas.83.8.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saraiva AA, Borges MM, Madeira MD, Tavares MA, Paula-Barbosa MM. Mitochondrial abnormalities in cortical dendrites from patients with Alzheimer’s disease. J Submicrosc Cytol. 1985;17:459–464. [PubMed] [Google Scholar]
- 104.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 105.Blass JP, Gibson GE, Hoyer S. The role of the metabolic lesion in Alzheimer’s disease. J Alzheimers Dis. 2002;4:225–232. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- 106.Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 108.Ahlemeyer B, Hühne R, Krieglstein J. Retinoic acid potentiated the protective effect of NGF against staurosporine-induced apoptosis in cultured chick neurons by increasing the trkA protein expression. J Neurosci Res. 2000;60:767–778. doi: 10.1002/1097-4547(20000615)60:6<767::AID-JNR9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 109.Ahlemeyer B, Bauerbach E, Plath M, Steuber M, Heers C, Tegtmeier F, Krieglstein J. Retinoic acid reduces apoptosis and oxidative stress by preservation of SOD protein level. Free Radic Biol Med. 2001;30:1067–1077. doi: 10.1016/s0891-5849(01)00495-6. [DOI] [PubMed] [Google Scholar]
- 110.St George-Hyslop PH. Piecing together Alzheimer’s disease. Sci Am. 2000;283:76–83. doi: 10.1038/scientificamerican1200-76. [DOI] [PubMed] [Google Scholar]
- 111.Wenk GL. Neuropathologic changes in Alzheimer’s disease. J Clin Psychiatry. 2003;64:7–10. [PubMed] [Google Scholar]
- 112.Trillo L, Das D, Hsieh W, Medina B, Moghadam S, Lin B, Dang V, Sanchez MM, De Miguel Z, Ashford JW, Salehi A. Ascending monoaminergic systems alterations in Alzheimer’s disease. translating basic science into clinical care. Neurosci Biobehav Rev. 2013;37:1363–1379. doi: 10.1016/j.neubiorev.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 113.Veng LM, Granholm AC, Rose GM. Age-related sex differences in spatial learning and basal forebrain cholinergic neurons in F344 rats. Physiol Behav. 2003;80:27–36. doi: 10.1016/s0031-9384(03)00219-1. [DOI] [PubMed] [Google Scholar]
- 114.Hunter CL, Bimonte-Nelson HA, Nelson M, Eckman CB, Granholm AC. Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: Effects of estrogen. Neurobiol Aging. 2004;25:873–884. doi: 10.1016/j.neurobiolaging.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 115.Santucci AC, Kanof PD, Haroutunian V. Effect of physostigmine on memory consolidation and retrieval processes in intact and nucleus basalislesioned rats. Psychopharmacology (Berl) 1989;99:70–74. doi: 10.1007/BF00634455. [DOI] [PubMed] [Google Scholar]
- 116.Berse B, Blusztajn JK. Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor alpha, cAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line. J Biol Chem. 1995;270:22101–22104. doi: 10.1074/jbc.270.38.22101. [DOI] [PubMed] [Google Scholar]
- 117.Kobayashi M, Matsuoka I, Kurihara K. Cholinergic differentiation of cultured sympathetic neurons induced by retinoic acid. Induction of choline acetyltransferase-mRNA and suppression of tyrosine hydroxylase-mRNA levels. FEBS Lett. 1994;337:259–264. doi: 10.1016/0014-5793(94)80204-1. [DOI] [PubMed] [Google Scholar]
- 118.Pedersen WA, Berse B, Schüler U, Wainer BH, Blusztajn JK. All-trans- and 9-cis-retinoic acid enhance the cholinergic properties of a murine septal cell line: Evidence that the effects are mediated by activation of retinoic acid receptor-alpha. J Neurochem. 1995;65:50–58. doi: 10.1046/j.1471-4159.1995.65010050.x. [DOI] [PubMed] [Google Scholar]
- 119.Mann DM, Lincoln J, Yates PO, Stamp JE, Toper S. Changes in the monoamine containing neurones of the human CNS in senile dementia. Br J Psychiatry. 1980;136:533–541. doi: 10.1192/bjp.136.6.533. [DOI] [PubMed] [Google Scholar]
- 120.Iversen LL, Rossor MN, Reynolds GP, Hills R, Roth M, Mountjoy CQ, Foote SL, Morrison JH, Bloom FE. Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer’s type. Neurosci Lett. 1983;39:95–100. doi: 10.1016/0304-3940(83)90171-4. [DOI] [PubMed] [Google Scholar]
- 121.Reinikainen KJ, Paljärvi L, Huuskonen M, Soininen H, Laakso M, Riekkinen PJ. A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer’s disease. J Neurol Sci. 1988;84:101–116. doi: 10.1016/0022-510x(88)90179-7. [DOI] [PubMed] [Google Scholar]
- 122.Storga D, Vrecko K, Birkmayer JG, Reibnegger G. Monoaminergic neurotransmitters,their precursors and metabolites in brains of Alzheimer patients. Neurosci Lett. 1996;203:29–32. doi: 10.1016/0304-3940(95)12256-7. [DOI] [PubMed] [Google Scholar]
- 123.Pinessi L, Rainero I, De Gennaro T, Gentile S, Portaleone P, Bergamasco B. Biogenic amines in cerebrospinal fluid and plasma of patients with dementia of Alzheimer’s type. Funct Neurol. 1987;2:51–58. [PubMed] [Google Scholar]
- 124.Cross AJ, Crow TJ, Ferrier IN, Johnson JA, Markakis D. Striatal dopamine receptors in Alzheimer-type dementia. Neurosci Lett. 1984;52:1–6. doi: 10.1016/0304-3940(84)90341-0. [DOI] [PubMed] [Google Scholar]
- 125.Pizzolato G, Chierichetti F, Fabbri M, Cagnin A, Dam M, Ferlin G, Battistin L. Reduced striatal dopamine receptors in Alzheimer’s disease: Single photon emission tomography study with the D2 tracer [123I]-IBZM. Neurology. 1996;47:1065–1068. doi: 10.1212/wnl.47.4.1065. [DOI] [PubMed] [Google Scholar]
- 126.Kemppainen N, Ruottinen H, Nâgren K, Rinne JO. PET shows that striatal dopamine D1 and D2 receptors are differentially affected in AD. Neurology. 2000;55:205–209. doi: 10.1212/wnl.55.2.205. [DOI] [PubMed] [Google Scholar]
- 127.Kim HS, Hong SJ, LeDoux MS, Kim KS. Regulation of the tyrosine hydroxylase and dopamine beta-hydroxylase genes by the transcription factor AP-2. J Neurochem. 2001;76:280–294. doi: 10.1046/j.1471-4159.2001.00044.x. [DOI] [PubMed] [Google Scholar]
- 128.Samad TA, Krezel W, Chambon P, Borrelli E. Regulation of dopaminergic pathways by retinoids: Activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family. Proc Natl Acad Sci U S A. 1997;94:14349–14354. doi: 10.1073/pnas.94.26.14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lalumiere RT, Nguyen LT, McGaugh JL. Post-training intrabasolateral amygdala infusions of dopamine modulate consolidation of inhibitory avoidance memory: Involvement of noradrenergic and cholinergic systems. Eur J Neurosci. 2004;20:2804–2810. doi: 10.1111/j.1460-9568.2004.03744.x. [DOI] [PubMed] [Google Scholar]
- 130.McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- 131.Maden M. Retinoids and spinal cord development. J Neurobiol. 2006;66:726–738. doi: 10.1002/neu.20248. [DOI] [PubMed] [Google Scholar]
- 132.Strickland S, Mahdavi V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell. 1978;15:393–403. doi: 10.1016/0092-8674(78)90008-9. [DOI] [PubMed] [Google Scholar]
- 133.Wong LF, Yip PK, Battaglia A, Grist J, Corcoran J, Maden M, Azzouz M, Kingsman SM, Kingsman AJ, Mazarakis ND, McMahon SB. Retinoic acid receptor beta2 promotes functional regeneration of sensory axons in the spinal cord. Nat Neurosci. 2006;9:243–250. doi: 10.1038/nn1622. [DOI] [PubMed] [Google Scholar]
- 134.Corcoran J, So PL, Barber RD, Vincent KJ, Mazarakis ND, Mitrophanous KA, Kingsman SM, Maden M. Retinoic acid receptor beta2 and neurite outgrowth in the adult mouse spinal cord in vitro. J Cell Sci. 2002;115:3779–3786. doi: 10.1242/jcs.00046. [DOI] [PubMed] [Google Scholar]
- 135.Miwako I, Kagechika H. Tamibarotene. Drugs Today (Barc) 2007;43:563–568. doi: 10.1358/dot.2007.43.8.1072615. [DOI] [PubMed] [Google Scholar]
- 136.Dubie T, Admassu B, Sisay T, Shiferaw H. Basic biology and therapeutic application of stem cells in various human and animal diseases. J Cell Biol Genet. 2014;4:40–52. [Google Scholar]
- 137.Yokoo T, Fukui A, Kobayashi E. Application of regenerative medicine for kidney diseases. Organogenesis. 2007;3:34–43. doi: 10.4161/org.3.1.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Christie VB, Barnard JH, Batsanov AS, Bridgens CE, Cart-mell EB, Collings JC, Maltman DJ, Redfern CP, Marder TB, Przyborski S, Whiting A. Synthesis and evaluation of synthetic retinoid derivatives as inducers of stem cell differentiation. Org Biomol Chem. 2008;6:3497–3507. doi: 10.1039/b808574a. [DOI] [PubMed] [Google Scholar]
- 139.Daftarian N, Kiani S, Zahabi A. Regenerative therapy for retinal disorders. J Ophthalmic Vis Res. 2010;5:250–264. [PMC free article] [PubMed] [Google Scholar]
- 140.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peng J, Zeng X. The role of induced pluripotent stem cells in regenerative medicine: Neurodegenerative diseases. Stem Cell Res Ther. 2011;2:32. doi: 10.1186/scrt73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Uitto J. Regenerative medicine for skin diseases: IPS cells to the rescue. J Invest Dermatol. 2011;131:812–814. doi: 10.1038/jid.2011.2. [DOI] [PubMed] [Google Scholar]
- 143.Ptaszek LM, Mansour M, Ruskin JN, Chien KR. Towards regenerative therapy for cardiac disease. Lancet. 2012;379:933–942. doi: 10.1016/S0140-6736(12)60075-0. [DOI] [PubMed] [Google Scholar]
- 144.Engberg N, Kahn M, Petersen DR, Hansson M, Serup P. Retinoic acid synthesis promotes development of neural progenitors from mouse embryonic stem cells by suppressing endogenous, Wnt-dependent nodal signaling. Stem Cells. 2010;28:1498–1509. doi: 10.1002/stem.479. [DOI] [PubMed] [Google Scholar]
- 145.Zhong Y, Wu Y, Liu R, Li Z, Chen Y, Evans T, Chuang P, Das B, He JC. Novel retinoic acid receptor α agonists for treatment of kidney disease. PLoS One. 2011;6:e27945. doi: 10.1371/journal.pone.0027945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jaturapatporn D, Isaac MG, McCleery J, Tabet N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst Rev. 2012;2:CD006378. doi: 10.1002/14651858.CD006378.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lull ME, Levesque S, Surace MJ, Block ML. Chronic apocynin treatment attenuates beta amyloid plaque size and microglial number in hAPP(751)(SL) mice. PLoS One. 2011;6:e20153. doi: 10.1371/journal.pone.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH, Rabinovitch PS, Reddy PH. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21:2973–2990. doi: 10.1093/hmg/dds128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li C, Ebrahimi A, Schluesener H. Drug pipeline in neurodegeneration based on transgenic mice models of Alzheimer’s disease. Ageing Res Rev. 2013;12:116–140. doi: 10.1016/j.arr.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 150.Shi Y, Kirwan P, Smith J, Maclean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4:124–129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Carreiras MC, Mendes E, Perry MJ, Francisco AP, Marco-Contelles J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr Top Med Chem. 2013;13:1745–1770. doi: 10.2174/15680266113139990135. [DOI] [PubMed] [Google Scholar]
- 152.Dias KS, Viegas C. Multi-target directed drugs: A modern approach for design of new drugs for treatment of Alzheimer’s disease. Curr Neuropharmacol. 2014;12:239–255. doi: 10.2174/1570159X1203140511153200. [DOI] [PMC free article] [PubMed] [Google Scholar]