

Abstract
Cav1.3 belongs to the family of voltage‐gated L‐type Ca2+ channels and is encoded by the CACNA1D gene. Cav1.3 channels are not only essential for cardiac pacemaking, hearing and hormone secretion but are also expressed postsynaptically in neurons, where they shape neuronal firing and plasticity. Recent findings provide evidence that human mutations in the CACNA1D gene can confer risk for the development of neuropsychiatric disease and perhaps also epilepsy. Loss of Cav1.3 function, as shown in knock‐out mouse models and by human mutations, does not result in neuropsychiatric or neurological disease symptoms, whereas their acute selective pharmacological activation results in a depressive‐like behaviour in mice. Therefore it is likely that CACNA1D mutations enhancing activity may be disease relevant also in humans. Indeed, whole exome sequencing studies, originally prompted to identify mutations in primary aldosteronism, revealed de novo CACNA1D missense mutations permitting enhanced Ca2+ signalling through Cav1.3. Remarkably, apart from primary aldosteronism, heterozygous carriers of these mutations also showed seizures and neurological abnormalities. Different missense mutations with very similar gain‐of‐function properties were recently reported in patients with autism spectrum disorders (ASD). These data strongly suggest that CACNA1D mutations enhancing Cav1.3 activity confer a strong risk for – or even cause – CNS disorders, such as ASD.

Abbreviations
- ASD
autism spectrum disorders
- APA
aldosterone producing adenoma
- CaM
calmodulin
- ExAC
Exome Aggregation Consortium
- PASNA
primary aldosteronism, seizures and neurological abnormalities
- SNPs
single nucleotide polymorphisms
- QON
ON‐gating charge movement
Introduction
L‐type channels are one of three classes of voltage‐gated Ca2+ channels characterized by high sensitivity to channel blockers (Ca2+ antagonists), such as dihydropyridines. Among the four L‐type channel isoforms (Cav1.1–1.4), Cav1.2 and Cav1.3 are expressed in most electrically excitable cells in mammals, including the brain (Koschak et al. 2014; Striessnig et al. 2014, 2015; Vandael et al. 2015, for recent reviews). Ca2+ entry through these isoforms supports physiological functions that are mediated by electrical activity, including neurotransmitter release in sensory cells, control of neuronal excitability and plasticity, hormone secretion, heart rhythm and contractility as well as smooth muscle contraction (Hofmann et al. 2014; Koschak et al. 2014; Striessnig et al. 2014, 2015; Vandael et al. 2015; Zamponi et al. 2015, for recent reviews). However, due to differences in their biophysical properties, protein interactions, expression patterns and modulation by other signalling pathways, they contribute to these physiological functions in different ways (Striessnig et al. 2014; Zamponi et al. 2015).
Insight into the physiological functions of Cav1.3 has mainly been obtained from Cav1.3‐deficient and mutant mice (Platzer et al. 2000; Sinnegger‐Brauns et al. 2004) and from homozygous loss‐of‐function mutations in humans (Baig et al. 2011). The essential role of Cav1.3 for different organ functions has recently been extensively reviewed (Koschak et al. 2014; Striessnig et al. 2014; Zamponi et al. 2015) and is therefore not discussed in detail here. In the past few years, the discovery of human gain‐of‐function mutations in CACNA1D, encoding the pore‐forming Cav1.3 α1‐subunit, revealed novel physiological and pathophysiological functions of this channel (Azizan et al. 2013; Scholl et al. 2013). Gain‐of‐function mutations in Cav1.2 (CACNA1C) have already been reported in 2004 as the cause for Timothy syndrome (Splawski et al. 2004). Cav1.2 is expressed at higher abundance than Cav1.3 in the brain and is the major L‐type channel isoform in the heart and vascular smooth muscle (Striessnig et al. 2014; Zamponi et al. 2015). Timothy syndrome patients exhibit cardiovascular (long‐QT syndrome) as well as developmental abnormalities (facial abnormalities, syndactyly), and a large percentage of the affected individuals also show autism, mental retardation and seizures (Splawski et al. 2004, 2005). This was the first indication that L‐type Ca2+ channels could contribute to neuropsychiatric and neurological disease. As summarized in recent reviews (Bhat et al. 2012; Smoller et al. 2013; Zamponi et al. 2015) substantial evidence has accumulated since then for a prominent role of CACNA1C single nucleotide polymorphisms (SNPs) for neuropsychiatric disease risk. In addition also auxiliary α2δ and β subunits as well as other Cav α1‐subunits have been implicated in autism spectrum disorders (ASD) (for review Breitenkamp et al. 2015; Soldatov, 2015).
Here we discuss recent evidence from human genetics, strongly pointing to an important role of Cav1.3 dysfunction in psychiatric disorders. As Cav1.3 channels are also expressed in many tissues outside the brain, CACNA1D missense mutations causing a gain‐of‐function are likely to be associated with other (syndromic) organ dysfunctions. Indeed, activating Cav1.3 missense mutations associated with CNS disorders were first discovered in whole exome sequencing studies identifying a causal role of CACNA1D mutations for primary aldosteronism. We will therefore discuss risk mutations for CNS disorders in the context of mutations causing primary aldosteronism.
Cav1.3 dysfunction in primary aldosteronism and CNS disease
The first disorders caused by mutations permitting enhanced Cav1.3 Ca2+ channel activity were discovered as somatic CACNA1D missense mutations in, typically benign, aldosterone‐producing adrenal adenomas (APAs) in patients with primary aldosteronism (Azizan et al. 2013; Scholl et al. 2013). APAs are found in about 5% of individuals referred to hypertension clinics. CACNA1D mutations were not the first somatic mutations to be described in APAs responsible for excess aldosterone production in these tumours. Also mutations in genes for the K+‐channel, KCNJ5, the Na+/K+‐ATPase, ATP1A1, and the Ca2+‐ATPase, ATP2B3, were identified and all mutation‐induced functional changes could be predicted to enhance intracellular Ca2+ signalling. For example, in the case of ATP1A1 mutations this is due to impaired Na+/K+‐ATPase activity which triggers depolarization‐induced Ca2+ entry through voltage‐dependent Ca2+ channels (Azizan et al. 2013; Beuschlein et al. 2013; Scholl et al. 2013).
Somatic mutations in CACNA1D and the other genes are not limited to APAs but are also found in the more frequent multinodular adrenals and in diffuse hyperplasia (Dekkers et al. 2014; Fernandes‐Rosa et al. 2015; Scholl et al. 2015 a). Only one nodule, usually the aldosterone synthase‐positive one, harbours a mutation in multinodular glands suggesting that the mutations are causative in aldosterone hypersecretion but not in nodule formation (Dekkers et al. 2014; Fernandes‐Rosa et al. 2015; Scholl et al. 2015 a).
Although T‐type (Cav3 family, Perez‐Reyes, 2003; Zamponi et al. 2015 for review) and L‐type channels were known to be present in zona glomerulosa cells (Hunyady et al. 1994; Rossier et al. 1996; Hu et al. 2012), activating mutations in CACNA1D and, more recently, also in CACNA1H (Cav3.2 T‐type channel, Scholl et al. 2015 b) outed these channels as the most relevant ones in human aldosterone producing cells. Since Ca2+ is the critical second messenger responsible for aldosterone production (for details see Barrett et al. in this series) it was likely that the CACNA1D missense mutations induce changes in either channel gating or channel expression that lead to enhanced Ca2+ entry during electrical activity of a zona glomerulosa cell. So far, at least 24 mutations in 20 different positions have been described in APAs (Fig. 1). Four of these mutations have been experimentally characterized and revealed functional changes compatible with a gain‐of‐function phenotype (Azizan et al. 2013; Scholl et al. 2013). This involved pronounced slowing of current inactivation during depolarizing stimuli, activation of channel current at more negative voltages and higher single channel open probability. For mutation I750M this will be discussed in more detail below.
Figure 1. Somatic and germline CACNA1D mutations reported in human disease .

A, CACNA1D missense mutations are illustrated by circles. Somatic missense mutations identified in APAs are highlighted in red (Azizan et al. 2013; Scholl et al. 2013; Fernandes‐Rosa et al. 2014; Akerstrom et al. 2015; Nishimoto et al. 2015; Wang et al. 2015; Scholl et al. 2015 a). So far, only mutations V259D, G403D/R, I750M and P1336R have been functionally characterized, and they show strong gain‐of‐function phenotypes (Azizan et al. 2013; Scholl et al. 2013). G403R and I750M were also identified as de novo germline mutations in two patients with primary aldosteronism, seizures and neurological abnormalities (PASNA), highlighted in blue (Scholl et al. 2013). Mutations identified in individuals with ASD are shown in green. Mutations G407R (exon 8a), A749G and V584I were identified in the affected individuals but absent in parents or unaffected siblings and therefore classified as de novo (Iossifov et al. 2012; O'Roak et al. 2012; De Rubeis et al. 2014). Mutations A59V, S1953L and R1997H were detected in a case‐control sample (De Rubeis et al. 2014). Mutations G407R and A749G have been characterized and show a pronounced gain‐of‐function (Fig. 2, Pinggera et al. 2015). B, alignment of amino acid sequence of transmembrane S6‐helices of repeats I and II containing the activation gates at their cytoplasmic ends. They are highly conserved among α1 subunits of the L‐type Ca2+ channel family. Cav1.3 undergoes alternative splicing in the activation gate of repeat I (exons 8a vs. 8b) (Baig et al. 2011). Positions mutated in ASD and PASNA are located in close proximity to each other as indicated in the alignment. Whereas ASD‐linked mutation G407R only occurs in exon 8a (Pinggera et al. 2015), G403D was identified in both exons in APAs but was present in exon 8b in PASNA (Scholl et al. 2013). (Alignment was performed by ClustalW using the following reference sequences: Cav1.3 EU363339, Cav1.1 Q02789, Cav1.2 NP_001153005.1, Cav1.4 Q9JIS7; conserved residues are highlighted in grey.)
Altogether these findings clearly demonstrate that the gating changes induced by the APA mutations result in enhanced Ca2+ signalling through Cav1.3 channels in these tumour cells. It is likely that increased Ca2+ entry occurs during T‐type (Cav3.2) Ca2+ channel‐sustained membrane oscillations (Hu et al. 2012). Moreover, this raises the important question whether such mutations could also contribute to the risk for neurological or neuropsychiatric disorders.
Cav1.3 channels are widely expressed in the CNS (Striessnig et al. 2014; Zamponi et al. 2015 for review) and in neurons with different electrical activity patterns. They affect neuronal activity in a complex manner. Through coupling to Ca2+‐activated K+ channels they contribute to spike‐frequency adaptation and post‐burst afterhyperpolarization (McKinney et al. 2009), thus decreasing excitability. Conversely, these channels activate at relatively low (subthreshold) voltages, which allows them to create dendritic plateau potentials, during which neurons can fire tonically (Olson et al. 2005). Moreover, Cav1.3 signalling is required for normal neuronal development, synapse maturation as well as synaptic pruning (Day et al. 2006; Hirtz et al. 2011, 2012). Cav1.3 deficiency in mice leads to a relatively benign CNS/sensory phenotype. This includes deafness (due to inner hair cell dysfunction), reduced drug‐taking (for details see article by Kabir et al. in this issue) and antidepressant‐like behaviour (for recent review see Striessnig et al. 2014; Zamponi et al. 2015). In contrast, acute selective pharmacological activation of Cav1.3 channels in vivo in mice (Sinnegger‐Brauns et al. 2004; Hetzenauer et al. 2006) induces a depression‐like phenotype and neuronal activation in a specific set of mainly limbic, hypothalamic and brainstem areas, which are associated with integration of emotion‐related behaviours (Hetzenauer et al. 2006). These experiments were performed in mice expressing dihydropyridine‐insensitive Cav1.2 channels (Cav1.2DHP−/−). Application of the Ca2+ channel activator BayK8644 therefore allowed the selective activation of Cav1.3 channels in the brain (Sinnegger‐Brauns et al. 2004). These animal studies suggest that Cav1.3 gain‐of‐function mutations could also increase risk for CNS abnormalities in humans presenting as neuropsychiatric disorders or epilepsy. Indeed, evidence that dysfunctional Cav1.3 channels with gating properties resembling those found in APA mutations (and thus capable of enhancing Ca2+ currents through Cav1.3 channels) provide a strong risk for CNS disease came from two patients with germline APA mutations (Scholl et al. 2013).
CACNA1D mutations associated with primary aldosteronism with seizures and neurological abnormalities
In the course of characterizing somatic CACNA1D APA mutations, Scholl et al. (2013) discovered two mutations identified in APAs as germline de novo mutations in two patients with primary aldosteronism seizures and neurological abnormalities, a syndrome which was termed PASNA.
Patient 1 carried a copy of the G403D mutation (in alternative exon 8b) and was a 3‐year old female of European ancestry with no significant family history (Scholl et al. 2013). After birth she developed sinus bradycardia (day 1), and was successfully treated for transient hypoglycaemia (day 2), respiratory distress (day 3), atrioventricular block with prolonged QT‐interval and elevated blood pressure. Echocardiograms demonstrated biventricular hypertrophy, a ventricular septal defect and a patent foramen ovale with mild persistent pulmonary hypertension of the newborn. Primary aldosteronism (elevated aldosterone, low plasma renin activity) and hypokalaemia were diagnosed at age 1 month. Treatment with amlodipine normalized blood pressure, and resolved biventricular hypertrophy. Brain imaging at age 1 month was suggestive of periventricular leukomalacia, two regions with previous haemorrhages (right occipital region and left periventricular parenchyma) and ventriculomegaly. Further development was characterized by failure to thrive and a global developmental delay. Cardiac abnormalities resolved and no evidence for gastrointestinal disease was obtained. The patient developed epilepsy and was diagnosed with cortical blindness, spasticity and cerebral palsy. The first generalized tonic‐clonic seizure (month 7) was not associated with brain CT changes and there was no history of febrile illness or evidence for serum electrolyte abnormalities. Despite pharmacotherapy one generalized tonic‐clonic seizure per month developed until age 12 months, when seizure frequency increased. EEG showed recurrent spikes emanating predominantly from the right temporoparietal occipital region, and to a lesser extent, independently, from the left temporal region without a clinical correlate. At 3 years she was not ambulatory and not verbal (global developmental delay). The blood pressure and serum K+ levels were normal; recent medications included rufinamide, valproic acid, ranitidine, baclofen and levetiracetam (Scholl et al. 2013).
Patient 2 carried a copy of the I750M mutation and was a 10‐year old African American female with no significant family history (Scholl et al. 2013). Delivery was by Caesarean section at 41 weeks gestational age after uterine rupture. She required resuscitation and was subsequently diagnosed with cerebral palsy, spastic quadriplegia and mild athetosis, severe generalized intellectual disability, complex partial seizures of likely right hemispheric origin, generalized seizures, a movement disorder with verbal outbursts, and a sleep disorder. Treatment included oxcarbazepine, risperidone and later levetiracetam. Brain MRI was normal. The patient developed hypertension before the age of 5 years and at the age of 8 years also significant hypokalaemia. In addition, high aldosterone levels were found despite suppressed plasma renin activity. Abdominal CT scan at the age of 9 years showed normal appearance of the adrenal glands. Mild left ventricular hypertrophy was noted on echocardiogram. Hypertension was treated with clonidine, and later spironolactone (Scholl et al. 2013).
There was no family history of early‐onset hypertension or seizures in these patients. A common finding is the presence of hyperaldosteronism (with hypertension and hypokalaemia), seizures and generalized intellectual disability and spasticity. Hyperaldosteronism is a symptom expected from these mutations. The presence of seizures in both patients suggests that Cav1.3 gain‐of‐function may also contribute to epilepsy risk.
Based on the published medical histories, it cannot be excluded that severe perinatal complications (asphyxia in patient 1, resuscitation in patient 2) caused the epilepsy in both patients. However, this neurological phenotype also prompted us to screen the literature for additional CACNA1D variants reported in epilepsy. Klassen et al. performed parallel exome sequencing of 237 ion channel genes in a well‐characterized human sample of epilepsy patients with presumed genetic origin (Klassen et al. 2011). Their study showed that disease risk from even deleterious ion channel mutations can be modified by variants in other ion channel proteins present in the same individual. In CACNA1D they found (in addition to coding synonymous, intronic mutations and one splice‐site mutation) one premature stop, and three missense mutations that occurred in probands but not in controls (Table 1). All of them occur in functionally sensitive regions of the channel. Two of them, A519P and Q1598X, are highly likely to affect channel function and have not been reported in > 120,000 alleles in the Exome Aggregation Consortium (ExAC) database. ExAC collects whole exome sequencing data from 60,706 unrelated individuals with no severe paediatric diseases as a useful reference set of allele frequencies for severe disease studies (Exome Aggregation Consortium (ExAC), Cambridge, MA, USA, http://exac.broadinstitute.org, accessed September 2015). A519P introduces a proline at the C‐terminal end of the cytoplasmic I–II linker. As recently shown this region forms a conserved polybasic helical structure that interacts with lipid headgroups at the plasma membrane. In Cav1.2 channels it stabilizes channel function and mediates channel inhibition by phospholipase C‐induced phosphoinositide breakdown (Kaur et al. 2015). The high conservation of this motif suggests a similar role in all L‐type channels (Kaur et al. 2015). Like charge neutralizing mutations, the A519P mutation is therefore expected to interfere with normal channel gating and/or inhibitory control by receptor‐mediated phospholipid breakdown (Hille et al. 2014). Even more intriguing is variant Q1598X, truncating the C‐terminus of the Cav1.3 α1 subunit. I1597 and Q1598 are part of the ‘IQ‐motif’ essential for calmodulin (CaM)‐mediated Ca2+‐induced inactivation of voltage gated Ca2+ channels, an important autoinhibitory mechanism preventing excessive Ca2+ influx (Van Petegem et al. 2005; Adams et al. 2014). Constitutively bound apo‐CaM enhances the Cav1.3 open probability which is reversed upon Ca2+ binding (Adams et al. 2014). If the allele carrying mutation Q1598X results in the expression of a functional α1 subunit protein then apo‐CaM binding to this region must be decreased. This should reduce open probability but also Ca2+‐dependent inactivation making Ca2+ influx more long lasting. In the brain the function of this IQ‐motif is already heavily regulated by RNA editing (Huang et al. 2012) and alternative splicing (Shen et al. 2006; Tan et al. 2011) creating Cav1.3 channels with reduced or absent CaM regulation. Mutant Q1598X channels could therefore enhance the number of channels with ‘CaM‐free’ gating properties, which may lead to enhanced Ca2+ signalling through persistent Cav1.3 current components (Huang et al. 2012). Currently, a contribution of these mutations to overall epilepsy risk in the affected patients remains speculative. However, if gating changes compatible with gain of channel function can be demonstrated in heterologous expression studies of these mutants, this would strongly support a role for CACNA1D in idiopathic epilepsy. As discussed below, this has potential clinical implications since L‐type Ca2+ channel blockers available for treatment of hypertension could be beneficial in patients identified with such mutations.
Table 1.
Missense mutations reported in CACNA1D in a patient cohort with idiopathic epilepsy (Klassen et al. 2011)
| Missense | Potential pathogenic | ||
|---|---|---|---|
| mutations | Location in Cav1.3 α1 | ExAC variant | relevance for CNS disorders |
| A519P | End of cytoplasmic I‐II linker: CRAA/PVK | Not reported; only variant introducing a Thr rather than a Pro is described: 3:53756390 G/A (1 of 121408 alleles reported) | High: located close to previously reported regulatory polybasic plasma membrane binding motif (Kaur et al. 2015) |
| P1318S | Cassette exon in IVS3‐S4 linker present in some transcripts: ENVP/SVPT | Variant: 3:53804021 C/T (584 of 121400 alleles) | Unlikely: 4 homozygote carriers in ExAC; affects only specific splice variants |
| I1759N | Ile1761Asn in C‐terminus: RPSI/NGNL | Variant: 3:53835365 T/A (1 of 121318 alleles); (Ile→Leu and Ile→Met also reported in 1 allele each) | Unknown: mutation positioned one residue after proposed PKA phosphorylation site |
| Q1598X | Stop after Ile1597 of ‘IQ’ domain, essential for calmodulin binding; expected to affect 80% of CACNA1D transcripts; 20% do not contain the mutated residue | No variant reported | High: mutation removes C‐terminus including regulatory site for calmodulin; may give rise to more persistent Cav1.3 currents |
CACNA1D mutations associated with autism spectrum disorders
Whole exome sequencing also revealed two de novo mutations in autistic children of the Simons Simplex Collection (Figs 1 and 2). Intriguingly, these mutations are localized in close proximity to PASNA and APA mutations, suggesting a high contribution to disease risk in these patients (Fig. 1). As discussed in recent reviews (Striessnig et al. 2014; Zamponi et al. 2015), this is further supported by the known functions of Cav1.3 for synapse formation and neuronal excitability as well as the fact that very similar mutations in structurally and functionally highly related Cav1.2 channels (CACNA1C gene; see above) can also cause autism (Splawski et al. 2004).
Figure 2. Gating changes induced by missense mutations A749G (ASD) vs. I750M (PASNA, APA) .
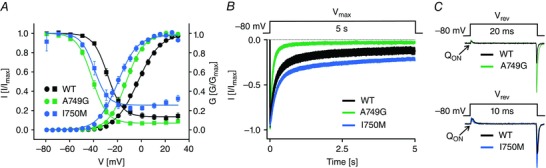
Mutations were introduced into the long isoform of Cav1.3 α1 subunits and heterologously expressed in tsA‐201 cells together with auxiliary α2δ‐1 and β3 subunits. The biophysical characterization of the mutants was performed by whole‐cell patch‐clamp recordings with 15 mm Ca2+ as charge carrier. A, steady state activation (circles) and inactivation (squares). Data shown as the mean ± SEM. Mutation A749G (n = 27) shifted voltage dependence of activation by 10 mV to more negative potentials in comparison to wild‐type (WT, n = 29). Mutation I750M (n = 11) resulted in an even stronger change (−15 mV shift compared to WT). In addition, A749G (n = 14) shifted steady state inactivation (measured after 5 s conditioning pulses to the indicated voltages) by 15 mV to more negative potential compared to WT (n = 18). This shift was less pronounced for I750M (−8 mV, n = 9). B, ICa inactivation upon 5 s depolarization to V max. Traces shown as the mean ± SEM. In contrast to I750M (n = 9) mutation A749G (n = 6) showed a more pronounced inactivation compared to WT (n = 15). The reduced inactivation of I750M is also evident in A. C, representative ICa traces upon depolarization to the reversal potential (V rev) normalized to the area of the ON‐gating charge movement (Q ON, as a measure of functional channels on the cell surface). Mutations A749G (upper panel, green) and I750M (lower panel blue) showed increased tail amplitudes when normalized to Q ON in comparison to WT (black), suggesting higher channel open probability or conductance. Modified from Azizan et al. (2013), I750M, and Pinggera et al. (2015), WT and A749G).
Patient 1 harbouring one copy of the A749G mutation (O'Roak et al. 2012) is a Caucasian female and was diagnosed with ASD at 94 months. Her full IQ score was 62, the verbal and non‐verbal IQ scores of 67 and 65, respectively. The patient carried another risk mutation in KATNAL2 with unknown physiological function (O'Roak et al. 2012) (patient information was obtained from the Simons Simplex collection, https://sfari.org/resources/autism‐cohorts/simons‐simplex‐collection, with permission and with approval by the local institutional review board).
Patient 2 carrying the G407R mutation (Iossifov et al. 2012) is a Hispanic male and was diagnosed at an age of 15 years with ASD. He has a full IQ score of 83, verbal and non‐verbal IQ scores of 81 and 88, respectively. In addition, a synonymous mutation in ADAMTSL1 was found in this patient (Iossifov et al. 2012). He was also diagnosed with a congenital heart problem at 1 month. Seizures, anxiety or depressive disorders and hyperaldosteronism were not reported in the patient records.
When heterologously expressed in tsA‐201 cells together with accessory β‐ and α2δ‐subunits, both mutations induced strong functional changes very similar to the two PASNA mutations. G407R closely resembled G403D (compare data in Azizan et al. 2013 vs. Pinggera et al. 2015) whereas A749G resulted in alterations comparable to I750M (Azizan et al. 2013; Pinggera et al. 2015; Fig. 2). Both shifted the voltage dependence of activation and inactivation to more hyperpolarized potentials. This enables mutant channels to activate at more negative membrane potentials thus representing a gain‐of‐function phenotype. The hyperpolarizing shift of the voltage dependence of inactivation on the other hand reduces the amount of active channels at physiological potentials. Especially in neurons with more depolarized membrane potentials (e.g. dopamine neurons in the substantia nigra) this could also reduce channel availability. In addition, A749G resulted in faster inactivation kinetics during depolarizing stimuli compared to I750M (Fig. 2). Faster inactivation in turn is expected to decrease Ca2+ influx through Cav1.3 channels in neurons during repetitive firing and might also reduce upstate potentials. Due to its non‐inactivating component, mutation I750M results in an increased window current, which represents the voltage range where the channels can be activated but are not completely inactivated, thus again representing a gain of function. A749G on the contrary reduces the window current due to the strong hyperpolarizing shift of the voltage dependence of inactivation and the faster inactivation kinetics. However, in fast spiking neurons a more negative activation range is expected to enhance Ca2+ current through A749G channels. Therefore the net effect of these mutations on Cav1.3 Ca2+ signalling may vary depending on the firing pattern and action potential shape of a neuron.
Using exome sequencing, De Rubeis et al. (2014) analysed rare coding variations in 3871 autism cases and 9937 ancestry‐matched or parental controls. Many of the identified risk variants encoded proteins implicated in synapse formation, transcriptional regulation and chromatin remodelling pathways. Amongst the critical synaptic components found to be mutated were also voltage‐gated ion channels, including missense variants in CACNA1D and two loss‐of‐function variants in CACNA2D3, which encodes the α2δ‐3 subunit (interacting with different L‐ and non‐L‐type Ca2+ channels; for review Dolphin, 2013). This study confirmed the de novo A749G and G407R risk mutations, but also identified an additional de novo mutation (V584I) and three other risk variants (Fig. 1) in a case‐control population. None of the newly identified mutations has been functionally characterized so far. A59V is located in an N‐terminal CaM interaction site (NSCaTE; Dick et al. 2008) and may thus affect the inactivation properties of the channel. This region is also highly conserved in Cav1.2 channels and the variant is not reported as a variant in the ExAC database.
Mutations S1953L and R1997H in the C‐terminal tail have been reported in the ExAC database (three S1953L, one S1953P; one R1997H, two R1997C). Moreover these residues are not conserved between Cav1.3 and Cav1.2. Their potential to contribute to ASD risk is therefore less obvious than for G407R and A749G. This is also true for the de novo mutation V584I for which two variants were reported in ExAC sequencing data.
If strong gain‐of‐function mutations as observed in APAs confer a high risk for PASNA and ASD, carriers are expected to present with CNS symptoms earlier in life and would therefore less likely show up in the ExAC control population, whereas this may not be true for mutations contributing lower risk. We screened the ExAC data for the presence of APA mutations and found variants corresponding to four mutated CACNA1D loci affected in APAs. One variant resulted in a glycine insertion after position 402 (p.Leu402_Gly403insGly) in exon 8b. An additional variant was reported in position 652, but in contrast to the corresponding APA mutation S652L (Fig. 2), this variant leads to mutation to a tryptophan. Mutation L655P was identified only once. Another mutation, V728I, was identified in 41 of > 121,400 alleles. All carriers were heterozygous for the variants. The glycine insertion at position 403 in CACNA1D leads to a non‐conducting channel due to impaired coupling of voltage sensing to pore opening (Baig et al. 2011). Therefore p.Leu402_Gly403insGly must result in a loss of function, which is not expected to cause a phenotype in the heterozygous state (Platzer et al. 2000; Baig et al. 2011). Mutations V728I, S652L and L655P have not been characterized so far. However, the fact that they all occur in the ExAC database suggests that the mutations, in particular V728I, might confer less risk to severe paediatric disease, perhaps by inducing more moderate gating changes.
Conclusion
Taken together we hypothesize that gain‐of‐function CACNA1D mutations differ with respect to their contribution to CNS disease risk. Mutations causing strong changes in channel gating, similar to those reported in PASNA and some of the APA mutations (e.g. G403R, V259D), may convey a very high disease risk. Abnormal neuronal Ca2+ signalling by these dysfunctional channels may not be easily compensated by variants in other genes (e.g. encoding ion channels; Klassen et al. 2011) and thus lead to disease symptoms in the vast majority of carriers. On the other side of the spectrum, APA mutations such as V728I may manifest as primary aldosteronism without CNS symptoms in humans when present in the germline. This would explain the high number of carriers in the ExAC database. In the brain such rare CACNA1D variants inducing more subtle changes in Cav1.3 function may only be of disease relevance in a permissive genetic background.
To prove this hypothesis more patients need to be identified with either PASNA, ASD with or without seizures or primary aldosteronism and strongly gating modifying de novo germline mutations in CACNA1D. If this is the case and the corresponding mutations (like G407R, A749G) retain their sensitivity to brain‐permeant L‐type Ca2+ channel blockers (such as isradipine, nifedipine or nimodipine), experimental therapies with these drugs in affected patients would be justified. If a contribution of such mutations to epileptic symptoms (in idiopathic epilepsy or as co‐morbidity in PASNA or, perhaps, autism) can also be demonstrated, these drugs may even be added to antiepileptic therapy.
In the last few years next generation sequencing has provided us with exciting novel insight into the aggregate burden of genetic variants. We are only at the beginning of understanding the role of de novo mutations in voltage‐gated L‐type Ca2+ channels for sporadic cases of CNS disorders. To better comprehend how distinct CACNA1D mutations contribute to different human disorders, further functional analysis of more high risk Cav1.3 mutations reported in affected patients is important. Mutations activating these channels may not only confer enhanced risk for neuropsychiatric disease, as described in this article, but also for neurodegenerative disorders, in particular Parkinson's disease. Recent work has provided strong evidence that L‐type channel mediated Ca2+ entry contributes to oxidative stress underlying the high vulnerability of substantia nigra neurons to cell death (Sulzer & Surmeier, 2013). Therefore studying these human mutations in mice must be the next step to understand their disease‐causing potential in humans.
Additional information
Competing interests
The authors declare no conflicts of interest.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors work is supported by the Austrian Science Fund (FWF F44020, W1101), the University of Innsbruck and the Tyrolean Government.
Acknowledgements
We thank Silvia De Rubeis for helpful discussions.
Biographies
Jörg Striessnig received his medical degree from the University of Innsbruck and is currently chairman of the Department of Pharmacology and Toxicology. He is interested in the molecular pharmacology of voltage‐gated L‐type Ca2+ channels. As a visiting assistant professor in the laboratory of Bill Catterall (University of Washington) he identified the drug binding regions for clinically used Ca2+ channel blockers. His group developed knockout and mutant mouse models to dissect the physiological role of various L‐type channel isoforms in different tissues and contributed to our understanding of many human Ca2+‐channelopathies. Currently his group focuses on the role of Cav1.3 channels for CNS disease risk.
Alexandra Pinggera is a doctoral student at the same department. She uses electrophysiological, immunological and biochemical techniques to study the consequences of human mutations on Ca2+ channel function. Her work has contributed to the identification of channel gain‐of‐function mutations in autism patients and new lipid‐ interaction domains on the channel protein.

This review was presented at the symposium “Voltage‐gated calcium channels ‐ from basic mechanisms to disease”, which took place at Physiology 2015 in Cardiff, UK, 6–8 July 2015.
References
- Adams PJ, Ben‐Johny M, Dick IE, Inoue T & Yue DT (2014). Apocalmodulin itself promotes ion channel opening and Ca2+ regulation. Cell 159, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerstrom T, Willenberg HS, Cupisti K, Ip J, Backman S, Moser A, Maharjan R, Robinson B, Iwen KA, Dralle H, Volpe CD, Backdahl M, Botling J, Stalberg P, Westin G, Walz MK, Lehnert H, Sidhu S, Zedenius J, Bjorklund P & Hellman P (2015). Novel somatic mutations and distinct molecular signature in aldosterone‐producing adenomas. Endocr Relat Cancer 22, 735–744. [DOI] [PubMed] [Google Scholar]
- Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AE, Davenport AP, Dekkers T, Tops B, Kusters B, Ceral J, Yeo GS, Neogi SG, McFarlane I, Rosenfeld N, Marass F, Hadfield J, Margas W, Chaggar K, Solar M, Deinum J, Dolphin AC, Farooqi IS, Striessnig J, Nissen P & Brown MJ (2013). Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45, 1055–1060. [DOI] [PubMed] [Google Scholar]
- Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger‐Brauns MJ, Brandt N, Engel J, Mangoni ME, Farooq M, Khan HU, Nurnberg P, Striessnig J & Bolz HJ (2011). Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci 14, 77–84. [DOI] [PubMed] [Google Scholar]
- Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson‐Couterie B, Benecke A, Quinkler M, Fallo F, Plouin PF, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro MC, Strom TM & Reincke M (2013). Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone‐producing adenomas and secondary hypertension. Nat Genet 45, 440–444. [DOI] [PubMed] [Google Scholar]
- Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM & Gould TD (2012). CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol 99, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenkamp AF, Matthes J & Herzig S (2015). Voltage‐gated calcium channels and autism spectrum disorders. Curr Mol Pharmacol 8, 123–132. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW & Surmeier DJ (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 9, 251–259. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Fu SC, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Crooks L, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita‐Laza I, Jimenez Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei J, Lehtimaki T, Lin CF, Ma'ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnstrom K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte‐Ruther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Wang LS, Weiss LA, Willsey AJ, Yu TW, Yuen RK, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barrett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ & Buxbaum JD (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers T, ter Meer M, Lenders JW, Hermus AR, Schultze Kool L, Langenhuijsen JF, Nishimoto K, Ogishima T, Mukai K, Azizan EA, Tops B, Deinum J & Kusters B (2014). Adrenal nodularity and somatic mutations in primary aldosteronism: one node is the culprit? J Clin Endocrinol Metab 99, E1341–E1351. [DOI] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W & Yue DT (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of Cav channels. Nature 451, 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2013). The α2δ subunits of voltage‐gated calcium channels. Biochim Biophys Acta 1828, 1541–1549. [DOI] [PubMed] [Google Scholar]
- Fernandes‐Rosa FL, Giscos‐Douriez I, Amar L, Gomez‐Sanchez CE, Meatchi T, Boulkroun S & Zennaro MC (2015). Different somatic mutations in multinodular adrenals with aldosterone‐producing adenoma. Hypertension 66, 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes‐Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, Strom TM, Monticone S, Amar L, Meatchi T, Mantero F, Cicala MV, Quinkler M, Fallo F, Allolio B, Bernini G, Maccario M, Giacchetti G, Jeunemaitre X, Mulatero P, Reincke M & Zennaro MC (2014). Genetic spectrum and clinical correlates of somatic mutations in aldosterone‐producing adenoma. Hypertension 64, 354–361. [DOI] [PubMed] [Google Scholar]
- Hetzenauer A, Sinnegger‐Brauns MJ, Striessnig J & Singewald N (2006). Brain activation pattern induced by stimulation of L‐type Ca2+‐channels: contribution of Cav1.3 and Cav1.2 isoforms. Neuroscience 139, 1005–1015. [DOI] [PubMed] [Google Scholar]
- Hille B, Dickson EJ, Kruse M, Vivas O & Suh BC (2014). Phosphoinositides regulate ion channels. Biochim Biophys Acta 1851, 844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirtz JJ, Boesen M, Braun N, Deitmer JW, Kramer F, Lohr C, Muller B, Nothwang HG, Striessnig J, Lohrke S & Friauf E (2011). Cav1.3 calcium channels are required for normal development of the auditory brainstem. J Neurosci 31, 8280–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirtz JJ, Braun N, Griesemer D, Hannes C, Janz K, Lohrke S, Muller B & Friauf E (2012). Synaptic refinement of an inhibitory topographic map in the auditory brainstem requires functional Cav1.3 calcium channels. J Neurosci 32, 14602–14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Flockerzi V, Kahl S & Wegener JW (2014). L‐type Cav1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev 94, 303–326. [DOI] [PubMed] [Google Scholar]
- Hu C, Rusin CG, Tan Z, Guagliardo NA & Barrett PQ (2012). Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J Clin Invest 122, 2046–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Kohr G, Higuchi M, Fatemi‐Shariatpanahi H, Harden B, Yue DT & Soong TW (2012). RNA editing of the IQ domain in Cav1.3 channels modulates their Ca2+‐dependent inactivation. Neuron 73, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunyady L, Rohacs T, Bago A, Deak F & Spat A (1994). Dihydropyridine‐sensitive initial component of the ANG II‐induced Ca2+ response in rat adrenal glomerulosa cells. Am J Physiol Cell Physiol 266, C67–C72. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR & Wigler M (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Pinggera A, Ortner NJ, Lieb A, Sinnegger‐Brauns MJ, Yarov‐Yarovoy V, Obermair GJ, Flucher BE & Striessnig J (2015). A polybasic plasma membrane binding motif in the I‐II linker stabilizes voltage‐gated Cav1.2 calcium channel function. J Biol Chem 290, 21086–21100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, Morgan M, Muzny D, Gibbs R & Noebels J (2011). Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145, 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Pinggera A, Schicker K & Striessnig J (2014). Role of L‐type Ca2+ channels in sensory cells In Pathologies of Calcium Channels, ed. Weiss N. & Koschak A, pp. 47–75. Springer, Berlin, Heidelberg. [Google Scholar]
- McKinney BC, Sze W, Lee B & Murphy GG (2009). Impaired long‐term potentiation and enhanced neuronal excitability in the amygdala of Cav1.3 knockout mice. Neurobiol Learn Mem 92, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez‐Sanchez CE, Nanba K & Rainey WE (2015). Aldosterone‐stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci USA 112, E4591–E4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J & Eichler EE (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez‐Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I & Surmeier DJ (2005). G‐protein‐coupled receptor modulation of striatal Cav1.3 L‐type Ca2+ channels is dependent on a Shank‐binding domain. J Neurosci 25, 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E ( 2003). Molecular physiology of low‐voltage‐activated T‐type calcium channels. Physiol Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, Tuluc P & Striessnig J (2015). CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L‐type calcium channels. Biol Psychiatry 77, 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott‐Fischer A, Stephan K, Bova S, Chen H, Zheng H & Striessnig J (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell 102, 89–97. [DOI] [PubMed] [Google Scholar]
- Rossier MF, Burnay MM, Vallotton MB & Capponi AM (1996). Distinct functions of T‐ and L‐type calcium channels during activation of bovine adrenal glomerulosa cells. Endocrinology 137, 4817–4826. [DOI] [PubMed] [Google Scholar]
- Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson‐Williams C, Libutti SK, Mane S, Hellman P, Westin G, Akerstrom G, Bjorklund P, Carling T, Fahlke C, Hidalgo P & Lifton RP (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone‐producing adenomas and primary aldosteronism. Nat Genet 45, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, Horne MJ, Dietrich D, Riemer J, Kucukkoylu S, Reimer EN, Reis AC, Goh G, Kristiansen G, Mahajan A, Korah R, Lifton RP, Prasad ML & Carling T (2015. a). Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin Endocrinol (Oxf) 83, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Stolting G, Nelson‐Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC, Quack I, Rump LC, Thiel A, Lande M, Frazier BG, Rasoulpour M, Bowlin DL, Sethna CB, Trachtman H, Fahlke C & Lifton RP (2015. b). Recurrent gain of function mutation in calcium channel CACNA1H causes early‐onset hypertension with primary aldosteronism. Elife 4, e06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yu D, Hiel H, Liao P, Yue DT, Fuchs PA & Soong TW (2006). Alternative splicing of the Cav1.3 channel IQ domain, a molecular switch for Ca2+‐dependent inactivation within auditory hair cells. J Neurosci 26, 10690–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger‐Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N & Striessnig J (2004). Isoform‐specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L‐type Calcium channels. J Clin Invest 113, 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI, Ripke S, Santangelo S & Sullivan PF (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome‐wide analysis. Lancet 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM ( 2015). CACNB2: An emerging pharmacological target for hypertension, heart failure, arrhythmia and mental disorders. Curr Mol Pharmacol 8, 32–42. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC & Keating MT (2005). Severe arrhythmia disorder caused by cardiac L‐type calcium channel mutations. Proc Natl Acad Sci USA 102, 8089–8096; discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager‐Flusberg H, Priori SG, Sanguinetti MC & Keating MT (2004). Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Ortner NJ & Pinggera A (2015). Pharmacology of L‐type calcium channels: novel drugs for old targets? Curr Mol Pharmacol 8, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Pinggera A, Kaur G, Bock G & Tuluc P (2014). L‐type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3, 15–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D & Surmeier DJ (2013). Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord 28, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BZ, Jiang F, Tan MY, Yu D, Huang H, Shen Y & Soong TW (2011). Functional characterization of alternative splicing in the C terminus of L‐type CaV1.3 channels. J Biol Chem 286, 42725–42735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC & Minor DL Jr (2005). Insights into voltage‐gated calcium channel regulation from the structure of the Cav1.2 IQ domain‐Ca2+/calmodulin complex. Nat Struct Mol Biol 12, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A & Carbone E (2015). Cav1.3 channels as key regulators of neuron‐like firings and catecholamine release in chromaffin cells. Curr Mol Pharmacol 8, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li X, Zhang X, Ma X, Chen L, Zhang Y, Lyu X, Tang Y, Huang Q, Gao Y, Fan Y & Ouyang J (2015). Prevalence and characterization of somatic mutations in chinese aldosterone‐producing adenoma patients. Medicine (Baltimore) 94, e708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]