

Abstract
AIM: To investigate the mechanism of calcyclin binding protein/Siah-1 interacting protein (CacyBP/SIP) nuclear translocation in promoting the proliferation of gastric cancer (GC) cells.
METHODS: The effect of CacyBP/SIP nuclear translocation on cell cycle was investigated by cell cycle analysis. Western blot analysis was used to assess the change in expression of cell cycle regulatory proteins and proteasome-mediated degradation of p27Kip1. Co-immunoprecipitation (co-IP) analysis was performed to examine the binding of CacyBP/SIP with Skp1. A CacyBP/SIP truncation mutant which lacked the Skp1 binding site was constructed and fused to a fluorescent protein. Subsequently, the effect on Skp1 binding with the fusion protein was examined by co-IP, while localization of fluorescent fusion protein observed by confocal laser microscopy, and change in p27Kip1 protein expression assessed by Western blot analysis.
RESULTS: CacyBP/SIP nuclear translocation induced by gastrin promoted progression of GC cells from G1 phase. However, while CacyBP/SIP nuclear translocation was inhibited using siRNA to suppress CacyBP/SIP expression, cell cycle was clearly inhibited. CacyBP/SIP nuclear translocation significantly decreased the level of cell cycle inhibitor p27Kip1, increased Cyclin E protein expression whereas the levels of Skp1, Skp2, and CDK2 were not affected. Upon inhibition of CacyBP/SIP nuclear translocation, there were no changes in protein levels of p27Kip1 and Cyclin E, while p27Kip1 decrease could be prevented by the proteasome inhibitor MG132. Moreover, CacyBP/SIP was found to bind to Skp1 by immunoprecipitation, an event that was abolished by mutant CacyBP/SIP, which also failed to stimulate p27Kip1 degradation, even though the mutant could still translocate into the nucleus.
CONCLUSION: CacyBP/SIP nuclear translocation contributes to the proliferation of GC cells, and CacyBP/SIP exerts this effect, at least in part, by stimulating ubiquitin-mediated degradation of p27Kip1.
Keywords: Calcyclin binding protein/Siah-1 interacting protein, Gastric cancer, Cell cycle, P27kip1, Ubiquitin
Core tip: Calcyclin binding protein/Siah-1 interacting protein (CacyBP/SIP) is a component of the ubiquitination pathway. Our study showed that defects in G1 arrest led to an increase in embryonic fibroblast growth rate in SIP-/- mice, and CacyBP/SIP could promote G1/S transition of pancreatic cancer cells and regulate the glucose limitation-induced p27 degradation. In gastric cancer (GC) tissue, CacyBP/SIP was identified to be expressed in the nuclei and could translocate into the nucleus after induction with gastrin and promote cell proliferation; however, the mechanism remains unclear. In the present investigation, the mechanism of CacyBP/SIP nuclear translocation in promoting the growth of GC cells was studied.
INTRODUCTION
Calcyclin binding protein (CacyBP) was originally discovered in Ehrlich ascites tumor cells and mouse brain as a calcyclin (S100A6) target protein[1], and later identified as a Siah-1 interacting protein (SIP)[2]. SIP, a component of ubiquitin-mediated proteolysis, binds the Skp1-Cul1-F box protein complex, thus called CacyBP/SIP.
Further investigations showed that CacyBP/SIP could bind other S100 proteins, including S100A1, S100A12, S100B, and S100P[3], which are the members of EF-hand Ca2+-binding proteins. Meanwhile, CacyBP/SIP can also bind other target proteins including Skp1[2], tubulin[4] and ERK1/2[5]. Functionally, some reports have suggested the role of CacyBP/SIP in cellular processes such as ubiquitination, proliferation, differentiation, tumorigenesis, cytoskeletal rearrangement, and regulation of transcription. Most of all, CacyBP/SIP was found to be closely associated with the malignant phenotypes of gastric cancer (GC)[6,7], renal cancer[8], pancreatic cancer[9], as well as breast cancer[10]. However, the exact function of CacyBP/SIP has not been clarified.
Matsuzawa et al[2] were the first to report that CacyBP/SIP is a component of the ubiquitin pathway. P53 induces β-catenin degradation through the Siah-1-CacyBP/SIP-SCF (Skp1/Cullin1/F-box) complex pathway, in which CacyBP/SIP exerts its function by associating with Skp1. An additional study showed that defects in G1 arrest led to an increase in embryonic fibroblast growth rate in SIP-/- mice[11]. Recently, it was shown that CacyBP/SIP could regulate the glucose limitation-induced p27 degradation[12].
Our laboratory was the first to show that CacyBP/SIP was involved in the multidrug resistance capacity of GC cells[13,14]. In our previous studies, CacyBP/SIP expression profile in a broad range of normal and malignant human tissues was analyzed by immunohistochemistry (IHC) analysis using an anti-CacyBP/SIP monoclonal antibody first produced in our laboratory[15]. Weak staining was observed in various normal tissues. However, CacyBP/SIP was ubiquitously detected in most tumor tissues, especially in GC tissues[16]. A subsequent study showed that CacyBP/SIP could translocate into the nucleus after induction with gastrin[17]. However, the functional role of CacyBP/SIP nuclear translocation in GC cells remains unclear.
In the present investigation, the role of CacyBP/SIP nuclear translocation in GC cells was studied. Our results indicate that CacyBP/SIP nuclear translocation promotes G1 phase progression by stimulating ubiquitin-mediated degradation of p27Kip1.
MATERIALS AND METHODS
Cell culture, reagents, and treatment of cells
The human GC cell line SGC7901 (derived from poorly differentiated adenocarcinoma) was obtained from China Cell Resource Center of Academy of Life Sciences (Shanghai). Cells were cultured in RPMI 1640 (HyClone, Logan, UT) supplemented with 10% FBS (Sijiqing, China), penicillin (100 units/mL) and streptomycin (100 μg/mL). Stably transfected SGC7901-CacyBP/SIPsi cells were cultured in RPMI 1640 medium containing 10% FBS and 200 μg/ML G418 (Invitrogen, Carlsbad, CA). Nocodazole (Alexis Corporation, Switzerland) was added to the culture medium at 0.2 μg/mL to freeze cell cultures in mitosis. Gastrin (Sigma, St. Louis, MO) was dissolved in RPMI 1640 and used for cell treatment.
Cell cycle analysis
Cells were plated at a density of 50000 cells/well in 2 mL of complete RPMI1640 media in 6-well plates and allowed to grow for 24 h until 60%-70% confluence. To achieve synchronization, cells were starved in serum-free medium for 24 h. Upon return to regular growth medium, cells were treated or untreated with 10-8 mol/L gastrin. After 48 h of culture, cells were fixed overnight in 70% ethanol at 4 °C, and then re-suspended in a buffer containing propidium iodide (PI). After 30 min of incubation, cells were subjected to DNA content analysis by flow cytometry (Coulter EPICS XL) using CellQuest software. This cell cycle analysis was performed in three independent experiments.
Western blot analysis
Treated and untreated cells were lysed in 300 μL of freshly prepared extraction buffer [1% SDS, 1 mmol/L Na3VO4, 150 mmol/L NaCl, 0.1 mol/L Tris (pH 7.4)]. To distinguish cytosolic from nuclear CacyBP/SIP, cell fractions were extracted using the NE-PER nuclear and cytoplasmic extraction kit (Pierce Biotechnology, Rockford, IL). Proteins were resolved at 40 μg/lane on 12% SDS-polyacrylamide gels and electrophoretically transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) for 20-50 min at 20 V. Membranes were incubated at 4 °C overnight with one of the following primary antibodies: monoclonal antibodies against CDK2 (1:100), Cyclin E (1:100), Skp1 (1:100) and p27Kip1 (1:200) (all from NeoMarkers, United States); polyclonal antibodies against Skp2 (1:1000) and PARP (1:1000) (all from Cell Signaling Technology, Boston, MA); and monoclonal antibodies against CacyBP/SIP (1:1000) and β-actin (1:2000) (Sigma Chemical, St. Louis, MO). Membranes were then incubated with anti-mouse IgG or anti-rabbit IgG (Amersham Biosciences, Piscataway, NJ) and were detected by SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology Inc., Rockford, IL). For each Western blot result, at least three independent experiments were conducted, and representative images are shown.
Immunoprecipitation assays
For immunoprecipitation, lysates were obtained from untreated or treated GC cells. When the cells had reached approximately 80% confluence, the cell layer was washed with ice-cold phosphate-buffered saline (PBS). Nuclear extracts were prepared using the NE-PER nuclear and cytoplasmic extraction kit. Then salts were removed from the nuclear extract using the Pierce Slide-A-Lyzer MINI Dialysis Unit (Pierce Biotechnology, Rockford, IL). The supernatant was collected and total protein (300 μg) was used for immunoprecipitation. The supernatant was incubated at 4 °C for 2 h with 1 μg of monoclonal anti-CacyBP/SIP or anti-GFP (Sigma Chemical, St. Louis, MO) and 40 μL of 25% protein A/G agarose slurry. The protein A/G agarose was recovered by centrifugation at 5000 rpm and washed four times with ice-cold lysis buffer. Proteins were eluted with 20 μL of SDS loading buffer by boiling for 5 min and subjected to immunoblot analysis with anti-Skp1 antibodies.
Construction and transfection
The cDNA coding for full-length human CacyBP/SIP was generated from the plasmid pET28-CacyBP/SIP constructed and described previously[14]. The cDNA was generated by PCR amplification using the following primers: a forward primer containing a Pst I site, 5’-aactgcagtcATGGCTTCAGAAGAGCTACAGA-3’, and a reverse primer with a BamH I site, 5’-cgggatccTCAAAATTCCGTGTCTCCTTTG-3’. The PCR product was sub-cloned into the pEGFP/C1 vector (Invitrogen, Carlsbad, CA) using Pst I and BamH I, producing a fusion protein pEGFP/C1-CacyBP/SIP.
The cDNA encoding a truncated form of CacyBP/SIP (1-72) (i.e., the N-terminal fragment of CacyBP/SIP containing residues 1-72) was derived from the full-length human CacyBP/SIP gene in plasmid pET28-CacyBP/SIP. This cDNA was generated by PCR amplification with the following primers: a forward primer with a Pst I site, 5’-aactgcagtcATGGCTTCAGAAGAGCTACAGA-3’; and a reverse primer with a BamH I site, 5’-cgggatcctcaCGTATAGCCCGTTGTAATGGG-3’. The PCR product was sub-cloned into the pEGFP/C1 vector using Pst I and BamH I, producing a fusion protein pEGFP/C1-CacyBP/SIP (1-72).
Stably-transfected GC cells SGC7901-CacyBP/SIP-siRNA were described previously and used in later experiments[17]. Cell transfection was performed with Lipofectamine2000 (Invitrogen, Carlsbad, CA) as described in the manufacturer’s protocol. For transient transfection of pEGFP/C1, pEGFP/C1-CacyBP/SIP or pEGFP/C1-CacyBP/SIP (1-72), cells were harvested for further experiments after 48 h of transfection. Cells were harvested at different time points (0, 4, 8, 12, and 24 h) for Western blot, cell cycle analysis, co-immunoprecipitation (co-IP), and confocal laser microscopy (Bio-Rad Laboratories, United States).
Statistical analysis
Bands from Western blots were quantified with Quantity One software (Bio-Rad). Relative protein levels were calculated by comparing absolute protein levels to the amount of β-actin. Numerical data are presented as mean ± SD. Calculation of the difference between means was performed by ANOVA and then a post-hoc test. All statistical analyses were performed using SPSS software (version 11.0; Chicago, IL, United States). Differences with P < 0.05 were considered statistically significant.
RESULTS
Effect of CacyBP/SIP nuclear translocation on cell cycle in GC cells
The effect of CacyBP/SIP nuclear translocation on cell cycle phase distribution was investigated in SGC7901 cells with or without 2-d exposure to gastrin (10-8 mol/L). After 2 d of culture, 69.70% ± 0.46% of untreated and 65.80% ± 0.60% of gastrin-treated SGC7901 cells were observed in the G1 peak. The analysis showed that the G1 phase of gastrin-treated cells was shorter than that of untreated cells (P = 0.008; Figure 1).
Figure 1.
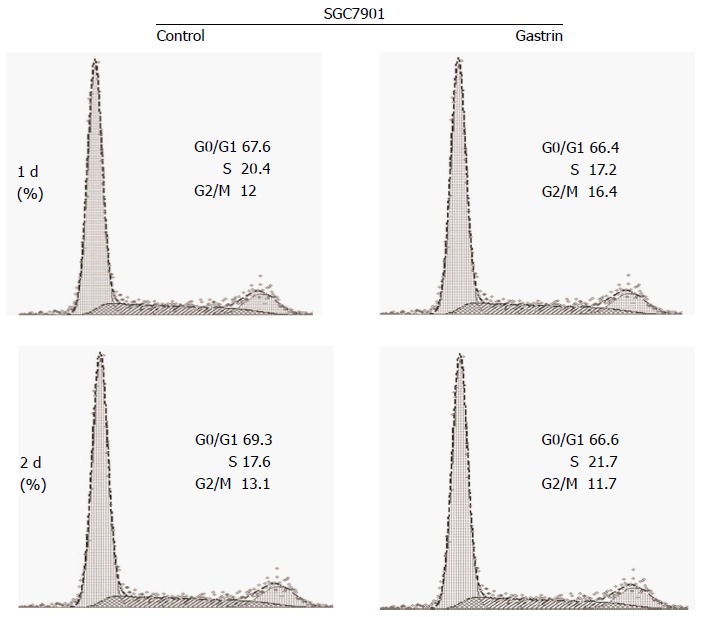
Gastrin-stimulated translocation of calcyclin binding protein/Siah-1 interacting protein into nucleus decreases the number of SGC7901 gastric cancer cell in the G0-G1 phases of the cell cycle. Cells were treated with gastrin (10-8 mol/L) for the indicated times and cell cycle variables were investigated by flow cytometry after propidium iodide (PI) staining. Data are presented as mean ± SD (n = 3), and graphs shown are representative of the three experiments.
Cells stably transfected with SGC7901-CacyBP/SIPsi1 which inhibited CacyBP/SIP expression to reduce the nuclear translocation of CacyBP/SIP were chosen for cell cycle assay. After 2 d of treatment, 71.09% ± 0.16% of untreated and 70.86% ± 0.25% of gastrin-treated SGC7901-CacyBP/SIPsi1 cells were observed in the G1 peak. Cell cycle analyses showed that no change was evident in the percentage of cells in G0-G1 phase in either cell line, whether untreated or treated with gastrin (P = 0.101; Figure 2).
Figure 2.
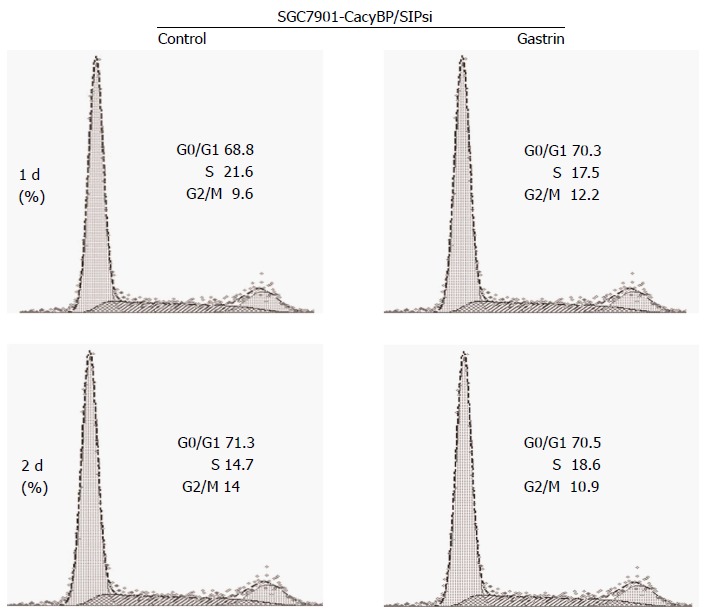
Treatment with gastrin increases the number of SGC7901-calcyclin binding protein/Siah-1si1 cells in the G0-G1 phases of the cell cycle. Cells were treated with gastrin (10-8 mol/L) for the indicated times and cell cycle variables were investigated by flow cytometry. Data are presented as mean ± SD (n = 3), and graphs shown are representative of the three experiments.
Effects of CacyBP/SIP nuclear translocation on cell cycle regulatory proteins
To correlate the effect of CacyBP/SIP on cell cycle progression with some molecular effectors of the restriction point, SGC7901 cells were treated with nocodazole for 15 h to synchronize cells in G2-M phase. After nocodazole was washed away, cells were incubated in fresh serum-free media in the presence or absence of gastrin. From 4 to 24 h, gastrin treatment (10-8 mol/L for 0, 4, 8, 12, or 24 h) induced an increase in the amount of Cyclin E protein, whereas the levels of Skp1, Skp2, and CDK2 were not affected (Figure 3). Conversely, a significant decrease in the level of p27Kip1 protein was detected during the first 8 h of treatment.
Figure 3.
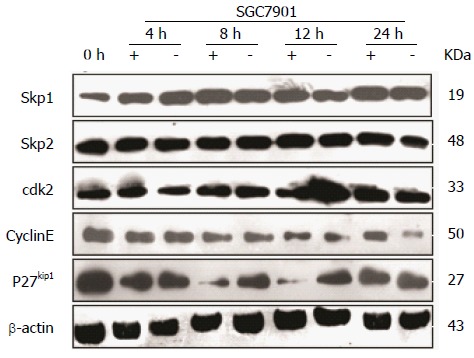
Effects of calcyclin binding protein/Siah-1 on cell cycle regulatory proteins. Cells were synchronized in G2-M phase with 0.2 μg/mL nocodazole for 15 h and nocodazole was removed by washing; cells were then incubated in fresh medium with (+) or without (-) gastrin for the indicated times. After treatment, cellular lysates were prepared and loaded per lane. Different blots with the same samples were detected with the indicated antibodies: Cyclin E, CDK2, p27Kip1, Skp1, Skp2, and β-actin as an internal control. Gastrin treatment induced an increase in the amount of Cyclin E protein and a decrease in the level of p27Kip1 protein during the first 8 h of treatment, whereas the levels of Skp1, Skp2, and CDK2 were not affected.
Furthermore, SGC7901-CacyBP/SIPsi1 cells were also synchronized in G2-M phase with nocodazole. After stimulation by gastrin, no change in protein levels of p27Kip1 or Cyclin E was observed in SGC7901-CacyBP/SIPsi1 (Figure 4).
Figure 4.
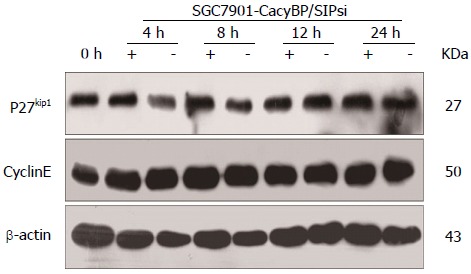
No change in protein levels of p27Kip1 and Cyclin E was observed in SGC7901-calcyclin binding protein/Siah-1si1 cells in which expression of calcyclin binding protein/Siah-1 was silenced. Cells were synchronized in G2-M phase with nocodazole for 15 h and nocodazole was removed by washing; and then cells were incubated in fresh medium with (+) or without (-) gastrin (10-8 mol/L). Different blots with the same samples were detected with the indicated antibodies: Cyclin E, p27Kip1, and β-actin as an internal control.
Proteasome-mediated degradation of p27Kip1 in GC cells
Our results shown in Figure 3 prompted us to investigate the mechanism of the decrease in p27Kip1 in more detail. It is well known that low p27Kip1 levels are attributed to increased rates of proteasome-mediated degradation. With this in mind, we studied the effect of CacyBP/SIP on p27Kip1 in the presence of the 26S proteasome inhibitor MG132. As shown in Figure 5, CacyBP/SIP nuclear translocation markedly decreased the stability of p27Kip1. Pre-incubation of growing cells with MG132 prevented p27Kip1 removal, which confirmed that the degradation of p27Kip1 in GC cells requires proteasome-mediated systems.
Figure 5.
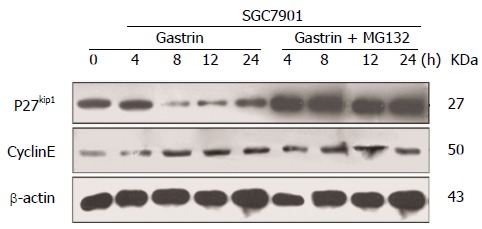
Proteasome-mediated degradation of p27Kip1 in gastric cancer cells. Western blot analysis of p27Kip1, Cyclin E and β-actin in cells 4, 8, 12, and 24 h after treatment with 10-8 mol/L gastrin in the absence or presence of MG132 (5 mmol/L, 15 min pre-treatment and co-treatment with gastrin).
CacyBP/SIP promotes proteasome-dependent degradation of p27Kip1 in GC cells through interaction with the SCF component Skp1
Additionally, we examined whether degradation of p27Kip1 is also associated with changes in ubiquitination. P27Kip1 is the primary target of the SCF complex, which is involved in the proteolysis of core components of the cell cycle machinery. Recently, based on a domain mapping study, the central region of CacyBP/SIP was confirmed to interact with Skp1, the adaptor protein of the SCF complex[2]. Therefore, we explored whether CacyBP/SIP could bind Skp1 to trigger the degradation of p27Kip1. The nuclear fractions of cell lysates were immunoprecipitated with anti-CacyBP/SIP, and the immunoprecipitated complexes were analyzed by anti-Skp1 immunoblotting. As shown in Figure 6, CacyBP/SIP bound to Skp1 after exposure to gastrin in SGC7901 cells, in agreement with the cyclical proteasomal degradation of p27Kip1.
Figure 6.
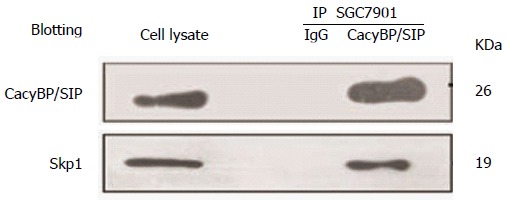
Physical interactions between Skp1 and calcyclin binding protein/Siah-1 in SGC7901 cells. Nuclear protein extracts from gastric cancer cells treated with 10-8 mol/L gastrin were immunoprecipitated with calcyclin binding protein/Siah-1 (CacyBP/SIP) MAb or control IgG. Immune complexes were analyzed by immunoblotting using an anti-Skp1 antibody with ECL-based detection.
To explore the possibility that CacyBP/SIP increased the degradation of p27Kip1 through interaction with Skp1, a truncated form of CacyBP/SIP was constructed. CacyBP/SIP (1-72), the N-terminal fragment of CacyBP/SIP which deleted the binding site with Skp1, was constructed and fused into pEGFP/C1. To exclude any interference from internal CacyBP/SIP in GC cells, SGC7901-CacyBP/SIPsi cells in which the expression of CacyBP/SIP was suppressed were used for the latter experiment. In SGC7901-CacyBP/SIPsi cells, transiently transfected pEGFP/C1-CacyBP/SIP (1-72), the truncated protein was capable of translocating into the nucleus after gastrin induction (Figure 7). However, CacyBP/SIP (1-72) abolished interactions with Skp1 as assessed by co-IP assays (Figure 8). In contrast, no change was detected in the protein level of p27Kip1 after gastrin induction (Figure 9). The diagram containing the interaction and mechanism of gastrin, CacyBP/SIP, P27kip1 and Skp1 in gastric cancer is shown in Figure 10.
Figure 7.
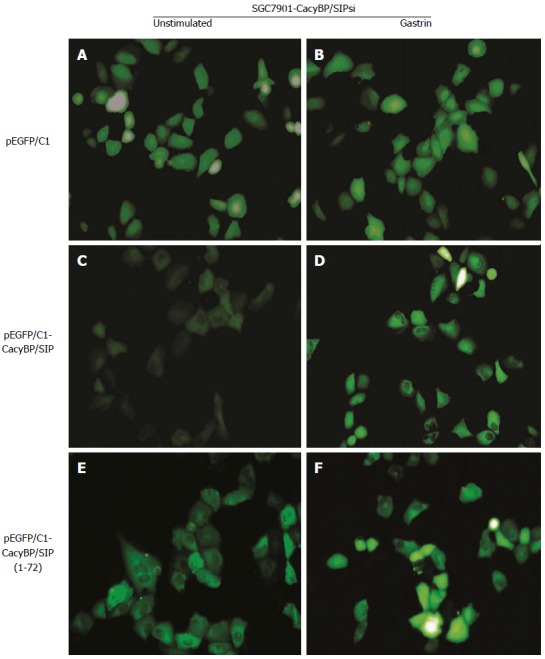
Immunofluorescent localization of calcyclin binding protein/Siah-1 (1-72) in SGC7901-calcyclin binding protein/Siah-1si cells. SGC7901-CacyBP/SIPsi cells were transfected with pEGFP/C1 vector, producing either GFP-tagged CacyBP/SIP or CacyBP/SIP (1-72) lacking the 155 central and C-terminal amino acids. Cells transfected with GFP-tagged pEGFP/C1 lacking a cDNA insert served as controls. Transfectants were treated or untreated with gastrin (10-8 mol/L), and EGFP fluorescence was analyzed under a confocal laser microscope. A, C, E: Unstimulated cells; B, D, F: Cells 8 h after gastrin stimulation.
Figure 8.
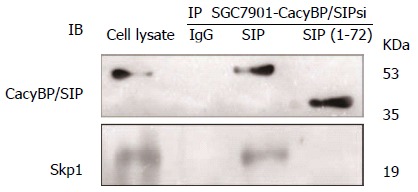
Interaction between Skp1 and calcyclin binding protein/Siah-1 (1-72) in SGC7901-calcyclin binding protein/Siah-1si transfected with pEGFP/C1-calcyclin binding protein/Siah-1 (1-72). Nuclear protein extracts from cells transfected with pEGFP/C1-CacyBP/SIP or pEGFP/C1- CacyBP/SIP (1-72) after treatment with 10-8 mol/L gastrin were immunoprecipitated with anti-GFP antibody or control IgG. Immune complexes were analyzed by immunoblotting using an anti-Skp1/GFP antibody with ECL-based detection. CacyBP/SIP: Calcyclin binding protein/Siah-1 interacting protein.
Figure 9.
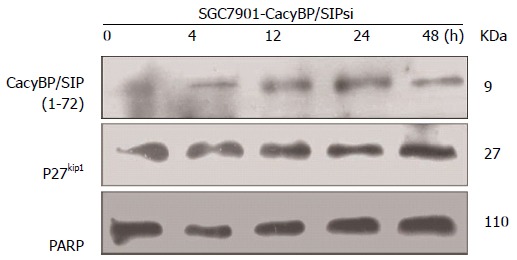
Lysates of the transfected cells were harvested for Western blot analysis 0, 4, 12, 24, and 48 h after treatment with gastrin (10-8 mol/L). Equal amounts of cellular protein (40 μg) were subjected to SDS-PAGE, followed by Western blot analysis for p27Kip1. PARP was used as a nuclear protein loading standard.
Figure 10.
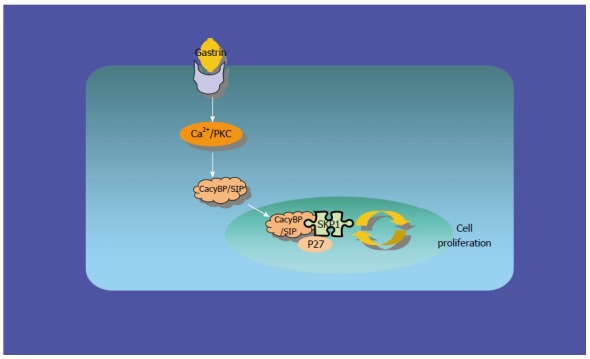
Interaction of gastrin, CacyBP/SIP, P27kip1 and Skp1 in gastric cancer.
DISCUSSION
CacyBP/SIP nuclear translocation has been suggested as a promoter of GC, but the mechanism remains unknown[17]. In this study, we examined the interaction of CacyBP/SIP nuclear translocation and p27Kip1 in GC cells. Our results suggested that CacyBP/SIP nuclear translocation promotes GC cell growth through enhancing the ubiquitin-mediated degradation of p27Kip1.
Our previous studies showed that CacyBP/SIP could translocate to the nucleus in colon cancer cells upon stimulation with KCl, gastrin, epidermal growth factor, prostaglandin E2, and hypoxia[18-21]. A previous study also found that CacyBP/SIP could translocate into the nucleus after induction with gastrin and promote the proliferation of GC CELLS[17]. However, the mechanism underlying this observation is unclear.
The study by Fukushima et al[11] was the first to identify the potential function of CacyBP/SIP in thymocyte development and G1 checkpoint. The authors observed that SIP-/- embryonic fibroblasts showed a growth-rate increase resulting from defects in G1 arrest. Another study determined that CacyBP/SIP could promote proliferation and G1/S transition of human pancreatic cancer cells[9]. In our study, cell cycle analysis showed an increase in the number of cells in S phase at the expense of those in G1 phase. This result is consistent with previous findings[17].
In eukaryotic cells, progression of the cell cycle is controlled by a series of cyclin-dependent kinases (CDKs). The transition from G1 to S is regulated mainly by the G1-specific kinases, consisting of CDK2 and Cyclin E, which are controlled by a regulatory molecule, p27Kip1, the CDK inhibitor. P27Kip1 normally partners with Cyclin E/CDK2 and inhibits CDK2 activity. When p27Kip1 is phosphorylated at Thr187 and targeted for ubiquitin-dependent proteolysis by SCFSkp2, CDK2/Cyclin E is released, allowing the transition of cells from G1 to S phase[22]. In the present study, we found that CacyBP/SIP nuclear translocation induced by gastrin was accompanied by decreased p27Kip1 and elevated Cyclin E protein levels. However, the level of cell cycle regulator p27Kip1 did not change when CacyBP/SIP nuclear translocation was reduced. These results indicate that the decreased level of p27Kip1, which led to the progression from G1 to S phase in GC cells, is the result of CacyBP/SIP nuclear translocation.
Recently, it was shown that CacyBP/SIP could regulate the glucose limitation-induced p27Kip1 degradation[12]. We supposed that CacyBP/SIP nuclear translocation affected the degradation of p27Kip1. It is widely accepted that the deregulation of p27Kip1 expression in human tumors is often due to post-transcriptional mechanisms[23]. Phosphorylation and ubiquitination are the two major types of post-translational protein modifications at work in the cell cycle. We know that low p27Kip1 levels in tumors are attributed to increased rates of proteasome-mediated degradation[24]. Our study showed that treatment with proteasome inhibitors such as MG132 restored p27Kip1 expression levels. Therefore we posit that decrease in p27Kip1 in GC cells is the result of proteasome-mediated degradation.
Studies have shown that p27Kip1 is the target of the SCF complex of E3 ubiquitin ligases. The Skp1-Cul1-F-box protein (SCF) complex is responsible for the specific ubiquitination of many regulators, and plays an integral role in regulating the G1/S phase transition in mammalian cells[25]. The SCF complex targets p27Kip1 for degradation through the F-box protein and Skp2[26,27]. Our results showed that the protein levels of Skp1 and Skp2 were unchanged when the level of p27Kip1 decreased. These results indicate that the decrease in p27Kip1 is not associated with the elevated activity of SCF.
CacyBP/SIP has been reported to bind Skp1, which led us to speculate that CacyBP/SIP was induced by gastrin to translocate to the nucleus, where it binds to Skp1 in order to promote p27Kip1 degradation. Our further experiments confirmed this hypothesis. Using Co-IP assays, we found that CacyBP/SIP could interact with Skp1, which is consistent with reports from other studies[2]. The central and the C-terminal truncated CacyBP/SIP mutant, in which the Skp1 binding site was deleted, was unable to induce any change in p27Kip1 levels, although it still translocated to the nucleus upon gastrin stimulation. Therefore, our results suggest that CacyBP/SIP nuclear translocation promotes proliferation of GC cells, at least in part, due to enhanced ubiquitin-mediated degradation of p27Kip1.
In conclusion, the present study provides evidence that CacyBP/SIP nuclear translocation stimulates the progression of cell cycle in human GC cells, an effect that is at least partially mediated by enhancing the ubiquitin-mediated degradation of p27Kip1.
ACKNOWLEDGMENTS
The authors acknowledge the valuable technical help of Dr. Yuanfei Li at the State Key Laboratory of Cancer Biology, Institute of Digestive Diseases, Xijing Hospital, and Fourth Military Medical University, Xi’an, China.
COMMENTS
Background
Calcyclin binding protein (CacyBP) was originally discovered as a calcyclin (S100A6) target protein, and later as a Siah-1 interacting protein (SIP), a component of ubiquitin-mediated proteolysis, thus named CacyBP/SIP. It has been reported that P53 induces β-catenin degradation through the Siah-1-CacyBP/SIP-SCF (Skp1/Cullin1/F-box) complex pathway, in which CacyBP/SIP exerts its function by associating with Skp1. A key study showed that defects in G1 arrest led to an increase in embryonic fibroblast growth rate in SIP-/- mice. It was also found that CacyBP/SIP could promote G1/S transition of pancreatic cancer cells. Recently, it was found that CacyBP/SIP could regulate the glucose limitation-induced P27 kip1 degradation. The authors’ previous study showed that CacyBP/SIP could translocate to the nucleus in colon cancer cells upon stimulation with KCl, gastrin, epidermal growth factor, prostaglandin E2, and hypoxia. In gastric cancer (gc) tissue, CacyBP/SIP was highly expressed in the nuclei. Further studies showed that CacyBP/SIP translocated into nucleus induced by gastrin and promote the proliferation of gc cells. However, the mechanisms underlying these observations have been unclear. In the present investigation, the role of CacyBP/SIP nuclear translocation in gc cells was studied. These results indicate that CacyBP/SIP nuclear translocation promotes G1 phase progression by enhancing the degradation of p27Kip1.
Research frontiers
CacyBP/SIP nuclear translocation decreased the number of gc cells in the G0-G1 phases of the cell cycle through increasing Cyclin E protein and decreasing p27Kip1 protein expression, whereas the levels of Skp1, Skp2 and CDK2 were unaffected. Inhibiting CacyBP/SIP nuclear translocation increased the number of cells in the G0-G1 phases, while no change in protein levels of p27Kip1 and Cyclin E was observed. Next, the authors studied the effect of CacyBP/SIP on p27Kip1 in the presence of the 26S proteasome inhibitor MG132. Pre-incubation with MG132 prevented p27Kip1 removal. The authors explored whether CacyBP/SIP could bind Skp1 to trigger the degradation of p27Kip1. Those results showed that CacyBP/SIP indeed bound Skp1. The authors also constructed CacyBP/SIP (1-72), the N-terminal fragment of CacyBP/SIP which deleted the binding site with Skp1 and fused it into pEGFP/C1. Transiently transfected pEGFP/C1-CacyBP/SIP (1-72) truncated protein was still capable of translocating into the nucleus. However, CacyBP/SIP (1-72) abolished interactions with Skp1 in co-immunoprecipitation assays. In contrast, no change was detected in the protein level of p27Kip1 after gastrin induction.
Innovations and breakthroughs
To explore the role of CacyBP/SIP nuclear translocation in gc cells, effect of CacyBP/SIP nuclear translocation on cell cycle of gc cells was studied. These studies showed that CacyBP/SIP nuclear translocation can shorten G1 phase. Using Western blot to explore the change of cell cycle regulatory proteins, the authors observed a decrease in p27Kip1 protein. Pre-incubation with proteasome inhibitor MG132 determined that the degradation of p27Kip1 was mediated by proteasome in gc cells. Using co-IP, the authors found that CacyBP/SIP promoted proteasome-dependent degradation of p27Kip1 in gc cells through interaction with the SCF component Skp1.
Applications
The study provides evidence that CacyBP/SIP nuclear translocation promotes the proteasome-mediated degradation of p27Kip1 through interaction with the SCF component Skp1. CacyBP/SIP may be a potential therapeutic target in gc.
Peer-review
In this study, the authors report the role of CacyBP/SIP nuclear translocation in GC. The study is technical sound and the results are properly analyzed. This study was based on the expression of cell cycle proteins such as CDK2, Cyclin E, Skp1, Skp2 and CacyBP/SIP detected by Western blot with or without gastrin. CacyBP/SIP was found to bind Skp1 which can trigger the degradation of P27kip1.
Footnotes
Supported by the National Natural Science Foundation of China, No. 81072040; and the Specialized Research Fund for the Doctoral Program of Ningxia Medical University.
Institutional review board statement: The study was reviewed and approved by General Hospital of Ningxia Medical University Review Board.
Conflict-of-interest statement: The authors declare that they have no conflicts of interest.
Data sharing statement: Technical appendix, statistical code and dataset available from the corresponding author at zhaihuihong@263.net. Participants have informed consent for data sharing.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 25, 2015
First decision: August 26, 2015
Article in press: December 14, 2015
P- Reviewer: Zhang J S- Editor: Ma YJ L- Editor: Wang TQ E- Editor: Wang CH
References
- 1.Filipek A, Kuźnicki J. Molecular cloning and expression of a mouse brain cDNA encoding a novel protein target of calcyclin. J Neurochem. 1998;70:1793–1798. doi: 10.1046/j.1471-4159.1998.70051793.x. [DOI] [PubMed] [Google Scholar]
- 2.Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- 3.Filipek A, Jastrzebska B, Nowotny M, Kuznicki J. CacyBP/SIP, a calcyclin and Siah-1-interacting protein, binds EF-hand proteins of the S100 family. J Biol Chem. 2002;277:28848–28852. doi: 10.1074/jbc.M203602200. [DOI] [PubMed] [Google Scholar]
- 4.Schneider G, Nieznanski K, Kilanczyk E, Bieganowski P, Kuznicki J, Filipek A. CacyBP/SIP interacts with tubulin in neuroblastoma NB2a cells and induces formation of globular tubulin assemblies. Biochim Biophys Acta. 2007;1773:1628–1636. doi: 10.1016/j.bbamcr.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Kilanczyk E, Filipek S, Jastrzebska B, Filipek A. CacyBP/SIP binds ERK1/2 and affects transcriptional activity of Elk-1. Biochem Biophys Res Commun. 2009;380:54–59. doi: 10.1016/j.bbrc.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 6.Ning X, Sun S, Hong L, Liang J, Liu L, Han S, Liu Z, Shi Y, Li Y, Gong W, et al. Calcyclin-binding protein inhibits proliferation, tumorigenicity, and invasion of gastric cancer. Mol Cancer Res. 2007;5:1254–1262. doi: 10.1158/1541-7786.MCR-06-0426. [DOI] [PubMed] [Google Scholar]
- 7.Zhai H, Meng J, Jin H, Li Y, Wang J. Role of the CacyBP/SIP protein in gastric cancer. Oncol Lett. 2015;9:2031–2035. doi: 10.3892/ol.2015.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun S, Ning X, Liu J, Liu L, Chen Y, Han S, Zhang Y, Liang J, Wu K, Fan D. Overexpressed CacyBP/SIP leads to the suppression of growth in renal cell carcinoma. Biochem Biophys Res Commun. 2007;356:864–871. doi: 10.1016/j.bbrc.2007.03.080. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Mo P, Li X, Zheng P, Zhao L, Xue Z, Ren G, Han G, Wang X, Fan D. CacyBP/SIP protein promotes proliferation and G1/S transition of human pancreatic cancer cells. Mol Carcinog. 2011;50:804–810. doi: 10.1002/mc.20737. [DOI] [PubMed] [Google Scholar]
- 10.Wang N, Ma Q, Wang Y, Ma G, Zhai H. CacyBP/SIP expression is involved in the clinical progression of breast cancer. World J Surg. 2010;34:2545–2552. doi: 10.1007/s00268-010-0690-2. [DOI] [PubMed] [Google Scholar]
- 11.Fukushima T, Zapata JM, Singha NC, Thomas M, Kress CL, Krajewska M, Krajewski S, Ronai Z, Reed JC, Matsuzawa S. Critical function for SIP, a ubiquitin E3 ligase component of the beta-catenin degradation pathway, for thymocyte development and G1 checkpoint. Immunity. 2006;24:29–39. doi: 10.1016/j.immuni.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Nagano Y, Fukushima T, Okemoto K, Tanaka K, Bowtell DD, Ronai Z, Reed JC, Matsuzawa S. Siah1/SIP regulates p27(kip1) stability and cell migration under metabolic stress. Cell Cycle. 2011;10:2592–2602. doi: 10.4161/cc.10.15.16912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang J, Luo G, Ning X, Shi Y, Zhai H, Sun S, Jin H, Liu Z, Zhang F, Lu Y, et al. Differential expression of calcium-related genes in gastric cancer cells transfected with cellular prion protein. Biochem Cell Biol. 2007;85:375–383. doi: 10.1139/o07-052. [DOI] [PubMed] [Google Scholar]
- 14.Shi Y, Hu W, Yin F, Sun L, Liu C, Lan M, Fan D. Regulation of drug sensitivity of gastric cancer cells by human calcyclin-binding protein (CacyBP) Gastric Cancer. 2004;7:160–166. doi: 10.1007/s10120-004-0286-3. [DOI] [PubMed] [Google Scholar]
- 15.Zhai H, Shi Y, Yu J, Hong L, Tang H, Wang J, Hu S, Bai F, Fan D. Establishment and characterization of calcyclin binding protein (CacyBP) monoclonal antibody. Hybridoma (Larchmt) 2006;25:91–94. doi: 10.1089/hyb.2006.25.91. [DOI] [PubMed] [Google Scholar]
- 16.Zhai H, Shi Y, Jin H, Li Y, Lu Y, Chen X, Wang J, Ding L, Wang X, Fan D. Expression of calcyclin-binding protein/Siah-1 interacting protein in normal and malignant human tissues: an immunohistochemical survey. J Histochem Cytochem. 2008;56:765–772. doi: 10.1369/jhc.2008.950519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhai HH, Meng J, Wang JB, Liu ZX, Li YF, Feng SS. CacyBP/SIP nuclear translocation induced by gastrin promotes gastric cancer cell proliferation. World J Gastroenterol. 2014;20:10062–10070. doi: 10.3748/wjg.v20.i29.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhai HH, Chen X, Lu YY, Wang X, Fan DN. Expression and nucleus translcation of calcyclin binding protein in colon cancer cells. Shijie Huaren Xiaohua Zazhi. 2008;16:3953–3957. [Google Scholar]
- 19.Xie FL, Zhai HH. Effects of epidermal growth factor on the nuclear translocation of CacyBP/SIP in colon cancer cells transfected by over expressed lentivirus vector of CacyBP/SIP. Shandong Yixue Zazhi. 2012;52:1–4. [Google Scholar]
- 20.Xie FL, Zhai HH. Study on the effect of Prostaglandin E2 on nuclear translocation of CacyBP/SIP. Chongqing Yixue. 2012;41:3020–3023. [Google Scholar]
- 21.Xie FL, Qiu CQ, Zhao YY, Yang B, Feng SS, Zhai HH. Effect of CoCl2 on CacyBP/SIP nuclear translocation of colon carcinoma cell lines. Jichu Yixue Yu Linchuang. 2012;32:1312–1317. [Google Scholar]
- 22.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 23.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 24.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323–333. doi: 10.1016/j.semcdb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 27.Sutterlüty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Müller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]