

Abstract
The rapid expansion of next-generation sequencing has yielded a powerful array of tools to address fundamental biological questions at a scale that was inconceivable just a few years ago. Various genome partitioning strategies to sequence select subsets of the genome have emerged as powerful alternatives to whole genome sequencing in ecological and evolutionary genomic studies. High throughput targeted capture is one such strategy that involves the parallel enrichment of pre-selected genomic regions of interest. The growing use of targeted capture demonstrates its potential power to address a range of research questions, yet these approaches have yet to expand broadly across labs focused on evolutionary and ecological genomics. In part, the use of targeted capture has been hindered by the logistics of capture design and implementation in species without established reference genomes. Here we aim to 1) increase the accessibility of targeted capture to researchers working in non-model taxa by discussing capture methods that circumvent the need of a reference genome, 2) highlight the evolutionary and ecological applications where this approach is emerging as a powerful sequencing strategy, and 3) discuss the future of targeted capture and other genome partitioning approaches in light of the increasing accessibility of whole genome sequencing. Given the practical advantages and increasing feasibility of high-throughput targeted capture, we anticipate an ongoing expansion of capture-based approaches in evolutionary and ecological research, synergistic with an expansion of whole genome sequencing.
Keywords: ancient DNA, detecting selection, genetic mapping, metagenomics, next generation sequencing, phylogenomics
Introduction
The ability to address many fundamental evolutionary and ecological questions is no longer constrained simply by the generation of sequence data. Instead, as next generation sequencing (NGS) has become more accessible, a major challenge has become choosing which sequencing strategies to pursue (Ekblom & Galindo 2011; Davey et al. 2011; McCormack et al. 2013b; Ellegren 2014). The power of a given NGS experiment to address a central research question would ideally drive such decisions. However, often these choices come down to more practical considerations, such as cost, ease of use, or researcher expertise level (Ekblom & Galindo 2011). Reference genomes remain integral to most NGS analytical frameworks, yet de novo whole genome sequencing (WGS) and assembly remains prohibitively costly, time-consuming, and computationally difficult for widespread adoption by individual labs. Thus, the challenges of NGS data can be particularly acute for biologists interested in species without established reference genomes (hereafter non-reference species).
Fortunately, diverse genome partitioning approaches have also been developed that enable the collection of genome-wide data at substantially reduced effort and cost compared to WGS (Davey et al. 2011). Two approaches, restriction-site-associated DNA sequencing (RAD-seq and related approaches; Miller et al. 2007; Baird et al. 2008; Elshire et al. 2011; Peterson et al. 2012; Wang et al. 2012) and whole transcriptome shotgun sequencing (RNA-seq; Wang et al. 2009), have quickly become the predominant genome partitioning methods used in evolutionary studies. Both RAD-seq and RNA-seq are relatively simple to implement and can be applied to an array of evolutionary questions within and between species (Davey & Blaxter 2010; Ekblom & Galindo 2011). As a simple NGS derivative of more traditional marker based approaches (Miller et al. 2007), RAD-seq in particular has emerged as the gateway genomic approach for most non-reference species.
Although these partitioning approaches are providing a wealth of insights in non-reference species, they can also be strongly limiting for some research questions (Ku et al. 2012; Rubin et al. 2012; Arnold et al. 2013; Henning et al. 2014) or even more effective when used in concert with other NGS strategies. High-throughput targeted capture is a general class of methods that achieves genome partitioning through selective enrichment of specific subsets of the genome prior to NGS. Targeted capture approaches were developed as more cost-effective and high-throughput alternatives to WGS and multiplex PCR, respectively, to obtain large datasets of orthologous loci across many individuals (Olson 2007). The first proof-of-principle high throughput capture studies targeted large subsets of the human genome using arrays (6726 exons, ~5 Mb, Albert et al. 2007; 204,490 exons, 42.7 Mb, Hodges et al. 2007; ~10,000 exons, 6.7 Mb, Porreca et al. 2007; 304 kb of the X chromosome, Okou et al. 2007), demonstrating the massive scaling potential of this approach. The subsequent development of in-solution targeted capture (Gnirke et al. 2009) provided numerous technical improvements over array-based platforms (Gnirke et al. 2009; Tewhey et al. 2009a; Mamanova et al. 2010) and has emerged as the industry standard. In addition to advantages in scalability and cost-effectiveness, targeted capture generally provides enhanced data quality relative to alternative genome partitioning approaches, including lower variance in target coverage, more accurate SNP calling, higher reproducibility, and longer assembled contigs (Gnirke et al. 2009; Tewhey et al. 2009a; Ku et al. 2012; Harvey et al. 2013).
The benefits of high throughput targeted capture were immediately apparent in biomedical fields (Olson 2007; Hodges et al. 2007). By focusing NGS efforts on “high-value genomic regions” (Hodges et al. 2007), such as exons or structural variants, targeted capture has yielded tremendous power to identify genetic variants associated with simple and complex human diseases (Choi et al. 2009; Ng. et al. 2009; O’Roak et al. 2011; Worthey et al. 2011; Calvo et al. 2012; Rivière et al. 2012; Zaidi et al. 2013; Gee et al. 2014; Iossifov et al. 2014; Guo et al. 2015). In parallel with its medical applications, targeted capture began to emerge as a powerful approach to address evolutionary questions in humans (Briggs et al. 2009; Burbano et al. 2010; Krause et al. 2010; Li et al. 2010; Yi et al. 2010). Although initially restricted to species with sequenced genomes (e.g., chimpanzee, Perry et al. 2010; Drosophila melanogaster, Wang et al. 2010; maize, Fu et al. 2010), more recent extensions to non-reference species have demonstrated the potential to effectively use targeted capture across diverse taxa (Cosart et al. 2011; Mason et al. 2011; Vallender 2011; Bi et al., 2012, 2013; Good et al. 2015). Nonetheless, targeted capture remains relatively uncommon in evolutionary biology studies.
A major impediment to the widespread use of targeted capture in non-reference species is the challenge of designing a capture probe set (Elshire et al. 2011), which by definition requires a priori knowledge of target sequences. Fulfilling this simple requirement can be daunting. The goal of this review is to discuss how some of these practical challenges can be overcome in non-reference species and to draw attention to the utility of targeted capture in addressing central questions in evolutionary and ecological genomics. We assume a basic understanding of hybridization-based capture methods, the details of which have been reviewed elsewhere (e.g., Mamanova et al. 2010). Finally, given the ongoing trend of decreasing NGS costs, we consider the extent to which targeted capture and other genome partitioning approaches are transitory genomic tools on the path to routine whole genome sequencing.
Targeted capture without a reference genome
Targeted capture approaches rely on prior sequence knowledge. Thus, the first major implementation challenge is identifying the genomic sequences to be used for capture design. Several studies have focused on technical aspects of capture design and implementation, including probe design (Tewhey et al. 2009a; Ávila-Arcos et al. 2011; Clark et al. 2011), library preparation protocols (Mamanova et al. 2010; Meyer & Kircher 2010; Harakalova et al. 2011; Kircher et al. 2012; Rohland & Reich 2012), and bioinformatics (Sulonen et al. 2011; Asmann et al. 2012; Cosart et al. 2014; Jiang et al. 2015). Few have discussed complexities that arise with identifying genomic regions of interest in non-reference species (but see Bi et al. 2012). Several solutions that circumvent the need of a reference genome for focal species have now emerged, enabling an expansion of targeted capture to a wide array of taxa. Broadly, these solutions fall into PCR capture, de novo assembly capture, and divergent reference capture (Fig. 1, Table 1).
Figure 1.

The steps to synthesize a targeted capture probe set in non-reference taxa using four alternative approaches. In PCR capture, target sequences are identified and amplified with PCR. Amplicons are then biotinylated to create long single-stranded probes to capture these sequences across a diverse range of species. For de novo assembly capture, initial transcriptomic, RAD-seq, or WGS experiments are used to generate de novo assemblies from which a targeted capture probe set is designed. In transcriptome-based approaches, probes are designed to target regions within a single exon corresponding to a transcript or set of transcripts of a gene. For RAD-seq or WGS capture, probes may target population-informative markers from the assembly. Finally, using a reference genome assembly of a divergent species, genomic sequences of interest and their locations (e.g., often the chromosome and the start and stop position of the interval) are identified and a probe set is designed to tile across the targeted regions.
Table 1.
The scalability, cost, taxonomic range of application (the level of species divergence from probes), and required genomic knowledge associated with three approaches of targeted capture in non-reference species.
|
|
|||
|---|---|---|---|
| Non-reference targeted capture methods | |||
| PCR | de novo assembly | Divergent genome | |
| Feasible range of targeted loci | up to ~100 | up to hundreds of thousands | up to hundreds of thousands |
| Total probe synthesis cost (maximum number of targets) | low | high | intermediate |
| Probe synthesis cost per probe | high | intermediate | low |
| Taxonomic range of application | Depends on targets, but relatively high | Transcriptome: high RAD-seq: moderate WGS: high to moderate depending on target | Depends on targets (high for exons or UCEs, low for quickly evolving loci) |
| Genomic knowledge required | PCR primer sequences | De novo transcriptome, RAD, or whole genome assembly | Annotated genome assembly or multisequence alignment |
Under the PCR-based approach, PCR products can be used as capture probes for high throughput sequencing of a relatively small number of genomic regions (Maricic et al. 2010; Mariac et al. 2014; Peñalba et al. 2014; Tsangaras et al. 2014). Here PCR is performed on a set of target loci, those PCR products are biotinylated or affixed to an array and used to capture homologous sequences for subsequent NGS (Fig. 1). This approach leverages the extensive availability of primer sequences for many non-reference species (Portik et al. 2012; Peñalba et al. 2014). In addition, probes generated from long-range PCR may increase capture efficiency of divergent sequences (e.g., up to 27% mitochondrial divergence; Peñalba et al. 2014), thus increasing the taxonomic range of this approach (Table 1). PCR-based capture is perhaps the simplest and least expensive application of targeted capture (assuming capture of a modest numbers of loci) because it avoids the most cost-intensive aspect of target capture: the synthesis of large probe sets. However, this approach is relatively low throughput and most appropriate for questions that require only a handful of loci (e.g., up to 100 loci, Table 1), such as phylogenetic or phylogeographic studies (Mariac et al. 2014; Peñalba et al. 2014).
Several capture approaches for non-model taxa leverage other sequencing strategies to develop de novo sequence assemblies from which to design a capture (Fig. 1). These approaches are appealing because they can theoretically be applied to any species where a de novo sequence assembly has been or could be generated. For instance, de novo RNA-seq transcriptomes or expressed sequence tag (EST) data may be used to design probes corresponding to exonic regions (Bi et al. 2012; Salmon et al. 2012; Bi et al. 2013; Hebert et al. 2013; Neves et al. 2013; Müller et al. 2014; Good et al. 2015). A minor technical challenge lies in identifying exon boundaries, because capture probes that span exon boundaries will result in low coverage of these regions and lower overall capture performance (Bi et al. 2012; Neves et al. 2013). Exon boundaries tend to be conserved and can usually be identified through comparison to annotation from fairly divergent reference genomes (Bi et al. 2012). Similarly, a low coverage WGS experiment could be used to generate a de novo assembly with probes designed to target anonymous or informative genomic regions (Linnen et al. 2013). Low coverage WGS data may also be aligned to a closely related reference genome to identify conserved exonic regions for capture. Shotgun WGS may be less tractable for species with large, low complexity genomes (see Box 2), though repetitive regions can be masked to avoid non-specific capture. As the costs of sequencing continue to drop, WGS will likely become an increasingly attractive alternative for capture design.
Box 2. Genome and locus-specific characteristics.
Intrinsic qualities of study organisms, such as genome size and complexity, should weigh heavily in decisions on sequencing strategy. Small, compact genomes present relatively few challenges to WGS (Davey et al. 2011; Riberio et al. 2012), while large genomes characterized by a high proportion repetitive elements (e.g., many eukaryotic genomes) may be more amenable to reduced representation sequencing approaches (Hirsch et al. 2014; Sims et al. 2014). However, targeted capture of repetitive sequences can result in substantial off-target capture and interfere with efficient on-target capture (Mercer et al. 2014; Saintenac et al. 2014). Approaches have been developed to de-enrichment for repetitive sequences, which may help to alleviate these problems (e.g., Fu et al. 2010); regardless, for many studies it is preferable to avoid these regions in favor of functional elements within unique genomic sequences. To this end, targeted capture has become synonymous with exome capture and has been used extensively to characterize protein-coding variation in plants species with large polyploid genomes and high transposable element abundance (Saintenac et al. 2011; Winfield et al. 2012; Zhou & Holliday 2012; Uitdewilligen et al. 2013; Neves et al. 2014). These studies demonstrate the utility of targeted capture to generate genome-wide datasets in species that have previously been intractable for WGS efforts.
Although targeted capture has primarily been applied to protein-coding exons (Hodges et al. 2007), regulatory regions such as UTR, miRNA, promoter regions, and other non-coding elements can also be targeted in a highly parallel fashion (Bainbridge et al. 2011; Hebert et al. 2013; Linnen et al. 2013; Carneiro et al. 2014a; Carneiro et al. 2014b; Schiessl et al. 2014). In addition, non-coding regions flanking exons and other baited regions are often indirectly targeted (Samuels et al. 2013). For instance, often regions up to 250 bp from baited sequences are recovered in targeted capture studies, albeit generally at lower coverage (Yi et al. 2010; Chevalier et al. 2014). The extent of flanking coverage is simply a function of genomic library insert size. The performance of targeted capture on non-coding regions has not been explored in detail, although some studies indicate reduced capture efficiency (Bainbridge et al. 2011; Vallender 2011). Bainbridge et al. (2011) found that both conserved UTR and other regulatory regions tended to have substantially lower coverage depth compared to coding DNA sequence (CDS), which may be correlated with GC-content (see inset Figure following Bainbridge et al. 2011). Across different capture studies, loci with intermediate GC-content are often most highly represented (45%, Tewhey et al. 2009a; 53%, Saintenac et al. 2011; 55%, Ávila-Arcos et al. 2011) because these loci amplify well and hybridize most efficiently to probes (Sims et al. 2014). Introns and intergenic regions often contain a higher frequency of repetitive elements (Tewhey et al. 2009a), however non-coding capture probes can avoid annotated repetitive DNA (Carneiro et al. 2014a). An increasing number of studies are including non-coding regions in their capture designs, however the potential costs associated with capturing these regions should be considered when designing a capture.
Finally, the uniformity in coverage across targeted loci may be leveraged to infer copy number variation (CNV) based on relative coverage (Saintenac et al. 2011; Schiessl et al. 2014; Jiang et al. 2015). The utility of targeted capture in this respect is a tremendous advantage because structural variation (e.g., whole genome or gene duplications and chromosomal rearrangements) can play an important role in adaptation and speciation (Lowry & Willis 2010; Kondrashov 2012), yet identifying and characterizing structural variation is often difficult. However, stochastic variation in coverage due to biases introduced during library preparation or hybridization can lead to incorrect CNV inferences (Sims et al. 2014), and care should be taken to correct for these issues.
Figure.
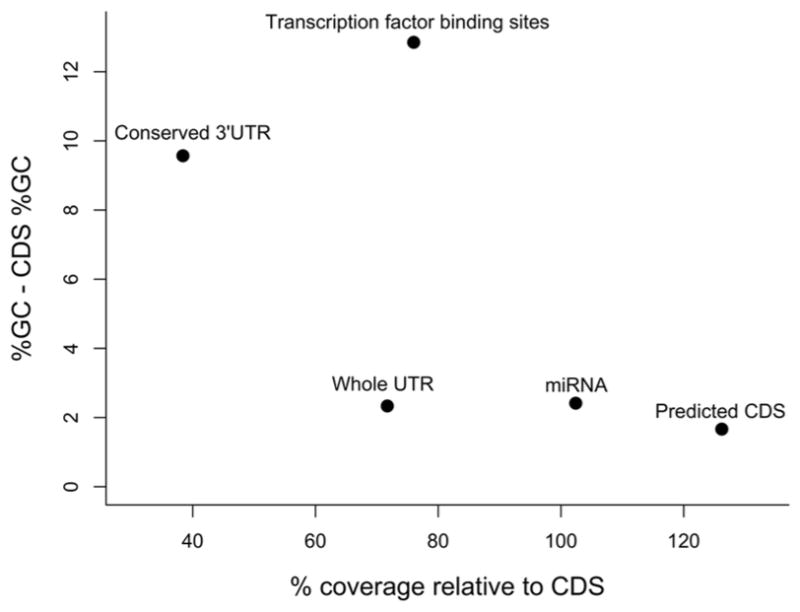
The performance of targeted capture across various genomic regions in humans. Depicted is the percent coverage relative to coding DNA sequence (CDS) as a function of deviation from GC-content of CDS (55.9%) (adapted from Bainbridge et al. 2011).
A capture could also be developed to target a panel of informative RAD markers (RAD-tags) identified from an initial RAD-seq experiment. This two-step approach, sometimes referred to as Rapture (M. Miller, pers. comm.), combines the power and ease of RAD-seq SNP discovery with the more robust technical performance of capture (i.e., higher repeatability and lower variance among samples) thereby enabling efficient and cost-effective generation of large population datasets. It is worth noting that several alternative technologies have also been developed for high-throughput genotyping of pre-defined genomic regions (e.g., see Fluidigm Access Array and other microfluidic approaches; Tewhey et al. 2009b). The details of these alternatives are beyond the scope of this review, but the choice to use capture-based resequencing versus an alternative genotyping approach usually depends on inherent cost tradeoffs between sample throughput and the total number of markers that can be efficiently assayed.
Capture approaches based on de novo sequence assemblies require performing two separate NGS experiments, decreasing cost-effectiveness (see Table 1). However, these initial experiments may be performed on a limited set of samples. For instance, a reference transcriptome can be constructed from a set of tissues from a single individual and then used to capture orthologous sequences from species across moderate evolutionary scales (Bi et al. 2012). Sequencing pools of individuals may also be preferable to identify the most informative or interesting genomic regions to sequence at a fraction of the cost of sequencing individual libraries (Schlötterer et al. 2014). However, capture approaches that first require de novo SNP discovery in a small subsample may suffer from ascertainment biases, including dropout of rare SNPs, which may affect downstream population genetic inferences (e.g., estimation of site frequency spectrum and related statistics; Clark et al. 2005; Lachance & Tishkoff 2013; McTavish & Hillis 2015). Steps may be taken to mitigate ascertainment biases by including as many samples as possible in the initial SNP discovery phase, correcting for SNP detection probabilities, or incorporating SNP ascertainment bias in population genetic analyses (Clark et al. 2005; Lachange & Tishkoff 2013).
A final approach is to design target probes based on a divergent reference genome (Cosart et al. 2011; George et al. 2011; Saintenac et al. 2011; Jin et al. 2012; Nadeau et al. 2012; Good et al. 2013; Hancock-Hanser et al. 2013; Hedtke et al. 2013; Li et al. 2013; Ilves & Lopez-Fernandez 2014) or a multisequence alignment (e.g., ultraconserved element capture; Faircloth et al. 2012; Lemmon et al. 2012; McCormack et al. 2012; Faircloth et al. 2013; McCormack et al. 2013a; Crawford et al. 2015; Leaché et al. 2015). The principle behind this strategy is straightforward: orthologous regions of the genome between divergent species can be captured if their sequences are sufficiently conserved. An appealing aspect of this approach is that annotated genomes can be exploited to interpret capture data from non-reference species without relying upon the biased representation inherent to transcriptomes. Furthermore, this approach is highly scalable and allows for capture of a handful of genome regions or even whole genomes, such as in metagenomic applications (Bos et al. 2011, Table 1). Unlike other non-reference capture approaches, divergent reference capture incurs no cost until the actual synthesis of probes because target sequence identification relies on pre-established genomic resources. Nonetheless, quantifying capture performance relative to reference divergence is critical for understanding the utility of divergent reference-based capture.
Several studies have examined the ability of probes designed from a single reference genome to capture DNA from a range of species (Vallender 2011; Jin et al. 2012; Lemmon et al. 2012; Good et al. 2013; Hancock-Hanser et al. 2013; Hedtke et al. 2013). For instance, commercially available human exome kits perform equivalently in humans and chimpanzees (~90–91% capture sensitivity) yet decline in sensitivity and depth of coverage when applied to species with >5% sequence divergence, such as macaques (Vallender 2011; Jin et al. 2012). A capture based on de novo transcriptomes from the alpine chipmunk (Tamias alpinus) was used to capture T. alpinus and three other species of chipmunks ranging up to 1.5% sequence divergence (Bi et al. 2012; 2013; Good et al. 2015). In these experiments, capture specificity and capture sensitivity were similar across all species. This same experiment included probes targeting anonymous regions from the genome of a more divergent species of squirrel (Ictidomys tridecemlineatus; ~9% sequence divergence). These divergent targets showed declines in normalized coverage in chipmunk species, consistent with the above primate studies, but high levels of overall target recovery nonetheless (~90% capture sensitivity). Hedtke et al. (2013) designed a targeted exon capture from the western clawed frog (Xenopus tropicalis) reference genome and applied it to 16 frog species spanning ~250 million years of divergence (up to ~10% sequence divergence) from the reference. Fold enrichment and capture sensitivity were strongly associated with species divergence time and loci with higher sequence divergence performed most poorly (Hedtke et al. 2013). Capture sensitivity for species >200 million years diverged was also highly variable (range 0% to ~100%), but generally less than 60%. These studies demonstrate that capture success does not seem to be compromised between closely related species but it declines abruptly at moderate levels of divergence (5–9%).
When should targeted capture be used?
No single NGS approach is ideal for all research applications. Rather, sequencing strategies should be tailored to specific research aims (Davey et al. 2011; Good 2011; Ekblom & Galindo 2011). Thus, the benefits and limits of sequencing approaches are best discussed in the context of a specific research aim. Below we highlight several active areas of evolutionary and ecological research that may benefit from targeted capture approaches, often when used in tandem with other genome partitioning approaches.
Genetic mapping of phenotypic traits
Genetic mapping remains one of the primary approaches to identify the genetic mechanisms underlying phenotypic evolution (Lynch & Walsh 1998; Stinchcombe & Hoekstra 2008). The development of NGS-based genotyping has led to tremendous advances in mapping quantitative trait loci (QTL; Baird et al. 2008; Andolfatto et al. 2011; Baxter et al. 2011; Martin et al. 2012; Weber et al. 2013; Henning et al. 2014). QTL mapping through crossing experiments or association/admixture studies requires a genetic map of the locations and relative positions of markers in the genome. To this end, RAD-seq and related approaches provide simple and inexpensive means to generate a dense map of anonymous markers (i.e., RAD-tags; Baird et al. 2008; Narum et al. 2013). However, mapping studies rarely yield single gene or marker resolution. In species with annotated reference genomes, RAD-tags can be aligned to the genome sequence to facilitate identification of candidate functional elements associated with traits (Fig. 2; Andolfatto et al. 2011). In contrast, it is very difficult to assess the proximity of QTL-associated RAD-tags to candidate genes in non-reference species (Fig. 2; Mascher & Stein 2014). In these instances targeted capture can be used to anchor anonymous genetic maps to known genes (or other functional elements), thereby informing the detection of causative loci (Fig. 2).
Figure 2.

The process of identifying causal loci underlying a phenotypic trait using different genetic mapping and sequencing strategies. With an available reference genome, RAD-seq is used to create a high-density linkage map that can be aligned to the genome to anchor RAD-tags (black ticks) to a physical map (genes are shown as red ticks and the causal locus is shown as a blue tick). To precisely localize candidate loci, initial genetic mapping can be followed by fine-scale RAD-seq mapping. Candidate loci found within the fine-mapped region can be assayed to verify their phenotypic function. Genetic mapping with RAD-seq is less powerful without a reference genome because of the difficulty identifying candidate loci. In lieu of a reference genome, high-resolution mapping with RAD-tags can be combined with low-resolution anchoring of known genes to identify candidate genes associated with a QTL and permit follow-up functional assays. High-resolution mapping with highly multiplexed exome capture genotyping could also directly reveal candidate genes to functionally test.
Several previous studies have demonstrated the power of genetic mapping with a combination of anonymous markers and a priori candidate genes (e.g., Brown et al. 2003; Steiner et al. 2007; Martin et al. 2012). Targeted capture can be used to quickly anchor RAD-tag maps to hundreds of candidate genes, providing a powerful surrogate for a whole genome reference (Baxter et al. 2011; Mascher et al. 2014; Neves et al. 2014; Fig. 2). Anchoring a RAD-based genetic map could be achieved though genotyping a relatively small subset of a larger QTL mapping panel with exon capture (i.e., low-resolution anchoring, see Fig. 2). This would still require performing exon capture on a large number of individuals, which is cost-prohibitive if only one or a few samples are captured at a time. However, standard capture platforms have the capacity for extensive custom multiplexing of individuals (Kircher et al. 2012; Rohland & Reich 2012), greatly reducing the cost of anchoring a map. Indeed, constructing high-density anchored linkage maps with only exome genotyping would also be a feasible, albeit more expensive, option that could provide many of the benefits of RAD-tags (i.e., high marker density and rapid genotyping capability; Fig. 2).
Exome data can also simplify additional fine-scale mapping to identify actual causal variants associated with phenotypic traits (Linnen et al. 2013; Chevalier et al. 2014). In contrast to anonymous RAD-tags that may lie in repetitive regions far from genes, causal variants underlying QTL may often be linked to gene coding sequences or their proximal regulatory sequences (Chevalier et al. 2014). Thus, exome genotyping used in isolation or in combination with genotyping of anonymous regions can provide a powerful and efficient means of determining which genomic regions are associated with a trait of interest (Linnen et al. 2013). To date, few studies have used targeted capture for genetic mapping of phenotypic traits outside of human disease (although see del Viso et al. 2012, Linnen et al. 2013, Tennessen et al. 2013, and Chevalier et al. 2014). Given the success of targeted capture in mapping genetic variants associated with human disease (Bamshad et al. 2011), we anticipate the capture-assisted QTL mapping of ecologically important phenotypic traits will emerge as a parallel utility.
Detecting selection in the genome
The relative importance of natural selection as a force governing evolutionary change has been a continuous source of debate amongst biologists (Fisher 1930; Wright 1932; Ford 1964; Lynch 2007). Genome technologies have finally begun to allow us to understand the frequency, mode, and distribution of selection across the genomes of diverse organisms (Begun et al. 2007; Drosophila 12 Genomes Consortium 2007; Jensen et al. 2008; Kosiol et al. 2008; Hohenlohe et al. 2010; Rubin et al. 2010; Hernandez et al. 2011; Lohmueller et al. 2011), providing important insights into this debate. However, the massive amounts of data required to assess broad-scale patterns of selection has limited the above studies to taxa with well-developed reference genomes. As a result, we still know very little about how selection shapes genomic variation across that majority of life. To advance our understanding of the evolutionary processes shaping genetic diversity it is imperative to extend these investigations to diverse natural populations. Targeted capture provides several benefits over alternative sequencing strategies to achieve this goal.
Quantifying molecular evolution using genome-wide patterns of protein divergence between species (dN/dS) is one powerful approach to detect selection that has been mostly restricted to species with reference genomes (e.g., Bazykin et al. 2004; Nielsen et al. 2005; Drosophila 12 Genomes Consortium 2007; Kosiol et al. 2008). Tests for selection based on protein divergence are appealing because they rely upon the well-established foundations of molecular evolution, are relatively robust to confounding demographic effects (Nielsen 2001), and enable functional inferences regarding the targets of selection. Targeted exome capture offers a highly cost-effective approach to extend these tests beyond classic model systems to a broader range of taxa (Burbano et al. 2010; Aagard et al. 2013; Vilstrup et al. 2013; George et al. 2011; Good et al. 2013). For instance, George et al. (2011) performed an exome capture of several Old and New World monkeys and revealed novel targets of positive selection on genes associated with keratinization or the conversion of epithelial cells to keratin. Moreover, comparative exome data can be used to move beyond the post hoc inferences provided by standard genome-wide scans for selection towards a priori hypothesis testing of specific genetic pathways or classes of genes (e.g., Smadja et al. 2012; Nadeau et al. 2012; Aagaard et al. 2013; Good et al. 2013). Thus, in addition to elucidating the frequency and mode of selection, comparative molecular evolutionary studies of targeted exome data can provide essential information on the ecological or life history drivers of molecular evolution.
Tests of selection using species divergence do not rely on population sampling within species; however, population genetic approaches to infer selection based on patterns of polymorphism within species (e.g., FST, site-frequency spectrum, linkage disequilibrium) or polymorphism compared to divergence between species (e.g., HKA test, McDonald-Kreitman test) require sampling many individuals to accurately estimate allele frequencies (Li et al. 2010; Hvilsom et al. 2012; Tennessen et al. 2012; Fu et al. 2013). While this level of population sampling is often not feasible with WGS or RNA-seq, exome capture provides a powerful platform to collect population-level genomic data. Targeted capture has frequently been used to estimate the efficacy of selection in populations (Hvilson et al. 2012; Veeramah et al. 2014; Bataillon et al. 2015) and to infer local adaptation by identifying highly differentiated loci between populations (Yi et al. 2010; Nadeau et al. 2012; Smadja et al. 2012; Hebert et al. 2013; Müller et al. 2014) or signatures of selective sweeps (Aagaard et al. 2013; Linnen et al. 2013; Tsangaras et al. 2014). The potential power of capture-based population genomic studies is exemplified by Yi et al. (2010) who used whole exome capture to investigate the genetic basis of high altitude adaptation in Tibetans. They found strong evidence that a transcription factor, EPAS1, involved in red blood cell production was involved in local adaptation to hypoxia based on patterns of differentiation between low elevation Han Chinese and high elevation Tibetan populations.
Finally, targeted capture is easily scalable from hundreds (Smadja et al. 2012) to hundreds of thousands of genomic regions (Hodges et al. 2007; Yi et al. 2010) and can include both coding and linked non-coding regions (Bainbridge et al. 2011; Hebert et al. 2013; Carneiro et al. 2014a; Carneiro et al. 2014b; Schiessl et al. 2014). Non-coding capture data sets (e.g., introns, intergenic regions) can serve as appropriate null models of genomic variation for inferring selective sweeps within the genome (e.g., Carneiro et al. 2014b) or regions of elevated differentiation between species (e.g., Carneiro et al. 2014a). Alternatively, UTRs and conserved non-coding regions often show strong signatures of adaptive evolution (Halligan et al. 2013; Carneiro et al. 2014b) and may be targeted to investigate the history of selection within these regions. Sequencing long contiguous regions of exons, flanking introns, up- or downstream regulatory sequence or even large tracts on non-coding intergenic regions generally provides more power to detect selective sweeps (Thornton & Jensen 2007) and to estimate the overall strength and genomic rate of selective sweeps (Jensen et al. 2008) when compared to sequencing shorter regions. Furthermore, the lower coverage variance and general high quality of targeted capture data offers several advantages over alternative approaches. For example, RAD-seq data can suffer from numerous data quality issues, such as allelic bias or dropout, which can impact population genetic inferences (Gautier et al. 2013; Henning et al. 2014). Here the increased data quality and the dynamic flexibility of targeted capture to scale both number and size of targets can provide a decided advantage over RAD-seq and other genome partitioning approaches.
Phylogenetics
Until recently, NGS-based genomics has arguably had a less profound impact on phylogenetics compared to other fields of biology (McCormack et al. 2013b; Jarvis et al. 2014; Mirarab et al. 2014; Misof et al. 2014). In addition to the computational challenges of inferring phylogenies from millions of DNA base pairs, certain phylogenetic problems are not easily resolved with the addition of more sequence data (Philippe et al. 2011). One way to ameliorate these issues is to sequence a tailored subset of informative orthologous loci to maximize phylogenetic signal while minimizing non-phylogenetic signal and computational effort (Philippe et al. 2005; Philippe et al. 2011). Here, the flexibility of targeted capture offers a tremendous advantage. Customized capture designs can target hundreds or thousands of loci (Faircloth et al. 2012; Mandel et al. 2014), slowly or quickly evolving loci (Mason et al. 2011; Lemmon & Lemmon 2013; McCormack et al. 2013a), and nuclear or organelle loci (Hedtke et al. 2013; Vilstrup et al. 2013; Ilves & Lopez-Fernandez 2014) to resolve an array of phylogenetic questions. As such, phylogeneticists are increasingly turning to targeted capture approaches to resolve their questions.
To maximize phylogenetic information, it is important to select loci with evolutionary rates appropriate to resolve a given relationship (Philippe et al. 2011). For instance, deep phylogenetic nodes are resolved with slowly evolving sequences that retain signals of orthology across distant taxa (McCormack et al. 2012). Genomic data from restriction enzyme-based approaches are beginning to be used for phylogenetic inference mostly below the genus level (e.g., Emerson et al. 2010; Eaton & Ree 2013). However, these data seem to be unreliable beyond moderate phylogenetic depth because restriction cut sites are often not conserved across taxa, leading to high levels of missing data and incorrect topologies (Rubin et al. 2012; Wagner et al. 2013; Leaché et al. 2015). High throughput targeted capture of slowly evolving ultraconserved element (UCE) markers has emerged as a very promising method to resolve deep nodes in the vertebrate phylogeny (Faircloth et al. 2012; Lemmon et al. 2012; McCormack et al. 2012; Faircloth et al. 2013; McCormack et al. 2013a; Crawford et al. 2015; Leaché et al. 2015). Capture of UCEs is appealing because one core set of loci can be identified from highly divergent reference genomes and then broadly applied to diverse taxa, enhancing comparisons among datasets while obviating the need to continually re-design custom probes. This method also should work across more shallow evolutionary time scales because UCEs flank more variable regions (Faircloth et al. 2012) that can provide resolution of more recent splitting events (Smith et al. 2014).
One looming issue with UCE markers is the extent to which selection might impact their application to various evolutionary questions. By design, UCEs are expected to experience among the strongest levels of purifying selection in the genome. While most of the phylogenetic signal from UCE regions comes from less-constrained sites linked to UCEs, these linked regions should nevertheless be strongly influenced by background selection (Charlesworth et al. 1993; 1995; Hahn 2008; Charlesworth 2012; Halligan et al. 2013). Thus, UCE markers are expected to have small effective population sizes and highly skewed allele frequencies relative to other genomic regions. It is important to note that patterns of genetic variation near protein-coding regions are also likely to be strongly influenced by background selection (Halligan et al. 2013), though the context dependence is better understood. These issues might not matter for some phylogenetic questions, but background selection does have profound impacts on most inferences that depend on patterns of population-level variation (Hammer et al. 2010; Hahn 2008). Thus, the validity of using UCE-linked markers to address population genetic questions should be considered carefully (e.g., coalescent history, effective population size, gene flow, phylogeography, etc.).
Protein-coding sequence can also be used for phylogenetic reconstruction at moderate to deep evolutionary scales due to their increased conservation relative to most non-coding regions (e.g., ~20% of mammalian protein-coding bases are evolutionarily constrained compared to ~4% genome-wide; Lindblad-Toh et al. 2011). Protein-coding sequences may be more appropriate than UCEs for organisms such as plants with large, complex, and repetitive genomes where UCE identification may be difficult (Mandel et al. 2014). Phylogenetic studies in frogs (Hedtke et al. 2013), cichlids (Ilves & López-Fernández 2014), and flowering plants (Mandel et al. 2014) have used protein-coding regions to show the power of these datasets for resolving complex phylogenetic histories.
Ancient DNA and metagenomic applications
Ancient or historic DNA (hereafter aDNA) extracted from paleontological remains, sediments, and museum specimens can provide a detailed understanding of the evolutionary history of lineages (Krings et al. 1997; Willerslev & Cooper 2005; Briggs et al. 2009; Green et al. 2010; Stoneking & Krause 2011; Orlando et al. 2013). Ancient DNA studies were traditionally hindered by limitations of PCR and DNA sequencing (Fig. 3). For instance, the short and highly damaged fragments that characterize aDNA often impair traditional PCR amplification (Knapp & Hofreiter 2010; Rowe et al. 2011; Tin et al. 2014). However, aDNA samples are highly amenable to many NGS approaches where library preparation requires shearing genomic DNA to the size range generally observed in aDNA (~40–500 bp, Pääbo 1989; Green et al. 2008; Sawyer et al. 2012; Fig. 3). As such, the analysis of aDNA has been greatly enabled by NGS (Green et al. 2010; Krause et al. 2010; Rasmussen et al. 2010; Meyer et al. 2012).
Figure 3.

The relative suitability of different NGS approaches for nucleic acid sources of varying levels of degradation (ranging from fresh samples to ancient samples) and non-target contamination (ranging from no contamination to extremely low levels of target sequence). Darker colored parts of the bars indicate a higher suitability of the approach whereas light colors indicate a low suitability.
An enduring challenge of working with aDNA samples from paleontological samples is to isolate sequences of interest from a complex blend of bacterial DNA and target DNA (Green et al. 2006; Àvila-Arcos et al. 2011; Carpenter et al. 2013). For example, remains of ~55,000 year old bone fragments used to generate the Neandertal reference genome were comprised of ~99% contaminant environmental DNA (Green et al. 2010). Whole genome shotgun sequencing of these types of aDNA is highly inefficient (Fig. 3). To overcome this issue, ancient hominin researchers have used capture to sequence targets ranging from complete mtDNA genomes (Briggs et al. 2009; Krause et al. 2010) to thousands of exons (Burbano et al. 2010; Castellano et al. 2014). By designing probes that capture specific target sequences, the problem of sequencing large amount of contaminant DNA is greatly diminished. These studies were some of the first to employ targeted capture outside of biomedical research and targeted capture has continued to expand across aDNA research (e.g., Àvila-Arcos et al. 2011; Bos et al. 2011; Schuenemann et al. 2011; Carpenter et al. 2013; Castellano et al. 2014; Enk et al. 2014).
Natural history museum specimens provide a vast repository of biological data that suffer from largely the same problems that afflict aDNA from paleontological remains (e.g., DNA degradation and, to a lesser extent, contamination). However, the utility of natural history collections for targeted capture investigations is just beginning to be realized (Mason et al. 2011; Bi et al. 2013; Hedtke et al. 2013; Vilstrup et al. 2013; Tsangaras et al. 2014). Bi et al. (2013) provided an exciting advance in genomic analysis of museum samples by capturing ~12,000 exons from forty alpine chipmunk (Tamias alpinus) specimens, including 20 contemporary samples and 20 collected in 1915. Exome data from the historic samples were compared to modern samples to infer recent shifts in genetic diversity and population structure coincident with range restrictions associated with climate change. This study provides a glimpse into the potential role of targeted capture in expanding natural history museum collections for genomics research (Nachman 2013). Given the vast number of specimens currently archived in museums, efficient incorporation of museum collections into genomics represents one of the most promising, but as of yet underutilized, applications of targeted capture.
A technically related issue is the isolation and sequencing of DNA from an environmental sample (metagenomics) to study pathogen evolution or microbial community composition (e.g., microbiome research). In particular, targeted capture has greatly enabled the recovery of pathogen sequences from host tissues (Bos et al. 2011; Kent et al. 2011; Schuenemann et al. 2011; Geniez et al. 2012; Bent et al. 2013; Wagner et al. 2014; Bos et al. 2015). For instance, Bos et al. (2011) used probes from modern strains of the pathogenic bacterium Yersinia pestis to reconstruct whole genome sequences from ancient Y. pestis strains found within the teeth of Black Death victims from 1348–1350. They found that the ancient Y. pestis strain responsible for the Black Death gave rise to all modern Y. pestis lineages. Later, this approach was used to demonstrate that the Plague of Justinian (541–543 AD) was likely caused by an independent emergence of Y. pestis in humans from rodent vectors (Wagner et al. 2014).
A central goal of community genomics is to characterize species diversity within a community. While shotgun-sequencing approaches have provided detailed profiles of microbial genetic diversity in environmental samples (Breitbart et al. 2002; Tyson et al. 2004), WGS is still difficult and expensive when compared to traditional methods of 16S rRNA sequencing (Hugenholz et al. 1998). The use of one or a few DNA markers for species identification is limited in many ways but remains a common approach in metagenomics (Valentini et al. 2009; Portik et al. 2012). Targeted capture can provide a high-throughput approach to analyze community composition using species barcodes and to explore functional variation in a community (e.g., by targeting genes underlying specific ecological functions; Denonfoux et al. 2013). Gut microbiomes constitute one type of community that has gained substantial interest recently for its potential role governing health and contributing to broader ecological and evolutionary patterns (Muegge et al. 2011; The Human Microbiome Project Consortium 2012). Sufficient sequence data are accumulating for the human microbiome to facilitate targeted capture of large portions of the diversity that comprise this community. These general principles can be extended to any type of species detection, including non-invasive sampling (Perry et al. 2010; Kidd et al. 2014). While relatively few studies have delved into the use of targeted capture for species detection purposes, this also represents a promising avenue of future research. Indeed, genetic surveys of environmental DNA (eDNA) are emerging as a powerful tool for species detection and biodiversity monitoring (Schnell et al. 2012; Wilcox et al. 2013; Bohmann et al. 2014). Thus far eDNA studies have predominantly relied upon PCR-based approaches (Bohmann et al. 2014). However, further development of targeted capture for eDNA applications seems inevitable given the strong parallels in technical challenges presented by eDNA and aDNA.
Is target capture a fleeting method?
Targeted capture holds tremendous potential to advance evolutionary and ecological research (Fig. 4). Genome partitioning approaches predominate many evolutionary studies simply because WGS remains prohibitively expensive or difficult for problems that require large sample sizes and for species with large genomes (although see Nystedt et al. 2013 and Neale et al. 2014). But it would be naïve to assume that many of the current limitations of WGS will persist into the future. NGS costs continue to plummet, while sequencing technologies and assembly methods continue to evolve. The difficulties associated whole genome assembly are quickly being overcome with technologies that generate long reads spanning repetitive regions (Treangen & Salzberg 2012; Huddleston et al. 2014; Goodwin et al. 2015) and with powerful new assembly approaches (Putnam et al. 2015). Given this, it is reasonable to speculate that targeted capture and other genome partitioning approaches will soon become obsolete (Ku et al. 2012). What roles might targeted capture play, if any, in genomic studies once WGS becomes universally accessible? Are there intrinsic advantages to targeted capture over WGS?
Figure 4.

The relative suitability of targeted capture (purple), RAD-seq (blue), and transcriptomic approaches (green) for genetic mapping of phenotypic traits, population genetics (includes inferring population history and detecting population level signatures of selection), molecular evolution (e.g., rates of protein evolution), phylogenetics, ancient DNA, and metagenomics. Specific height of each bar is arbitrary but increasing height corresponds to increasing suitability.
Many evolutionary and ecological questions simply do not require whole genome sequences (Davey et al. 2011). Even as WGS becomes economically feasible, genome partitioning approaches will remain preferred if they are cheaper for broader sampling or the data are easier to analyze. The inherent tradeoff between sample size and genome coverage means that, regardless of sequencing costs, exome capture will always enable sequencing larger samples of individuals than WGS given the same sequencing effort. The latter strategy may be favored or required for addressing detailed population genetic questions, identifying rare variants associated with phenotypes (Tennessen et al. 2012), or identifying epistatic interactions among genes (Wei et al. 2014). Eventually, however, the costs of WGS and targeted capture may drop to the point where the large differences in throughput between the approaches outweigh minor differences in cost. Regardless, targeted capture approaches are likely to remain well suited for certain questions. In phylogenetic contexts targeted sequencing of loci with desired rates of evolution may often lead to better resolved topologies than whole genome data. Targeted capture also offers the ability to isolate specific sequences of interest from a blend of DNA of many organisms, which holds tremendous utility for aDNA research, metagenomics, and host-parasite or pathogen research. While we are careful to not disregard the importance of non-coding DNA (e.g., transposable elements) in evolution, for many questions we are specifically interested in coding variation and their regulatory regions and thus not the vast majority of the genome.
Ultimately, the increasing feasibility of WGS will enable rather than nullify targeted capture approaches (Teer & Mullikin 2010). We expect targeted capture and other genome partitioning approaches to remain a vital tool in evolutionary and ecological research in the near future. Our genomics tool set continues to diversify and we no longer have to rely on a single approach for all research questions. Rather, we are afforded the opportunity to customize methods to suit our specific research. Targeted capture is only one method to choose from, but given the flexibility of this approach we believe its broad implementation would benefit many evolutionary and ecological disciplines.
Box 1. Definitions.
- aDNA
ancient DNA, DNA isolated from ancient or historic sources including paleontological remains, fossil remains, sediment or ice cores, and museum specimens.
- Array capture
targeted capture using probes fixed on a glass slide.
- Bait/probe
Single-stranded oligonucleotides with a complementary sequence to a portion of a targeted genomic interval.
- Capture sensitivity
the percentage of targets covered by at least one mapped read.
- Capture specificity
the percentage of unique reads mapping to target sequences.
- eDNA
environmental DNA, DNA isolated from an environmental sample (e.g., water, sediment, gut) containing a blend of DNA sequences from many organisms.
- Exome
the portion of the genome coding for exons.
- Exome capture
a targeted capture of the exome or component of the exome (e.g., often protein-coding exons).
- Fold enrichment
the fold-increase in the number of reads covering targeted regions over the expected number from a shotgun whole genome approach.
- Hybridization
the step of targeted capture where single-stranded genomic sequences bind to complementary probe sequences for enrichment.
- In-solution capture
targeted capture using biotinylated probes that allow hybridization to occur in a liquid solution. Biotin molecules binds with high affinity to streptavidin beads, which allows target sequences bound to probes to be isolated from non-target sequences.
- Non-reference species
a species lacking a reference genome.
- UCE
ultraconserved element, a region in the genome that shows extraordinary sequence conservation across deeply divergent taxa, likely as a result of strong purifying selection.
Box 3. Genomic samples.
The quality and quantity of available tissue or nucleic acid samples can be a major limiting factor in any genetic study. Low quality nucleic acid sources (e.g., aDNA and eDNA) often preclude transcriptome approaches because of the rapid rates of mRNA degradation (Sharova et al. 2009; Fig. 3) and changes in transcript profiles as a function of the time lag between tissue collection and preservation (Sanoudou et al. 2004; Birdsill et al. 2010). Post-mortem enzymatic, hydrolytic, and oxidative reactions degrade and damage genomic DNA over time (Pääbo et al. 2004; Briggs et al. 2007), but at a much slower rate compared to RNA. Whole genome sequencing, targeted capture, and restriction-enzyme digest approaches have all been applied to degraded genomic samples (Green et al. 2010; Meyer et al. 2012; Bi et al. 2013; Tin et al. 2014), although the extent of DNA degradation may dictate which sequencing approaches are most appropriate. For instance, additional DNA digestion with restriction enzymes may produce fragments that are too short for analysis and lead to extensive data loss (Tin et al. 2014, Burrell et al. 2015; Fig. 3). Shotgun sequencing and targeted capture can be applied to degraded DNA without additional fragmentation, although the choice of which of these strategies to pursue with degraded DNA depends largely on the study organism, the amount of sample, and the study question (Burrell et al. 2015).
Acknowledgments
We thank Ryan Bracewell, Tom Brekke, Colin Callahan, Zak Clare-Salzler, Findley Finseth, Erica Larson, Erin Nordquist, Brice Sarver, and Katie Zarn for valuable comments and insight on ideas and figures in this paper. Two anonymous reviewers provided helpful criticism on an earlier version of this manuscript. Matthew R. Jones is supported by a National Science Foundation Graduate Research Fellowship under Grant No. (DGE-1313190). Many of the concepts discussed in this review were developed from ongoing research in the Good lab, supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (award number R01HD73439) and the National Institute of General Medical Sciences (award number R01GM098536) of the National Institutes of Health.
References
- Aagaard JE, George RD, Fishman L, MacCoss MJ, Swanson WJ. Selection on plant male function genes identifies candidates from reproductive isolation of yellow monkeyflowers. PLoS Genetics. 2013;9:e1003965. doi: 10.1371/journal.pgen.1003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert TJ, Molla MN, Muzny DM, et al. Direct selection of human genomic loci by microarray hybridization. Nature Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- Andolfatto P, Davison D, Erezyilmaz D, et al. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Research. 2011;21:610–617. doi: 10.1101/gr.115402.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold B, Corbett-Detig RB, Hartle D, Bomblies K. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Molecular Ecology. 2013;22:3179–3190. doi: 10.1111/mec.12276. [DOI] [PubMed] [Google Scholar]
- Asmann YW, Middha S, Hossain A, et al. TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics. 2012;28:277–278. doi: 10.1093/bioinformatics/btr612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ávila-Arcos MC, Cappellini E, Romero-Navarro JA, et al. Application and comparison of large-scale solution-based DNA capture-enrichment methods on ancient DNA. Scientific Reports. 2011;1:74. doi: 10.1038/srep00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Wang M, Wu Y, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biology. 2011;12:R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Reviews Genetics. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Bataillon T, Duan J, Hvilsom C, et al. Inference of purifying and positive selection in three subspecies of chimpanzees (Pan troglodytes) from exome sequencing. Genome Biology and Evolution. 2015;7:1122–1132. doi: 10.1093/gbe/evv058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SW, Davey JW, Johnston JS, et al. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS One. 2011;6:e19315. doi: 10.1371/journal.pone.0019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazykin GA, Kondrashov FA, Ogurtsov AY, Sunyaev S, Kondrashov AS. Positive selection at sites of multiple amino acid replacements since rat-mouse divergence. Nature. 2004;429:558–562. doi: 10.1038/nature02601. [DOI] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biology. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bent ZW, Tran-Gyamfi MB, Langevin SA, et al. Enriching pathogen transcripts from infected samples: a capture-based approach to enhanced host-pathogen RNA sequencing. Analytical Biochemistry. 2013;438:90–96. doi: 10.1016/j.ab.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Bi K, Vanderpool D, Singhal S, Linderoth T, Moritz C, Good JM. Transcriptome based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics. 2012;13:403. doi: 10.1186/1471-2164-13-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Linderoth T, Vanderpool D, Good JM, Nielsen R, Moritz C. Unlocking the vault: next-generation museum population genomics. Molecular Ecology. 2013;24:6018–6032. doi: 10.1111/mec.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsill AC, Walker DG, Lue L, Sue LI, Beach TG. Postmortem interval effect on RNA and gene expression in human brain tissue. Cell and Tissue Banking. 2011;12:311–318. doi: 10.1007/s10561-010-9210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmann K, Evans A, Gilbert MTP, et al. Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology and Evolution. 2014;29:358–367. doi: 10.1016/j.tree.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Bos KI, Schuenemann VJ, Golding GB, et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011;478:506–510. doi: 10.1038/nature10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos KI, Jäger G, Schuenemann VJ, et al. Parallel detection of ancient pathogens via array-based DNA capture. Philosophical Transactions of the Royal Society B. 2015;370:20130375. doi: 10.1098/rstb.2013.0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Salamon P, Andresen B, et al. Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences USA. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Stenzel U, Johnson PFL, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proceedings of the National Academy of Sciences USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Good JM, Green RE, et al. Target retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- Brown GR, Bassoni DL, Gill GP, et al. Identification of quantitative trait loci influencing wood property traits in Loblolly Pine (Pinus taeda L.). III. QTL verification and candidate gene mapping. Genetics. 2003;164:1537–1546. doi: 10.1093/genetics/164.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell AS, Disotell TR, Bergey CM. The use of museum specimens with high throughput DNA sequencers. Journal of Human Evolution. 2015;79:35–44. doi: 10.1016/j.jhevol.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbano HA, Hodges E, Green RE, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328:723–725. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante CD, Fledel-Alon A, Williamson S, et al. Natural selection on protein coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Science Translational Medicine. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro M, Albert FW, Afonso S, et al. The genomic architecture of population divergence between subspecies of the European rabbit. PLoS Genetics. 2014a;10:e1003519. doi: 10.1371/journal.pgen.1003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro M, Rubin C-J, Di Palma F, et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science. 2014b;345:1074–1079. doi: 10.1126/science.1253714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter ML, Buenrostro JD, Valdiosera C, et al. Pulling out the 1%: whole genome capture for the targeted enrichment of ancient DNA sequencing libraries. The American Journal of Human Genetics. 2013;93:852–864. doi: 10.1016/j.ajhg.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano S, Parra G, Sánchez-Quinto FA, et al. Patterns of coding variation in the complete exomes of three Neandertals. Proceedings of the National Academy of Sciences USA. 2014;111:6666–6671. doi: 10.1073/pnas.1405138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Morgan MT. The pattern of neutral molecular variation under the background selection model. Genetics. 1995;141:1619–1632. doi: 10.1093/genetics/141.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The role of background selection in shaping patterns of molecular evolution and variation: evidence from variability on the Drosophila X chromosome. Genetics. 2012;191:233–246. doi: 10.1534/genetics.111.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier FD, Valentim CLL, LoVerde PT, Anderson TJC. Efficient linkage mapping using exome capture and extreme QTL in schistosome parasites. BMC Genomics. 2014;15:617. doi: 10.1186/1471-2164-15-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Research. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Chen R, Lam HYK, et al. Performance comparison of exome DNA sequencing technologies. Nature Biotechnology. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosart T, Beja-Pereira A, Chen S, Ng SB, Shendure J, Luikart G. Exome-wide DNA capture and next generation sequencing in domestic and wild species. BMC Genomics. 2011;12:347. doi: 10.1186/1471-2164-12-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosart T, Beja-Pereira A, Luikart G. EXONSAMPLER: a computer program for genome-wide and candidate gene exon sampling for targeted next-generation sequencing. Molecular Ecology Resources. 2014;14:1296–1301. doi: 10.1111/1755-0998.12267. [DOI] [PubMed] [Google Scholar]
- Crawford NG, Parham JF, Sellas AB, et al. A phylogenomic analysis of turtles. Molecular Phylogenetics and Evolution. 2015;83:250–257. doi: 10.1016/j.ympev.2014.10.021. [DOI] [PubMed] [Google Scholar]
- Davey JW, Blaxter ML. RADSeq: next-generation population genetics. Briefings in Functional Genomics. 2010;9:416–423. doi: 10.1093/bfgp/elq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- del Viso F, Bhattacharya D, Kong Y, Gilchrist MJ, Khokha MK. Exon capture and bulk segregant analysis: rapid discovery of causative mutations using high throughput sequencing. BMC Genomics. 2012;13:649. doi: 10.1186/1471-2164-13-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denonfoux J, Parisot N, Dugat-Bony E, et al. Gene capture coupled to high throughput sequencing as a strategy for targeted metagenome exploration. DNA Research. 2013;20:185–196. doi: 10.1093/dnares/dst001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Eaton DAR, Ree RH. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae) Systematic Biology. 2013;62:689–706. doi: 10.1093/sysbio/syt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. Genome sequencing and population genomics in non-model organisms. Trends in Ecology and Evolution. 2014;29:51–63. doi: 10.1016/j.tree.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson KJ, Merz CR, Catchen JM, et al. Resolving postglacial phylogeography using high-throughput sequencing. Proceedings of the National Academy of Sciences USA. 2010;107:16196–16200. doi: 10.1073/pnas.1006538107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enk JM, Devault AM, Kuch M, Murgha YE, Rouillard J-M, Poinar HN. Ancient whole genome enrichment using baits built from modern DNA. Molecular Biology and Evolution. 2014;31:1292–1294. doi: 10.1093/molbev/msu074. [DOI] [PubMed] [Google Scholar]
- Faircloth BC, McCormack JE, Crawford NG, Harvey MG, Brumfield RT, Glenn TC. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Systematic Biology. 2012;61:717–726. doi: 10.1093/sysbio/sys004. [DOI] [PubMed] [Google Scholar]
- Faircloth BC, Sorenson L, Santini F, Alfaro ME. A phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs) PLoS One. 2013;8:e65923. doi: 10.1371/journal.pone.0065923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu C-I. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature. 2002;415:1024–1026. doi: 10.1038/4151024a. [DOI] [PubMed] [Google Scholar]
- Fisher RA. The genetical theory of natural selection. Oxford Univeristy Press; Oxford, UK: 1930. [Google Scholar]
- Ford EB. Ecological genetics. Methuen; London, UK: 1964. [Google Scholar]
- Fu Y, Springer NM, Gerhardt DJ, et al. Repeat subtraction-mediated sequence capture from a complex genome. The Plant Journal. 2010;62:898–909. doi: 10.1111/j.1365-313X.2010.04196.x. [DOI] [PubMed] [Google Scholar]
- Fu W, O-Connor TD, Goo Jun, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Gharbi K, Cezard T, et al. The effect of RAD allele dropout on the estimation of genetic variation within and between populations. Molecular Ecology. 2013;22:3165–3178. doi: 10.1111/mec.12089. [DOI] [PubMed] [Google Scholar]
- Gee HY, Otto EA, Hurd TW, et al. Whole-exome resequencing distinguished cystic kidney diseases from phenocopies in renal ciliopathies. Kidney International. 2014;85:880–887. doi: 10.1038/ki.2013.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geniez S, Foster JM, Kumar S, et al. Targeted genome enrichment for efficient purification of endosymbiont DNA from host DNA. Symbiosis. 2012;58:201–207. doi: 10.1007/s13199-012-0215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George RD, McVicker G, Diederich R, et al. Trans genomic capture and sequencing of primate exomes reveals new targets of positive selection. Genome Research. 2011;21:1686–1694. doi: 10.1101/gr.121327.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM. Reduced representation methods for subgenomic enrichment and next generation sequencing. In: Orgogozo V, Rockman MV, editors. Molecular Methods in Evolutionary Genetics. Humana Press; New York, USA: 2011. [DOI] [PubMed] [Google Scholar]
- Good JM, Wiebe V, Albert FW, et al. Comparative population genomics of the ejaculate in humans and the great apes. Molecular Biology and Evolution. 2013;30:964–976. doi: 10.1093/molbev/mst005. [DOI] [PubMed] [Google Scholar]
- Good JM, Vanderpool D, Keeble S, Bi K. Negligible nuclear introgression despite complete mitochondrial capture between two species of chipmunks. Evolution. 2015 doi: 10.1111/evo.12712. in press. [DOI] [PubMed] [Google Scholar]
- Goodwin S, Gurtowski J, Ethe-Sayers S, Deshpande P, Schatz M, McCombie WR. Oxford nanopore sequencing and de novo assembly of a eukaryotic genome. 2015 doi: 10.1101/013490. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Ptak SE, et al. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- Green RE, Malaspinas A-S, Krause J, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, et al. A draft sequence of the Neanderthal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Chmielecki J, Goparaju C, et al. Whole-exome sequencing reveal frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Research. 2015;75:264. doi: 10.1158/0008-5472.CAN-14-1008. [DOI] [PubMed] [Google Scholar]
- Hahn MW. Toward a selection theory of molecular evolution. Evolution. 2008;62:255–265. doi: 10.1111/j.1558-5646.2007.00308.x. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Kousathanas A, Ness RW, et al. Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genetics. 2013;9:e1003995. doi: 10.1371/journal.pgen.1003995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF, Woerner AE, Mendez FL, Watkins JC, Cox MP, Wall JD. The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nature Genetics. 2010;42:830–831. doi: 10.1038/ng.651. [DOI] [PubMed] [Google Scholar]
- Hancock-Hanser BL, Frey A, Leslie MS, Dutton PH, Archer FI, Morin PA. Targeted multiplex next-generation sequencing: advances in techniques of mitochondrial and nuclear DNA sequencing for population genomics. Molecular Ecology Resources. 2013;13:254–268. doi: 10.1111/1755-0998.12059. [DOI] [PubMed] [Google Scholar]
- Harakalova M, Mokry M, Hrdlickova B, et al. Multiplexed array-based and in solution genomic enrichment for flexible and cost-effective targeted next generation sequencing. Nature Protocols. 2011;6:1870–1886. doi: 10.1038/nprot.2011.396. [DOI] [PubMed] [Google Scholar]
- Harvey MG, Smith BT, Glenn TC, Faircloth BC, Brumfield RT. Sequence capture versus restriction sited associated DNA sequencing for phylogeography. 2013 doi: 10.1093/sysbio/syw036. arXiv, 1312.6439. [DOI] [PubMed] [Google Scholar]
- Hebert FO, Renaut S, Bernatchez L. Targeted sequence capture and resequencing implies a predominant role of regulatory regions in the divergence of a sympatric lake whitefish species pair (Coregonus clupeaformis) Molecular Ecology. 2013;22:4896–4914. doi: 10.1111/mec.12447. [DOI] [PubMed] [Google Scholar]
- Hedtke SM, Morgan MJ, Cannatella DC, Hillis DM. Targeted enrichment: maximizing orthologous gene comparisons across deep evolutionary time. PLoS One. 2013;8:e67908. doi: 10.1371/journal.pone.0067908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning F, Lee HJ, Franchini P, Meyer A. Genetic mapping of horizontal stripes in Lake Victoria cichlid fishes: benefits and pitfalls of using RAD marker for dense linkage mapping. Molecular Ecology. 2014;23:5224–5240. doi: 10.1111/mec.12860. [DOI] [PubMed] [Google Scholar]
- Hernandez RD, Kelley JL, Elyashiv E, et al. Classic selective sweeps were rare in recent human evolution. Science. 2011;331:920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch CD, Evans J, Buell CR, Hirsch CN. Reduced representation approaches to interrogate genome diversity in large repetitive plant genomes. Briefings in Functional Genomics. 2014;13:257–267. doi: 10.1093/bfgp/elt051. [DOI] [PubMed] [Google Scholar]
- Hodges E, Xuan Z, Balija V, et al. Genome-wide in situ exon capture for selective resequencing. Nature Genetics. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genetics. 2010;6:e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Ranade S, Malig M, et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Research. 2014;24:688–696. doi: 10.1101/gr.168450.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. Journal of Bacteriology. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvilsom C, Qian Y, Bataillon T, et al. Extensive X-linked adaptive evolution in central chimpanzees. Proceedings of the National Academy of Sciences USA. 2012;109:2054–2059. doi: 10.1073/pnas.1106877109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves KL, López-Fernández H. A targeted next-generation sequencing toolkit for exon-based cichlid phylogenomics. Molecular Ecology Resources. 2014;14:802–811. doi: 10.1111/1755-0998.12222. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O-Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis ED, Mirarab S, Aberer AJ, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–1331. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JD, Thornton KR, Andolfatto P. An approximate bayesian estimator suggests strong, recurrent selective sweeps in Drosophila. PLoS Genetics. 2008;4:e1000198. doi: 10.1371/journal.pgen.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Oldridge DA, Diskin SJ, Zhang NR. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Research. 2015;43:e39. doi: 10.1093/nar/gku1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, He M, Ferguson B, et al. An effort to use human based exome capture methods to analyze chimpanzee and macaque exomes. PLoS One. 2012;7:e40637. doi: 10.1371/journal.pone.0040637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent BN, Salichos L, Gibbons JG, et al. Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture. Genome Biology and Evolution. 2011;3:209–218. doi: 10.1093/gbe/evr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Sharpton TJ, Bobo D, et al. Exome capture from saliva produces high quality genomic and metagenomic data. BMC Genomics. 2014;15:262. doi: 10.1186/1471-2164-15-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Swayer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Research. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp M, Hofreiter M. Next generation sequencing of ancient DNA: requirements, strategies and perspectives. Genes. 2010;1:227–243. doi: 10.3390/genes1020227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proceedings of the Royal Society of London B. 2012;279:5048–5057. doi: 10.1098/rspb.2012.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C, Vinař T, da Fonseca RR, et al. Patterns of positive selection in six mammalian genomes. PLoS Genetics. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J, Fu Q, Good JM, et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010;464:894–897. doi: 10.1038/nature08976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S. Neandertal DNA sequences and the origin of modern humans. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Ku C-S, Wu M, Cooper DN, et al. Exome versus transcriptome sequencing in identifying coding region variants. Expert Reviews of Molecular Diagnostics. 2012;12:241–251. doi: 10.1586/erm.12.10. [DOI] [PubMed] [Google Scholar]
- Lachance J, Tishkoff SA. SNP ascertainment bias in population genetic analyses: why it is important and how to correct it. Bioessays. 2013;35:780–786. doi: 10.1002/bies.201300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaché AD, Chavez AS, Jones LN, Grummer JA, Gottscho AD, Linkem CW. Phylogenomics of phrynosomatid lizards: conflicting signals from sequences capture versus restriction site associated DNA sequencing. Genome Biology and Evolution. 2015;7:706–719. doi: 10.1093/gbe/evv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon AR, Emme SA, Lemmon EM. Anchored hybrid enrichment for massively high-throughput phylogenomics. Systematic Biology. 2012;61:727–744. doi: 10.1093/sysbio/sys049. [DOI] [PubMed] [Google Scholar]
- Lemmon EM, Lemmon AR. High-throughput genomic data in systematics and phylogenetics. Annual Reviews of Ecology, Evolution, and Systematics. 2013;44:99–121. [Google Scholar]
- Li C, Hofreiter M, Straube N, Corrigan S, Naylor GJP. Capturing protein-coding genes across highly divergent species. BioTechniques. 2013;54:321–326. doi: 10.2144/000114039. [DOI] [PubMed] [Google Scholar]
- Li Y, Vinckenbosch N, Tian G, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nature Genetics. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnen CR, Poh Y-P, Peterson BK, et al. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science. 2013;339:1312–1316. doi: 10.1126/science.1233213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Albrechtsen A, Li Y, et al. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS Genetics. 2011;7:e1002326. doi: 10.1371/journal.pgen.1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Willis JH. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biology. 2010;8:e1000500. doi: 10.1371/journal.pbio.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The origins of genome architecture. Sinauer Associates, Inc; Sunderland, Massachusetts, USA: 2007. [Google Scholar]
- Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sinauer Associates, Inc; Sunderland, Massachusetts, USA: 1998. [Google Scholar]
- Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichment strategies for next- generation sequencing. Nature Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- Mandel JR, Dikow RB, Funk VA, et al. A target enrichment method for gathering phylogenetic information from hundreds of loci: an example from Compositae. Applications in Plant Sciences. 2014;2:1300085. doi: 10.3732/apps.1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariac C, Scarceli N, Pouzadou J, et al. Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeographic studies. Molecular Ecology Resources. 2014;14:1103–1113. doi: 10.1111/1755-0998.12258. [DOI] [PubMed] [Google Scholar]
- Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One. 2010;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Papa R, Nadeau NJ, et al. Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proceedings of the National Academy of Sciences USA. 2012;109:12632–12637. doi: 10.1073/pnas.1204800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher M, Jost M, Kuon J-E, et al. Mapping-by-sequencing accelerates forward genetics in barley. Genome Biology. 2014;15:R78. doi: 10.1186/gb-2014-15-6-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher M, Stein N. Genetic anchoring of whole-genome shotgun assemblies. Frontiers in Genetics. 2014;5:208. doi: 10.3389/fgene.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason VC, Li G, Helgen KM, Murphy WJ. Efficient cross-species capture hybridization and next-generation sequencing of mitochondrial genomes from noninvasively sampled museum specimens. Genome Research. 2011;21:1695–1704. doi: 10.1101/gr.120196.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Faricloth BC, Crawford NG, Gowaty PA, Brumfield RT, Glenn TC. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Research. 2012;22:746–754. doi: 10.1101/gr.125864.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Harvey MG, Faircloth BC, Crawford NG, Glenn TC, Brumfield RT. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS One. 2013a;8:e4848. doi: 10.1371/journal.pone.0054848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT. Applications of next-generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution. 2013b;66:526–538. doi: 10.1016/j.ympev.2011.12.007. [DOI] [PubMed] [Google Scholar]
- McTavish EJ, Hillis DM. How do SNP ascertainment schemes and population demographics affect inferences about population history? BMC Genomics. 2015;16:266. doi: 10.1186/s12864-015-1469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Crawford J, et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nature Protocols. 2014;9:989–1009. doi: 10.1038/nprot.2014.058. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Springs Harbor Protocols. 2010;2010 doi: 10.1101/pdb.prot5448. pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge M-T, et al. A high coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research. 2007;17:240–248. doi: 10.1101/gr.5681207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Bayzid MS, Boussau B, Warnow T. Statistical binning enables an accurate coalescent-based estimation of the avian tree. Science. 2014;346:1250463. doi: 10.1126/science.1250463. [DOI] [PubMed] [Google Scholar]
- Misof B, Liu S, Meusemann K, et al. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014;346:763–767. doi: 10.1126/science.1257570. [DOI] [PubMed] [Google Scholar]
- Muegge BD, Kuczynski J, Knights D, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Freund F, Wildhagen H, Schmid KJ. Targeted re-sequencing of five Doulas-fir provenances reveals population structure and putative target genes of positive selection. Tree Genetics & Genomes. 2015;11:816. [Google Scholar]
- Nachman MW. Genomics and museum specimens. Molecular Ecology. 2013;22:5966–5968. doi: 10.1111/mec.12563. [DOI] [PubMed] [Google Scholar]
- Nadeau NJ, Whibley A, Jones RT, et al. Genomic islands of divergence in hybridizing Heliconius butterflies identified by large scale targeted sequencing. Philosophical Transactions of the Royal Society B. 2012;367:343–353. doi: 10.1098/rstb.2011.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narum SR, Buerkle CA, Davey JW, Miller MR, Hohenlohe PA. Genotyping-by sequencing in ecological and conservation genomics. Molecular Ecology. 2013;22:2841–2847. doi: 10.1111/mec.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale DB, Wegrzyn JL, Stevens KA, et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biology. 2014;15:R59. doi: 10.1186/gb-2014-15-3-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves LG, Davis JM, Barbazuk WB, Kirst M. Whole-exome targeted sequencing of the uncharacterized pine genome. The Plant Journal. 2013;75:146–156. doi: 10.1111/tpj.12193. [DOI] [PubMed] [Google Scholar]
- Neves LG, Davis JM, Barbazuk WB, Kirst M. A high-density gene map of loblolly pine (Pinus taeda L.) based on exome sequence capture genotyping. G3. 2014;4:29–37. doi: 10.1534/g3.113.008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R. Statistical tests of selective neutrality in the age of genomics. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biology. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt B, Street NR, Wetterbom A, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–84. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ, Zwick ME. Microarray based genomic selection for high-throughput resequencing. Nature Methods. 2007;4:907–909. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- Olson M. Enrichment of super-sized resequencing targets from the human genome. Nature Methods. 2007;1:891–892. doi: 10.1038/nmeth1107-891. [DOI] [PubMed] [Google Scholar]
- Orlando L, Ginolhac A, Zhang G, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499:74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature Genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S. Ancient DNA: Extraction, characterization, molecular cloning and enzymatic amplification. Proceedings of the National Academy of Sciences USA. 1989;86:1936–1943. doi: 10.1073/pnas.86.6.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S, Poinar H, Seere D. Genetic analyses from ancient DNA. Annual Reviews of Genetics. 2004;38:645–679. doi: 10.1146/annurev.genet.37.110801.143214. [DOI] [PubMed] [Google Scholar]
- Peñalba JV, Smith LL, Tonione MA, et al. Sequence capture using PCR-generated probes: a cost-effective method of targeted high-throughput sequencing for nonmodel organisms. Molecular Ecology Resources. 2014;14:1000–1010. doi: 10.1111/1755-0998.12249. [DOI] [PubMed] [Google Scholar]
- Perry GH, Marioni JC, Melsted P, Gilad Y. Genomic-scale capture and sequencing of endogenous DNA from feces. Molecular Ecology. 2010;19:5332–5344. doi: 10.1111/j.1365-294X.2010.04888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Delsuc F, Brinkmann H, Lartillot N. Phylogenomics. Annual Reviews in Ecology, Evolution, and Systematics. 2005;36:541–562. [Google Scholar]
- Philippe H, Brinkmann H, Lavrov DV, et al. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biology. 2011;9:e1000602. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca GJ, Zhang K, Li JB, et al. Multiplex amplification of large sets of human exons. Nature Methods. 2007;4:931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- Portik DM, Wood PL, Jr, Grismer JL, Stanley EL, Jackman TR. Identification of 104 rapidly-evolving nuclear protein-coding markers for amplification across scaled reptiles using genomic resources. Conservation Genetics Resources. 2012;4:1–10. [Google Scholar]
- Putnam NH, O-Connell B, Stites JC, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. 2015 doi: 10.1101/gr.193474.115. arXiv, 1502.05331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Li Y, Lindgreen S, et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riberio FJ, Przybylski D, Yin S, et al. Finished bacterial genomes from shotgun sequence data. Genome Research. 2012;22:2270–2277. doi: 10.1101/gr.141515.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviére J-B, Mirzaa GM, O-Roak BJ, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nature Genetics. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Research. 2012;22:939–946. doi: 10.1101/gr.128124.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RC, Singhal S, MacManes MD, et al. Museum genomics: low-cost and high-accuracy genetic data from historical specimens. Molecular Ecology Resources. 2011;11:1082–1092. doi: 10.1111/j.1755-0998.2011.03052.x. [DOI] [PubMed] [Google Scholar]
- Rubin BER, Ree RH, Moreau CS. Inferring phylogenies from RAD sequence data. PLoS One. 2012;7:e33394. doi: 10.1371/journal.pone.0033394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin C-J, Zody MC, Eriksson J, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- Saintenac C, Jiang D, Akhunov ED. Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genome Biology. 2011;12:R88. doi: 10.1186/gb-2011-12-9-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon A, Udall JA, Jeddeloh JA, Wendel J. Targeted capture of homoeologous coding and noncoding sequence in polyploidy cotton. G3. 2012;2:921–930. doi: 10.1534/g3.112.003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DC, Han L, Li J, et al. Finding the lost treasures in exome sequencing data. Trends in Genetics. 2013;29:593–599. doi: 10.1016/j.tig.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanoudou D, Kang PB, Hanslett JN, Han M, Kunkel LM, Beggs AH. Transcriptional profile of postmortem skeletal muscle. Physiological Genomics. 2004;16:222–228. doi: 10.1152/physiolgenomics.00137.2003. [DOI] [PubMed] [Google Scholar]
- Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS One. 2012;7:e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiessl S, Samans B, Hüttel B, Reinhard R, Snowdon RJ. Capturing sequence variation among flowering-time regulatory gene homologs in the allopolyploid crop species Brassica napus. Frontiers in Plant Science. 2014;5:404. doi: 10.3389/fpls.2014.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C, Tobler R, Kofler R, Nolte V. Sequencing pools of individuals mining genome-wide polymorphism data without big funding. Nature Reviews Genetics. 2014;15:749–763. doi: 10.1038/nrg3803. [DOI] [PubMed] [Google Scholar]
- Schnell IB, Thomsen PF, Wilkinson N, et al. Screening mammal biodiversity using DNA from leeches. Current Biology. 2012;22:R262. doi: 10.1016/j.cub.2012.02.058. [DOI] [PubMed] [Google Scholar]
- Schuenemann VJ, Bos K, DeWitte S, et al. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death. Proceedings of the National Academy of Sciences USA. 2011;108:E746–E752. doi: 10.1073/pnas.1105107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MSH. Database for mRNA half-life of 19,977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Research. 2009;16:45–58. doi: 10.1093/dnares/dsn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nature Reviews Genetics. 2014;15:121–132. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- Smadja CM, Canbäck B, Vitalis R, et al. Large-scale candidate gene scan reveals the role of chemoreceptor genes in host plant specialization and speciation in the pea aphid. Evolution. 2012;6:2723–2738. doi: 10.1111/j.1558-5646.2012.01612.x. [DOI] [PubMed] [Google Scholar]
- Smith NGC, Eyer-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Smith BT, Harvey MG, Faircloth BC, Glenn TC, Brumfield RT. Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Systematic Biology. 2014;63:83–95. doi: 10.1093/sysbio/syt061. [DOI] [PubMed] [Google Scholar]
- Steiner CC, Weber JN, Hoesktra HE. Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biology. 2007;6:e36. doi: 10.1371/journal.pbio.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe JR, Hoekstra HE. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity. 2008;100:158–170. doi: 10.1038/sj.hdy.6800937. [DOI] [PubMed] [Google Scholar]
- Stoneking M, Krause J. Learning about human populations history from ancient and modern genomes. Nature Reviews Genetics. 2011;12:603–614. doi: 10.1038/nrg3029. [DOI] [PubMed] [Google Scholar]
- Sulonen A-M, Ellonen P, Almusa H, et al. Comparison of solution-based exome capture methods for next-generation sequencing. Genome Biology. 2011;12:R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Human Molecular Genetics. 2010;19:R145–R151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O-Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Govindarajulu R, Liston A, Ashman TL. Targeted sequence capture provides insight into genome structure and genetics of male sterility in a gynodioecious diploid strawberry, Fragaria vesca ssp. Bracteata (Rosaceae) G3. 2013;3:1341–1351. doi: 10.1534/g3.113.006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Nakano M, Wang X, et al. Enrichment of sequencing targets from the human genome by solution hybridization. Genome Biology. 2009a;10:R116. doi: 10.1186/gb-2009-10-10-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Warner JB, Nakano M, et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nature Biotechnology. 2009b;27:1025–1031. doi: 10.1038/nbt.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. Structure, function and diversity of a healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton KR, Jensen JD. Controlling the false-positive rate in multilocus genome scans for selection. Genetics. 2007;175:737–750. doi: 10.1534/genetics.106.064642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tin MM-T, Economo EP, Mikheyev AS. Sequencing degraded DNA from non destructively sampled museum specimens for RAD-tagging and low-coverage shotgun phylogenetics. PLoS One. 2014;9:e96793. doi: 10.1371/journal.pone.0096793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature Reviews Genetics. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsangaras K, Siracusa MC, Nikolaidis N, et al. Hybridization capture reveals evolution and conservation across the entire koala retrovirus genome. PLoS One. 2014;9:e95633. doi: 10.1371/journal.pone.0095633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, et al. Insights into community structure and metabolism by reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Uitdewilligen JGAML, Wolters A-MA, D’hoop BB, Borm TJA, Visser RGF, van Eck HJ. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS One. 2013;8:e62365. doi: 10.1371/journal.pone.0062355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini A, Pompanon F, Taberlet P. DNA barcoding for ecologists. Trends in Ecology and Evolution. 2009;24:110–117. doi: 10.1016/j.tree.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Vallender EJ. Expanding whole exome resequencing into non-human primates. Genome Biology. 2011;12:R87. doi: 10.1186/gb-2011-12-9-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, Gutenkunst RN, Woerner AE, Watkins JC, Hammer MF. Evidence for increased levels of positive and negative selection on the X chromosome versus autosomes in humans. Molecular Biology and Evolution. 2014;31:2267–2282. doi: 10.1093/molbev/msu166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilstrup JT, Seguin-Orlando A, Stiller M, et al. Mitochondrial phylogenomics of modern and ancient equids. PLoS One. 2013;8:e55950. doi: 10.1371/journal.pone.0055950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner CE, Keller I, Wittwer S, et al. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Molecular Ecology. 2013;22:787–798. doi: 10.1111/mec.12023. [DOI] [PubMed] [Google Scholar]
- Wagner DM, Klunk J, Harbeck M, et al. Yersinia pestis and the Plague of Justinian 541–543 AD: a genomics analysis. The Lancet. 2014;14:319–326. doi: 10.1016/S1473-3099(13)70323-2. [DOI] [PubMed] [Google Scholar]
- Wang H, Chattopadhyay A, Li Z, et al. Rapid identification of heterozygous mutations in Drosophila melanogaster suing genomic sequence capture. Genome Research. 2010;20:981–988. doi: 10.1101/gr.102921.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Meyer E, McKay JK, Matz MV. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nature Methods. 2012;9:808–810. doi: 10.1038/nmeth.2023. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JN, Peterson BK, Hoekstra H. Discrete genetic modules are responsible for complex burrow evolution in Peromyscus mice. Nature. 2013;493:402–405. doi: 10.1038/nature11816. [DOI] [PubMed] [Google Scholar]
- Wei W-H, Hemani G, Haley CS. Detecting epistasis in human complex traits. Nature Reviews Genetics. 2014;15:722–733. doi: 10.1038/nrg3747. [DOI] [PubMed] [Google Scholar]
- Wilcox TM, McKelvey KS, Young MK, et al. Robust detection of rare species using environmental DNA: the importance of primer specificity. PLoS One. 2013;8:e59520. doi: 10.1371/journal.pone.0059520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willerslev E, Cooper A. Ancient DNA. Proceedings of the Royal Society of London B. 2005;272:3–16. doi: 10.1098/rspb.2004.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfield MO, Wilkinson PA, Allen AM, et al. Targeted re-sequencing of the allohexaploid wheat exome. Plant Biotechnology Journal. 2012;10:733–742. doi: 10.1111/j.1467-7652.2012.00713.x. [DOI] [PubMed] [Google Scholar]
- Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genetics in Medicine. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- Wright S. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proceedings of the Sixth Annual Congress of Genetics. 1932;1:356–366. [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, et al. Sequencing of fifty human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaida S, Choi M, Wakimoto H, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Holliday JA. Targeted enrichment of the black cottonwood (Populus trichocarpa) gene space using sequence capture. BMC Genomics. 2012;13:703. doi: 10.1186/1471-2164-13-703. [DOI] [PMC free article] [PubMed] [Google Scholar]