

Abstract
Traumatic brain injury (TBI) can result in persistent cognitive, behavioral and emotional deficits. However, the vast majority of patients are not chronically hospitalized; rather they have to manage their disabilities once they are discharged to home. Promoting recovery to pre-injury level is important from a patient care as well as a societal perspective. Electrical neuromodulation is one approach that has shown promise in alleviating symptoms associated with neurological disorders such as in Parkinson’s disease (PD) and epilepsy. Consistent with this perspective, both animal and clinical studies have revealed that TBI alters physiological oscillatory rhythms. More recently several studies demonstrated that low frequency stimulation improves cognitive outcome in models of TBI. Specifically, stimulation of the septohippocampal circuit in the theta frequency entrained oscillations and improved spatial learning following TBI. In order to evaluate the potential of electrical deep brain stimulation for clinical translation we review the basic neurophysiology of oscillations, their role in cognition and how they are changed post-TBI. Furthermore, we highlight several factors for future pre-clinical and clinical studies to consider, with the hope that it will promote a hypothesis driven approach to subsequent experimental designs and ultimately successful translation to improve outcome in patients with TBI.
Keywords: traumatic brain injury, electrical neuromodulation, deep brain stimulation, oscillations, hippocampus, theta
Introduction
There are an estimated 3.8 million new traumatic brain injury (TBI) cases annually, and well over 5.3 million patients report chronic TBI-related deficits (Langlois et al., 2006; DeKosky et al., 2010). Ultimately, an estimated $221 billion (combined acute and chronic care) is spent to treat TBI annually (Coronado et al., 2012). Critically, however, only 7% ($14.6 billion) of the estimated $221 billion is spent on direct medical costs (Coronado et al., 2012) and therefore, the vast majority of the financial burden is related to the long-term care of patients with chronic disabilities. In addition to the financial cost, there is a significant and well-documented emotional toll of caring for chronic TBI patients both on caregivers and society at large (Roozenbeek et al., 2013). Therefore, there is a critical need to develop innovative strategies to specifically address and improve the quality of life for patients with chronic disability following TBI. In the following review, we propose that oscillations observed in the electroencephalogram (EEG) play a key role in cognitive function and that a TBI-induced change in oscillations can result in impaired behavioral function. Finally, we discuss the potential for electrical neurostimulation to improve chronic behavioral outcome in TBI patients.
A Review of the Immediate Effects of TBI
The application of mechanical force on the brain initiates a complex series of interacting (sometimes non-monotonic) biochemical cascades, which, along with the initial impact, characterize TBI pathophysiology. Disruption of the cell membrane can lead to an ionic disturbance of Na+, K+, Ca2+, Mg2+, and Zn2+ (Vink et al., 1988; Katayama et al., 1990; Soares et al., 1992; Nilsson et al., 1993; Smith et al., 1993). The high concentration of K+ and Ca2+ in the extracellular space triggers release of neurotransmitters (e.g., glutamate), which can further exacerbate the ionic disturbance creating a vicious cycle (Faden et al., 1989; Katayama et al., 1990; Nilsson et al., 1990; Lyeth et al., 1993; Reeves et al., 1997; Shin and Dixon, 2015). This wave of depolarization can lead to excitotoxic cell death beyond what is observed in the injury core and surrounding penumbra (Sullivan et al., 1976; Dixon et al., 1987; Lowenstein et al., 1992; Hicks et al., 1993; Yamaguchi et al., 1996; Leonard et al., 1997; Yakovlev et al., 1997; Floyd et al., 2002; Witgen et al., 2005; Fedor et al., 2010). Moreover, there is considerable evidence that the pathophysiological release of neurotransmitters can alter the function of glutamatergic (Faden et al., 1989; Miller et al., 1990; Smith et al., 1993; Schwarzbach et al., 2006), cholinergic (Robinson et al., 1990; Yamamoto et al., 1993; Jiang et al., 1994; Lyeth et al., 1994; Delahunty et al., 1995; Shin and Dixon, 2015), GABAergic (Reeves et al., 1997; Witgen et al., 2005; Bonislawski et al., 2007; Gupta et al., 2012), and dopaminergic receptor systems (Donnemiller et al., 2000; Massucci et al., 2004; Shin et al., 2011), resulting in potential long-term cellular and circuit dysfunction independent of cell death.
One specific change related to excessive activation of the nervous system following TBI is the accumulation of intracellular calcium and the subsequent activation of calcium dependent catalytic enzymes such as calpain (Kampfl et al., 1997; Khorchid and Ikura, 2002). While hyperactivation of calpains is commonly associated with apoptosis (Patel et al., 1996), calpains also advance cytoskeletal and plasma membrane breakdown as well as disruption of Na+ channel function (Hicks et al., 1995; Folkerts et al., 1998; Saatman et al., 1998; Johnson et al., 2013). Changes in the cytoskeleton and membrane can trigger further ionic imbalance and specifically lead to high intra-axonal Ca2+ levels, which further challenge the already damaged axons resulting from the primary injury (Graham et al., 2000; Kita et al., 2000; Baker et al., 2002; Johnson et al., 2013; Li et al., 2014). Not surprisingly, for a prolonged period after the initial injury, neurons in the corrupted neural network can have impaired neurophysiological responses (Reeves et al., 1997; Golarai et al., 2001; Santhakumar et al., 2001; Kao et al., 2004; Goforth et al., 2011) including long-term potentiation (LTP; Miyazaki et al., 1992; Reeves et al., 1995; D’Ambrosio et al., 1998; Sick et al., 1998; Sanders et al., 2000; Schwarzbach et al., 2006; Li et al., 2014). In addition, TBI results in deficits impacting certain forms of behavioral plasticity (Ip et al., 2002; Griesbach et al., 2004) and formation of long term memories (Rimel et al., 1981; Leininger et al., 1990; Fedor et al., 2010; Gurkoff et al., 2013; Zhang et al., 2015). Disruption to cognition and plasticity following TBI is of particular relevance to our research interests and will be the focus of this review. Specific emphasis will be placed on how oscillatory activity contributes to information processing and how modifying injury- perturbed EEG could be relevant to reversing deficits in the clinical population. To this end we will first elaborate on what local field oscillations are and how they are generated in the brain.
Defining an Oscillation
There is both growing evidence and excitement that neuromodulation, and specifically invasive electrical neurostimulation, can be used to improve function in patients with neurological disorders (Lozano and Lipsman, 2013; Suthana and Fried, 2014; Tekriwal and Baltuch, 2015). In the case of TBI it is clear that functional consequences can be severe and persist for many years after the insult (Jennett et al., 1981; Whiteneck et al., 2004; DeKosky et al., 2010; Ponsford et al., 2014). And at least some of these cognitive and behavioral deficits could be mitigated with neurostimulation (Buzsáki and Watson, 2012; Lozano and Lipsman, 2013; Shin et al., 2014). Although the precise mechanism is still being delineated, it is hypothesized that driving specific neural circuits can entrain physiological circuit activity ultimately improving behavioral outcomes. Stemming from this, based on our recent findings we hypothesize that TBI-induced alterations in neural connectivity result in altered oscillations, as observed in the EEG. Further, we hypothesize that stimulating the injured nervous system to restore or substitute these oscillations will improve outcome. However, essential to the implementation and assessment of any intervention is the knowledge of the underlying mechanisms involved. Therefore, the next sections will summarize the basic neurophysiology associated with brain oscillations observed in the EEG and how these oscillations contribute to neural function. Critically this overview will introduce concepts from the perspective of developing research strategies to determine whether electrical neurostimulation can be used to improve cognitive outcome in TBI patients.
EEG is the measurement of change in the extracellular field potential recorded from the scalp that is generated by the sum of ionic movements across synapses, dendrites, soma, axons and the electroconductive cerebral spinal fluid. Similar activity measured from intracranial electrodes is commonly referred to as intracranial EEG (iEEG) or electrocorticography (ECoG). For simplicity, we will refer to all recordings, scalp and intracranial, as EEG for the remainder of this manuscript. The average of ionic movements within the immediate surrounding volume of an implanted electrode is referred to as the local field potential (LFP). There are many cellular actions that sum together to contribute to the total change in the ionic balance measured by a depth electrode, such as synaptic activity, Ca2+ fluctuations, intrinsic currents and resonances, spike after-hyperpolarization, gap junctions and glial interaction (Berridge and Rapp, 1979; Buzsáki et al., 2012). The magnitude of the electric field detected in the EEG is related to the alignment of the electrode relative to the processes of cells in any given region (Buzsáki et al., 1986; Montgomery et al., 2009). Specifically, an electrode placed parallel to the dipole created by ionic movements will result in the highest amplitude recording (Kringelbach et al., 2007).
A synchronized and reoccurring change in ionic movements results in an oscillation that can be observed in the EEG. Oscillations may arise due to a variety of mechanisms, such alternating excitation-inhibition (or excitation-excitation or inhibition-inhibition) of neurons, pacemaker cells, resonance or subthreshold membrane oscillation (James et al., 1977; Buzsáki et al., 1983; McCormick and Bal, 1997; Marshall et al., 2002; Klausberger et al., 2003; Wang, 2010). There are multiple discrete oscillatory bands ranging from 0.05 to 500 Hz that have been operationally defined based on functional states of the brain (Klausberger et al., 2003; Penttonen and Buzsáki, 2003; Buzsáki and Watson, 2012). While the general structure of many oscillations is similar (e.g., alternating excitation-inhibition, pacemaker cell), granularly each rhythm is quite distinct from one other. How, when, and where an oscillation is generated defines its operation and contribution to information processing, and in the case of a brain injury, the pathophysiology of a disorder. Therefore, in order to understand how TBI might affect the generation or maintenance of oscillations, and how to develop and assess potential strategies to restore oscillations, it is critical to consider how mechanistically an oscillation is generated.
Understanding How Oscillations are Generated
One of the earliest and most studied examples of oscillations observed in the EEG is from studies of sleep progression. For example, a defining characteristic of early non-REM (NREM) sleep is the presence of spindle waves, which are 1–3 s bursts of activity in the 7–14 Hz range every 3–10 s (Brown et al., 2012). To describe spindle generation it is important to consider both which brain regions as well as which specific cellular mechanisms are responsible for generating rhythmicity. Spindles arise due to the thalamic reticular nucleus (TRN) hyperpolarizing thalamocortical neurons with a rhythmic burst of inhibitory synaptic potentials (IPSPs; Avanzini et al., 1989; Bal et al., 1995a,b). This hyperpolarization leads to the activation of low-threshold T-type Ca2+ channels (IT), which even at low, negative membrane potentials can generate an action potential. Subsequently thalamocortical neurons generate a burst of excitatory synaptic potentials (EPSPs) that activate the TRN as well as corticothalamic neurons giving rise to a spindle (Crunelli et al., 1989; Bal et al., 1995a,b). Convergence of excitatory input onto TRN activates low-threshold Ca2+ channels, which send prolonged IPSPs back to thalamocortical neurons starting the oscillatory cycle anew (Steriade and Deschenes, 1984; Avanzini et al., 1989; McCormick and Bal, 1997). Thus, the time to go through one full cycle prescribes the observed frequency of a spindle (Bal et al., 1995a).
Thalamocortical bursting activity gives rise to another dominant NREM sleep oscillation in the delta frequency band (0.5–4 Hz; McCormick and Bal, 1997; Brown et al., 2012). Unlike the spindle waves, delta oscillations are generated in a single cell by the interplay between ionic currents (Steriade et al., 1993b). Low-threshold Ca2+ bursting in thalamocortical neurons is followed by a hyperpolarizing overshoot. This de-inactivates IT and opens the hyperpolarization-activated cation channel causing an h-current (Ih). Ih slowly depolarizes the cell towards the threshold for a Ca2+ spike by activating IT. Depolarization past −65 mV and subsequently −35 mV inactivates IT and deactivates Ih, respectively, and leads to an action potential (Crunelli et al., 1989; McCormick and Bal, 1997). Repolarization overshoots start the cycle again. However, it should be noted that other mechanisms have been proposed to account for the thalamocortical delta oscillation (Ball et al., 1977; Steriade et al., 1993a).
Specific to our understanding of oscillations during sleep it is easy to imagine how the precise activity of a series of receptor systems and the related interaction of ionic currents would be sensitive to the large ionic imbalance that follows TBI (Vink et al., 1988; Katayama et al., 1990; Soares et al., 1992; Nilsson et al., 1993; Smith et al., 1993). Consistent with this assertion, TBI is associated with sleep disturbances (Mathias and Alvaro, 2012) and specifically a decrease in delta power during NREM sleep for at least 12 weeks post injury (Parsons et al., 1997). Therefore, when considering how TBI alters oscillations and the potential for neurostimulation one has to determine not only which circuits and specific mechanisms are affected, but also when one needs to stimulate.
Our primary interest related to TBI and EEG is how injury may alter hippocampal oscillations and cognitive function. This interest is driven by a rich history in TBI-induced spatial learning deficits, deficits that we now know are concurrent with altered hippocampal oscillations (Fedor et al., 2010; Lee et al., 2013, 2015). Unlike the previously described oscillations, hippocampal theta (3–12 Hz), and specifically in CA1 is generated and maintained by the interaction of multiple rhythm generators as well as intrinsic membrane properties of hippocampal neurons that contribute to the detected rhythmic slow wave (Green and Arduini, 1954; Vanderwolf, 1969; Buzsáki et al., 1986; Kirk, 1998; Kocsis et al., 1999; Mormann et al., 2008; Montgomery et al., 2009; Colgin, 2013; Watrous et al., 2013). In the hippocampal CA1 subfield there are two well characterized dipoles of theta: the distal dendrites and soma (Figure 1). The first dipole, measured strongest near the hippocampal fissure, is attributed to layer 3 entorhinal cortex (EC) and CA3 subfield rhythmic excitation of distal dendrites of CA1 (Bland, 1986; Alonso and García-Austt, 1987; Konopacki et al., 1987; Kamondi et al., 1998; Kocsis et al., 1999). This dendritic depolarization co-occurs with somatic hyperpolarization, which reflects inputs from the medial septum nucleus (MSN; Green and Arduini, 1954; Petsche et al., 1962; James et al., 1977; Bland, 1986; Vertes et al., 2004). The MSN consists of three types of neurons: GABAergic, cholinergic and glutamatergic. In fact, afferents from each of these neuronal subtypes play a role in the generation of the second dipole. Specifically, the interplay of phasic GABAergic inhibition, tonic cholinergic and glutamatergic excitation of hippocampal interneurons results in CA1 theta (Cole and Nicoll, 1984; Smythe et al., 1992; Tóth et al., 1997; Apartis et al., 1998; Wang, 2002; Hajszan et al., 2004; Colom et al., 2005; Vandecasteele et al., 2014; Fuhrmann et al., 2015). The septal GABAergic cells act as pacemakers of theta generation in CA1 pyramidal cells through disinhibiting hippocampal interneurons (Freund and Antal, 1988; Ylinen et al., 1995; Wang, 2002). In addition, MSN cholinergic and glutamatergic neurons directly modulate excitability in CA1 pyramidal cells, which in turn excite back projecting hippocamposeptal interneurons completing the septohippocampal loop (Figure 1; Gaykema et al., 1991; Tóth and Freund, 1992; Tóth et al., 1993; Manseau et al., 2008; Mattis et al., 2014; Sun et al., 2014). This interplay between septohippocampal interneurons has the added effect of disinhibiting and inhibiting the soma of CA1 pyramidal neurons at the theta frequency, which can be measured at or dorsal to the pyramidal layer. Interestingly, hippocampal interneurons are vulnerable to cell death after TBI (Tóth et al., 1997b; Almeida-Suhett et al., 2015; Huusko et al., 2015). In addition there is evidence that injury can alter function in these neurons (O’Dell et al., 2000; Ross and Soltesz, 2000; Mtchedlishvili et al., 2010; Gupta et al., 2012; Almeida-Suhett et al., 2015; Drexel et al., 2015). Any change in interneuronal number or function could contribute to changes seen in the theta band post injury. While it is well accepted that TBI can result in cell death and dysfunction in interneurons in general, in order to get a better understanding of the hippocampal pathophysiology it will be important for future studies to examine which specific classes of interneurons (Figure 1, e.g., O-LM, PV basket, axo-axonic) that contribute to CA1 theta generation are also affected by TBI (Klausberger et al., 2003).
Figure 1.

Schematic of CA1 theta generators. Illustrated is a CA1 pyramidal cell (blue triangle) and hippocampal GABAergic interneurons (peach circle) within each CA1 layer. Approximate CA1 layers are indicated by dashed horizontal lines (so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare). Interneurons within each layer represent a subclass of interneurons (e.g., O-LM, PV basket, axo-axonic) for that layer, which receive different inputs and have distinct projections (e.g., back projecting). Dashed boxes represent CA1 inputs from medial septal nucleus (MSN), entorhinal cortex (EC) and the CA3 subfield. Arrows represent excitatory (blue-Glu: glutamate, green: ACh- acetylcholine), whereas short vertical lines are inhibitory (red and peach: GABA) connections. Projections from the MSN are left out for clarity and are represented by halos (circular on interneurons, linear on CA1 pyramidal cell).
These theta rhythm generators also work in concert with the intrinsic properties of hippocampal neurons. Specifically, there are intrinsic resonant and subthreshold membrane oscillating events which contribute to the hippocampal oscillations. For example, CA1 pyramidal cells have resonance (preferred frequency for maximal response) at theta frequency due to the interplay between voltage gated ionic currents. Depolarizations activate IM (K+ current), which has the effect of hyperpolarizing the cell. Hyperpolarization activates Ih (mix Na+/K+ current), which brings the potential closer to spike threshold. With the addition of a persistent Na+ current (INAP) these currents oppose each other resulting in a membrane resonance in the theta frequency (Pike et al., 2000; Hu et al., 2002). This CA1 rhythmicity is further amplified with subthreshold membrane oscillations via persistent Na+ and Ca2+ currents (Leung and Yim, 1991; García-Muñoz et al., 1993; Fransén et al., 2004). In a similar vein, both EC cells projecting to the hippocampus (Alonso and Llinas, 1989; Alonso and Klink, 1993; Klink and Alonso, 1993; Dickson et al., 2000; Quilichini et al., 2010) and hippocampal inhibitory interneurons (Maccaferri and McBain, 1996; Chapman and Lacaille, 1999; Pike et al., 2000) have a natural resonance and subthreshold membrane oscillation in the theta frequency range due to a mix of voltage-sensitive Na+ and K+ currents. Furthermore, the MSN displays intrinsic bursting in the theta range (Vinogradova et al., 1980; Zhadina and Vinogradova, 1983). Thus, the magnitude of synaptically driven theta from the generators in the EC and septum is boosted by multiple intrinsic resonances from cells within the hippocampus as well as extrinsic to the hippocampal formation (Goutagny et al., 2009).
In summary, there are bands of oscillations starting as low as <0.1 Hz and ranging to as high as 600 Hz. Over the years we have operationally defined discrete ranges of oscillations (e.g., theta and gamma) based on specific cellular mechanisms as they relate to observed behavioral relationships. Research into individual oscillatory bands has revealed that mechanisms for the generation and maintenance of oscillations are complex and varied, with interactions of synaptic and intrinsic generators summing together to provide a single detected change in the LFP. However, it remains an open question to what extent TBI alters any, or perhaps all, of the specific mechanisms involved in the generation of individual oscillatory bands and ultimately the neural network underlying cognition.
How Oscillations Interact and Contribute to Information Processing
While each electrode yields a single LFP measure, that LFP is made up of several components. In fact, the combination of synaptic and intrinsic membrane events frequently leads to power in multiple oscillatory bands. Specifically, when one decomposes an individual LFP one can see that each oscillation occurs simultaneously (Figure 2). Figure 2 illustrates sinusoidal waves whose frequency corresponds to the slow, delta, theta and gamma oscillatory bands. A more exhaustive list of oscillatory frequencies was described by Penttonen and Buzsáki (2003). In general slow wave oscillations, and relevant to the current discussion those in the theta frequency range, are thought to synchronize distal regions of the brain promoting plasticity, while faster gamma oscillations are hypothesized to link and/or activate local neuronal ensembles (Bragin et al., 1995; Penttonen and Buzsáki, 2003; Buzsáki and Draguhn, 2004). There are several published reviews relating to a broader analysis of EEG and their role in plasticity and learning (Başar et al., 2001; Buzsáki, 2005; Lakatos et al., 2008; Knyazev, 2012; De Gennaro and Ferrara, 2003). However, based on the current level of understanding of these oscillations as they pertain to TBI is limited and therefore an in depth description of these findings is beyond the scope of this review. But, if we want to understand the extent of the effect of TBI on oscillations, it is important to not only consider one specific frequency band at a single electrode, for example hippocampal theta, but instead consider how multiple frequency bands are related at a single recording site (i.e., cross frequency coupling), and also how similar frequency bands are related between distal electrodes (i.e., phase coherence). Thus, in order to better understand the effects of brain injury on EEG it will be necessary to sample from multiple regions within a circuit as well as to investigate a range of frequency interactions in addition to a power analysis.
Figure 2.
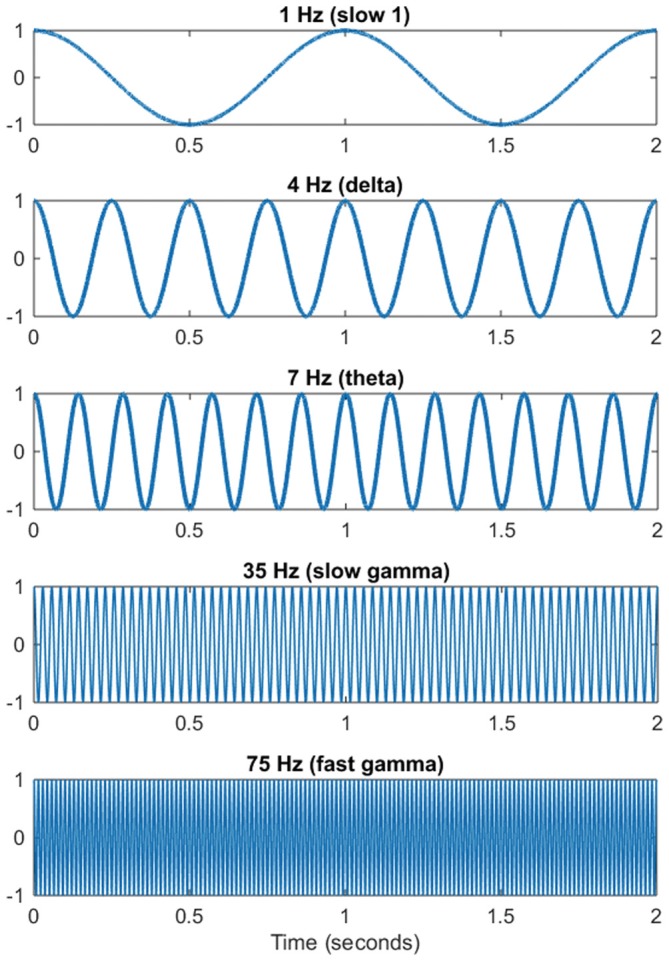
Oscillations of varying frequency. MATLAB generated sine curves to represent neuronal oscillations that make up the unfiltered electroencephalogram (EEG). Each panel represents a distinct frequency and in parentheses the corresponding oscillatory band name most often associated with it.
Interactions that take place across different frequencies at a single recording site are referred to as cross frequency coupling. The interplay between two frequencies could take place across several different domains (Figure 3). For example, cross-frequency power-power coupling (amplitude-amplitude) occurs when the power of the low frequency oscillation dictates the power of the high frequency oscillation; cross-frequency phase-phase coupling (n:m phase locking) refers to a fixed number of high frequency oscillations nested in each slower cycle; cross-frequency phase-frequency and phase-power coupling indicates that the frequency and power of the faster wave is modulated by the phase of the slower oscillation, respectively (Jensen and Colgin, 2007; Belluscio et al., 2012). For a more thorough review on the significance of each of these interactions as they relate to cognition see (Axmacher et al., 2006; Lisman and Buzsáki, 2008; Colgin, 2013; Lisman and Jensen, 2013).
Figure 3.

Illustrations of cross-frequency coupling. (A) A slow oscillation in the theta range, along with its power indicated by the red line. (B–E) Illustrate the different types of interplay that faster oscillations (e.g., gamma) can have with the slower trace in (A). (B) Power–power: the power (amplitude) of the faster wave correlates with the power of the slower wave. (C) Phase–phase: a fixed number of faster cycles within each phase of the slower oscillation. In this case there are four cycles within each phase. (D) Phase–frequency: the number of cycles in the faster wave correlates with specific phase of the slower wave. (E) Phase–power: the power of the faster wave correlates with the specific phase of the slower wave, independent of the power of the slower wave. Reproduced with permission from Elsevier (Jensen and Colgin, 2007).
Phase coherence is the relationship of two oscillations of the same frequency across different electrodes. For example, there can be phase-phase coupling of oscillations such that the phases of each ongoing oscillation are in sync (i.e., the peak of one oscillation always occurs in the same phase of a second oscillation measured at a second site). Likewise, two similar frequencies can correlate in their power, independent of the phase. Specifically, as the power of an oscillation increases at one recording site, a similar increase in power is observed at a second electrode. These types of specific interactions suggest that oscillations are not simply a local phenomenon but instead have a role in network activity.
In addition to phase coherence, oscillations can organize the firing of individual neurons by summing together subthreshold excitatory inputs or organizing the firing procession of assemblies of neurons (O’Keefe and Recce, 1993; Skaggs et al., 1996; Tukker et al., 2007). While there are different implications for the specific type of interaction observed, in general interaction between rhythms supports neural communication, plasticity, formation of functional ensembles and consolidation of long-term memories (Buzsáki and Draguhn, 2004; Fell and Axmacher, 2011; Belluscio et al., 2012). The common variable is that oscillatory patterns contribute to higher order information processing including the formation of neuronal ensembles.
Ensemble formation, or the linking of a group of cells, is at the heart of information processing (Fries, 2005; Buzsáki, 2010). Oscillations are capable of promoting ensembles partly through temporally precise segregation and boosting of communication between some groups of neurons. Theta-gamma phase modulation (cross frequency phase-amplitude coupling) can explain how one region of the hippocampus, say CA1, can be involved in multiple networks virtually simultaneously. For instance, CA3-CA1 shows the greatest coherence and phase locking of single unit firing in the slow gamma band (gammaS: 25–50 Hz), which peaks in the early descending phase of the CA1 theta. On the other hand, medial EC (MEC)-CA1 has high coherence at mid gamma frequency (gammaM 50–90 Hz) which is strongest at the peak of theta (Colgin et al., 2009; Belluscio et al., 2012; Schomburg et al., 2014). Thus, CA1 inputs are segregated across the phase of theta cycle and therefore individual pyramidal neurons have the potential to temporally align with multiple ensembles within a phase of theta (for detailed discussion, see Buzsáki and Schomburg, 2015). Alternating back and forth between functional networks could be important, as an example, for shifting between encoding new and retrieval of previous information (Montgomery and Buzsáki, 2007; Colgin et al., 2009), assembling discrete stimuli into a single representation (Gray et al., 1989; Engel et al., 1991), selective attention/gain modulation (Fries et al., 2001) and associative binding (Headley and Paré, 2013). Critically, in many cases theta provides the temporal structure for local gamma, while simultaneously coupling cell assemblies between regions and “chunking” (Buzsáki, 2010) information into discrete processing units (Senior et al., 2008; Buzsáki and Moser, 2013; Colgin, 2013). Given that theta is altered post-TBI (Fedor et al., 2010; Lee et al., 2013, 2015), it will be important for future studies to examine how TBI may affect these tightly coupled interactions and whether changes in these interactions might underlie injury-induced behavioral deficits.
Theta modulation can also strengthen synaptic connections and organize information flow. For instance, hippocampal theta phase locks local cortical gamma activity across multiple regions and the firing of individual cortical neurons (Sirota et al., 2003; Hyman et al., 2005; Jones and Wilson, 2005; Siapas et al., 2005; Fujisawa and Buzsáki, 2011). Convergence of cortical inputs onto the hippocampus coincides with a time (i.e., theta phase) that is optimized to support hippocampal synaptic plasticity (Berry and Thompson, 1978; Huerta and Lisman, 1995; Seager et al., 2002). In turn, local hippocampal plasticity is shaped by a difference in gamma phase-phase synchronization between subfields. During tonic REM sleep CA3-CA1 gamma coherence is decreased, while dentate gyrus-CA3 gamma is increased. This releases CA1 from CA3 recurrent collateral control and allows the dentate gyrus to modify CA3 synaptic activity. However, during brief interspersed periods of phasic REM theta and gamma coherence across all three subfields is increased and so is CA1 firing (Montgomery et al., 2008). Thus, it seems that CA1 is excluded until hippocampal information is transmitted back to the cortex (Buzsáki, 1989; Wilson and McNaughton, 1994; Ji and Wilson, 2007). Shifting between local cell assemblies ensures accurate transmission of information, discrete synaptic modifications free of interference and a receptive receiver to form an ensemble. Unfortunately, it is yet to be determined if these interactions are affected by TBI and if they are correlated with cognitive and behavioral deficits.
While to this point we have focused on theta, ensemble formation can be modulated at other frequencies. For example, during NREM sleep cortical slow oscillations (0.02–0.8 Hz) drive the cortex to alternate between a depolarized and a hyperpolarized (up/down) state (Steriade et al., 1993a; Cowan and Wilson, 1994; Timofeev et al., 2001). This slow oscillation also propagates to the thalamus and the hippocampus. A depolarized cortical state is associated with thalamacortical spindles which can bias high frequency CA1 burst activity, commonly referred to as sharp wave-ripples (Buzsáki et al., 1992; Battaglia et al., 2004; Mölle et al., 2006; Buzsáki and Silva, 2012). These ripples are synchronized to a particular phase of the spindles and drive the activation of specific cortical ensembles (Siapas and Wilson, 1998; Sirota et al., 2003; Isomura et al., 2006; Wierzynski et al., 2009). This hippocampo-cortical interplay may bind hippocampal output with coactive cortical ensembles. Together, these interactions have the added effect of associating two different networks in the spirit of coordinating information storage and promoting formation of long term memories through reciprocal excitation of ensembles. Ultimately, depending on the effect of TBI these ensembles could be as important of targets as those organized by theta oscillations.
It is important to note that the very functions ascribed to oscillations are perturbed in TBI patients. On a variety of neuropsychological exams patients score worse on tests of attention, concentration, working memory, reaction time, judgement and measures of effort (Rimel et al., 1981; Levin et al., 1988a; McDowell et al., 1997; Bales et al., 2009). These failures of information processing consolidate into deficiencies in verbal and visual memory, episodic memory, multitasking, executive function and cognition (Levin et al., 1988b; Hanks et al., 1999; Millis et al., 2001; Alvarez et al., 2003; Bales et al., 2009; Bootes and Chapparo, 2010; McCauley et al., 2014; Mäki-Marttunen et al., 2015). Deficits in higher order information processing are compounded by sleep-wake disturbances (Kempf et al., 2010; Shay et al., 2014; Skopin et al., 2015). TBI patients report a range of sleep-related disorders including difficulty falling asleep, more nighttime awakenings and daytime naps, increased fatigue, and prolonged sleep (Parcell et al., 2006; Kempf et al., 2010; Mathias and Alvaro, 2012). These sleep deficits are accompanied by altered REM and NREM sleep as detected by nighttime scalp recordings, as well as changes in nocturnal hormone secretion (Parsons et al., 1997; Frieboes et al., 1999). Given the overlap between functions associated with oscillations and observed deficits in TBI patients, additional research is critical to understand whether dysfunction can be ascribed to an alteration in oscillations. Moreover, if there is a relationship between oscillations and outcome in brain injured patients treatments aimed at modulating EEG are seemingly an appropriate starting point.
Altered EEG After TBI
There are data that clearly indicate that TBI alters oscillations both in pre-clinical models as well as in patients (for detailed table on altered EEG, see Thatcher et al., 1989; Wallace et al., 2001; Rapp et al., 2015). In rodent models, there is a pronounced decrease in alpha, beta, delta and theta power following mechanical injury (Dixon et al., 1987; Ishige et al., 1987; McIntosh et al., 1987; Paterno et al., 2016). While most of these reductions return to baseline levels within minutes to hours after the injury (Sullivan et al., 1976; Dixon et al., 1988; McIntosh et al., 1989), some, like diminished theta, can persists for as long as 8–10 weeks after the insult (Fedor et al., 2010). A prolonged decrease in theta power is accompanied by other neurophysiological irregularities, even in brain regions spared from significant cell death. One such area, the CA1 subfield, exhibits altered excitation and inhibition, reduced LTP and pathological spine anatomy days to weeks after injury (Reeves et al., 1997; Sick et al., 1998; Sanders et al., 2000; Schwarzbach et al., 2006).
Analysis of human TBI patients resembles the reported prolonged recovery of EEG in experimental models of TBI. While modifications in brain activity can be seen as early as 24 h even after a subconcussive head trauma (Johnson et al., 2014), altered EEG following TBI can last for years after injury (Thatcher et al., 1989; Alvarez et al., 2008; Kempf et al., 2010; Slobounov et al., 2012). The changes in the EEG are not confined to a single oscillatory band, as they have been reported for the alpha, beta, delta, theta and gamma range (Alvarez et al., 2008; Tomkins et al., 2011; Rapp et al., 2015). Alterations of EEG activity are not state dependent, as changes are observed when a patient is at rest (Virji-Babul et al., 2014; Borich et al., 2015), actively moving (Slobounov et al., 2012) and during sleep (Parsons et al., 1997; Frieboes et al., 1999). In fact, abnormalities in scalp EEG are so consistent in patients they have been used to differentiate between injured and non-injured subjects, classify the severity of the injury, and some suggest, predict long term outcome after TBI (Thatcher et al., 1989, 1991, 2001; Alvarez et al., 2003; Arciniegas, 2011). For example, one of the criteria used to diagnose mild TBI many months after injury is an increase in coherence and a decrease in phase offset between frontal and temporal lobes, along with a decrease in power between frontal and posterior cortical regions (Thatcher et al., 1989). Furthermore, reversal of pathological EEG power ratio with administration of a neurotrophic peptide correlated with improvement in attention and working memory (Alvarez et al., 2008). The persistence of an abnormal EEG after a head injury suggests a potential link to prolonged psychological symptoms.
As is clear from the previous sections, it is not rigorous enough to determine that EEG is altered following injury. Unfortunately for the patient, it is also unlikely that there is a single mechanism that can easily explain why the EEG has changed. Therefore, it is critical to identify a starting point for research. While many neural systems and processes may be affected by a head injury, of particular interest (in our laboratory) is hippocampal dysfunction and the generation/maintenance of theta. TBI alters hippocampal neurotransmitter systems involved in the generation of theta, including acetylcholine, glutamate and GABA (Saija et al., 1988; Faden et al., 1989; Katayama et al., 1990; Robinson et al., 1990; Marshall et al., 2002). Rapid and prolonged increases in neurotransmitter levels act on local receptors causing long-lasting adaptation (Miller et al., 1990; Delahunty, 1992; Jiang et al., 1994; Delahunty et al., 1995; Schwarzbach et al., 2006). Thus, even after the injury-induced alteration of extracellular concentration of neurotransmitters returns to basal levels, modified receptors may have an ectopic response to subsequent activation, potentially affecting the timing and strength of receptor coupled processes essential to rhythm generation (Lyeth et al., 1992; Fineman et al., 1993; Kato et al., 2007; Marcoux et al., 2008). Another consequence of an intense glutamate discharge is excitotoxicity (Choi, 1988). This cell loss is readily observed in CA3 and dentate gyrus (Hicks et al., 1993; Floyd et al., 2002; Witgen et al., 2005), both of which are contributing nodes to CA1 theta (Bland, 1986; Kocsis et al., 1999; Marshall et al., 2002). Within the dentate gyrus, GABAergic interneurons in the hilus seem to be especially vulnerable to excitotoxicity, due to an increased excitatory drive onto their glutamatergic receptors (Tóth et al., 1997b; Hunt et al., 2011). Consequently, GABAergic cell death profoundly alters the excitability of not only the dentate gyrus, but the hippocampus as a whole leading to deficits in LTP and theta generation (Reeves et al., 1995, 1997; van den Pol et al., 1996; Witgen et al., 2005; Mtchedlishvili et al., 2010; Dinocourt et al., 2011). This hippocampal pathological process results in delayed degeneration in the MSN, a critical pacemaker for theta generation (Leonard et al., 1997). More specifically, cholinergic neurons within the septum show a marked susceptibility to cell death days to weeks following mild/moderate fluid percussion (Leonard et al., 1994; Schmidt and Grady, 1995) and a controlled cortical impact injury (Dixon et al., 1997). Such neuronal atrophy leads to enlarged ventricles and a proliferation of astrocytes, detected up to a year after TBI (Smith et al., 1997). Furthermore, downstream structures to the MSN, such as the hippocampus, show changes consistent with cholinergic hypofunction. In order to compensate for a decrease in evoked cholinergic neurotransmission (Dixon et al., 1996), there is an increase in the protein responsible for packing acetylcholine into presynaptic vesicles, downregulation of inhibitory autoreceptors and a hypersensitivity hippocampal cholinergic receptors and subsequent response of 2nd messengers (Jiang et al., 1994; Delahunty et al., 1995; Ciallella et al., 1998). MSN function could be further encumbered by post-traumatic epilepsy (Santhakumar et al., 2001; Frey, 2003; Pitkänen and McIntosh, 2006). Chronic seizure activity is related to a decrease (Garrido Sanabria et al., 2006) and altered firing of putative theta generating GABAergic cells in the septum (Colom et al., 2006). These observations have fueled the hypothesis that at least temporal lobe epilepsy in part arises due to an imbalance in septohippocampal theta and that theta stimulation could potentially be used as antiepileptic (Kitchigina et al., 2013; Fisher, 2015).
Degeneration and white matter damage also likely interfere with normal patterns of brain oscillations. Axonal abnormalities may arise from the initial shearing forces from the impact and an ionic imbalance in the extracellular space (Hicks et al., 1995; Graham et al., 2000; Li et al., 2014). Loss of ionic equilibrium leads to axonal increase of Ca2+ permeability, calpain activation, mitochondrial dysfunction and eventually breakdown of the cytoskeleton (Maxwell et al., 1997; Johnson et al., 2013). These changes may culminate in deafferentation/denervation and inappropriate synaptic plasticity (Povlishock and Katz, 2005; Hunt et al., 2011). Many of these axonal changes can be detected weeks after the insult and correlate with behavioral abnormalities (Kempf et al., 2010; Spain et al., 2010). It is not surprising then that compromised axonal structure and function results in irregular oscillatory interactions, even years after the injury. These structural deformities along with neurochemical aberrations contribute significantly to the observed deficits in the propagation of functionally relevant hippocampal theta (Fedor et al., 2010), and subsequently brain function after TBI (Hanks et al., 1999; Millis et al., 2001). These data clearly indicate that multiple TBI-induced mechanisms can play a role in altered brain oscillations and their interactions thus contributing to long-term impaired cognition.
Theta DBS
Pathologies associated with TBI are wide-ranging, occurring at the molecular, physiological and structural level. These alterations in turn may lead to changes in network activity, affecting neural communication and plasticity. Abnormal rhythm generation could potentially hinder and prolong recovery after a TBI insult (Thatcher et al., 1991; Tomkins et al., 2011). Furthermore, once a patient has progressed out of the acute post-injury phase of the disease, neuroprotection is no longer a viable therapeutic option. Therefore, there is an urgency to develop treatment strategies for TBI patients who have chronic disability. DBS represents a potential intervention that can drive neural networks, improve neurophysiology and ultimately behavioral outcome in a subset of brain-injured patients. The advantage of neurostimulation, say over pharmacology, is its ability to target specific regions, inherent higher temporal resolution and ability to generate specific patterns of electrical input, all of which are critical factors in the generation and interaction of oscillations. Furthermore, neurostimulation has shown promise in alleviating symptoms in motor, cognitive, behavioral and psychiatric conditions (Brunoni et al., 2011; Lozano and Lipsman, 2013; Suthana and Fried, 2014). The relative success of DBS in each specific situation is determined by a growing body of parameters, beyond the scope of this article to survey (Kuncel and Grill, 2004; Butson and McIntyre, 2007; Birdno and Grill, 2008). Therefore, given the focus of this review we will highlight the potential use of one stimulation strategy, the use of low frequency stimulation within the theta range.
Exogenous induction of theta in structures like the hippocampus can improve cognitive processes in experimental animals. Hippocampal theta can be achieved either with direct hippocampal stimulation or by targeting afferent structures such as the fornix or MSN. Using rodent models, pre-training stimulation of MSN decreased the time to acquire discriminatory learning (Deupree et al., 1982) while post-training stimulation facilitated memory consolidation (Landfield, 1977; Wetzel et al., 1977). These results mirror the positive correlations observed between endogenous theta and enhanced acquisition and retention (Landfield et al., 1972; Berry and Thompson, 1978; Seager et al., 2002; Mandile et al., 2003; Mitchell et al., 2008). Importantly, the uniqueness of these findings lies not in the MSN per se, but in the theta oscillation. The critical role of theta oscillations specifically was revealed in studies were theta stimulation of the fornix was able to drive hippocampal theta oscillations following chemical inactivation of the MSN (Green and Arduini, 1954; Petsche et al., 1962; Vertes et al., 2004). Not only did stimulation drive theta, but also rescued the behavioral impairment (McNaughton et al., 2006). Further substantiating the selectivity of theta range, high frequency MSN stimulation does not facilitate mnemonic processes (Landfield, 1977; Wetzel et al., 1977). Therefore hippocampal theta, generated endogenously or extrinsically, plays a critical role in neural computations supporting animal cognition.
The beneficial effects of stimulation in the theta range are not limited to cognitive processes. For example, low frequency stimulation has been shown to be beneficial after an acute spinal cord contusion where an 8 Hz stimulation of the raphe nucleus improved motor coordination and sensory processing, increased white matter integrity and reduced astrocytosis (Hentall and Burns, 2009; Hentall and Gonzalez, 2012). Therefore it is possible that stimulation of theta following TBI might also have indirect effects that could improve the hippocampal milieu post-injury facilitating anatomical as well as physiological recovery. Epilepsy treatment is also closely associated with low frequency stimulation. Effective reduction of kindling from a 60 Hz induced seizure is achieved with 3–5 Hz stimulation (Gaito et al., 1980; Kile et al., 2010; Koubeissi et al., 2013). Building on this framework, Fisher recently proposed a novel hypothesis that MSN stimulation in the theta range may benefit patients with epilepsy (Fisher, 2015). Therefore stimulation following severe TBI might also have the added benefit of reducing or preventing post-traumatic epilepsy.
Theta DBS in TBI Models
Recently, several studies have described the restorative effect of theta stimulation after TBI injury. Lee et al. (2013, 2015) stimulated the MSN at 7.7 Hz and recorded an increase in hippocampal theta power along with better spatial performance in the Barnes maze following a moderate lateral fluid percussion injury (Figure 4). Several stimulation controls bolstered the hypothesis that theta band stimulation was specifically augmenting the septohippocampal system in TBI rats. In particular, successful MSN stimulation in the theta range was intensity specific, there was no effect on overall motor output (i.e., distance traveled) and MSN stimulation at 100 Hz did not rescue the deficit in spatial performance (i.e., spatial search strategy) in the maze (Lee et al., 2015). Moreover, the authors concluded the effect was restorative and not simply enhancing function as similarly stimulated sham animals experienced no improvement in spatial learning. Likewise, the Hentall group observed positive effects on spatial memory in the watermaze and forelimb reaching movements when, following lateral fluid percussion, they stimulated the raphe nucleus, part of the ascending system that generates theta, at 8 Hz (Vertes et al., 2004; Carballosa Gonzalez et al., 2013). In conjunction with behavioral outcomes, stimulation reversed cortical cell loss, white matter degeneration and decreases in cortical and hippocampal levels of cAMP, an intracellular second messenger (Carballosa Gonzalez et al., 2013). These proof of principle studies illustrate the potential of theta stimulation to augment physiological and behavioral outcome following TBI.
Figure 4.

Systems level overview of septohippocampal theta. Blue arrows represent proposed theta generators and essential modulators of hippocampal theta. Red arrows, stemming from the hippocampus, represent various structures that are known to be modulated/interact with hippocampal theta. *Raphe only projects to the hippocampus, not MSN.
Importantly, theta stimulation has not been the only successful stimulation paradigm observed in experimental models of TBI. In a model of mild TBI, theta burst stimulation (TBS) was able to rescue working memory in the T-maze delayed non-match to sample task. Rather than using a continuous single pulse (7.7 or 8 Hz) fixed stimulation (Sweet et al., 2014) used a TBS protocol to stimulate the fornix, specifically with five 50 ms long bursts of high frequency (200 Hz) pulses per second. The hypothetical advantage of TBS as compared to continuous theta stimulation is that the 200 Hz gamma stimulation partially recapitulates endogenous patterned firing at a physiologically relevant theta frequency. In fact TBS has been demonstrated to induce long lasting LTP (Rose and Dunwiddie, 1986; Staubli and Lynch, 1987; Diamond et al., 1988; Kirkwood et al., 1993). Accordingly, Sweet et al. (2014) reported that TBS, but not low (5 Hz) or high (130 Hz) frequency stimulation of the fornix, improved performance of TBI rats in the T-maze. Spatial memory in the water maze was also improved with TBS; however, 5 Hz stimulation was not tested.
There are several key takeaways from these two successful stimulation paradigms. The first is that task might matter. There is evidence that, depending on task, there is a shift in the frequency of the theta oscillation (Kramis et al., 1975; Watrous et al., 2013). Therefore, the specific frequency within the theta range may be critical to improving outcome and the target may be different for different behaviors. Following that reasoning, while 5 Hz stimulation may not have improved T-maze performance, it is possible that 7.7 (or some other frequency) may have. In fact, in a study by McNaughton et al. (2006), it was observed that to optimally restore behavior in the watermaze after chemical inactivation of the MSN, it was best to stimulate the fornix with an endogenous EEG pattern recorded from the supramamillary nucleus as compared to fixed 7.7 Hz or an irregular theta stimulation pattern (with an average frequency of 7.7 Hz). These data indicate that not all “theta” is the same, and that the specific frequency within the theta band may be highly relevant.
The fact that these initial reports of DBS in TBI did not report completely homogenous results is worth noting. The data highlights the need for additional research to investigate the large parametric space available for potential stimulation parameters. There are many additional variables to be considered: intermittent vs. constant stimulation; endogenous stimulation (where theta is recorded from an uninjured site and played back in the injured hippocampus as in McNaughton et al., 2006) vs. exogenous fixed frequency; theta burst vs. single pulse; variants in voltage, pulse width and square as compared to sinusoidal; which regions, nuclei or subfields to target; at which point during the task/behavior to stimulate or whether to stimulate offline during sleep or to stimulate relative to an endogenous oscillation independent of the behavior. A closed loop system where stimulation was based on the recorded EEG (from a different region) would subsequently be amenable to biofeedback (Wallace et al., 2001; Rosin et al., 2011; de Hemptinne et al., 2015).
Different stimulation parameters will not only influence the efficacy of the treatment, but also most likely the extent of unintended effects. The most commonly reported adverse events are related to the implant rather than stimulation and include inflammation, headache, pain at the implant site, and mild paresthesia surrounding the implant (Kenney et al., 2007; Fisher et al., 2010; Salanova et al., 2015). However, in studies of stimulation for treatment of epilepsy there are reports of cognitive dysfunction, depression and suicide in a small number of patients (Bergey et al., 2015; Salanova et al., 2015). Therefore it will be important to monitor which symptoms TBI patients report receiving low frequency stimulation of the septohippocampal system. While complications with the surgery and device itself are minimal, as argued by Fisher (2015) in his proposal to stimulate the MSN in epileptic patients, there are however potential risks of eliciting seizures or promoting addiction to constant stimulation (for discussion on long-term safety of DBS, see Kenney et al., 2007). These potential risks (e.g., prior epileptic activity, predisposition to addiction, mood/affect disorders) should be taken into consideration when enrolling patients so as to minimize potential harm. The inclusion criterion could be further refined based on the mechanism of action of neurostimulation in the theta range. If DBS is masking an enduring effect or if it is restorative will potentially influence the type of therapy one gets, such as; when should DBS be administered relative to the injury and in response to what type of injury? Will immediate intervention interfere with the healing process or will waiting too long make the system unamenable? How long will the benefits of DBS persist, if stimulation is discontinued? Should the treatment continue indefinitely or should there be a clinical marker/threshold to stop or augment the stimulation? Thus, there is a clear need for considerable pre-clinical animal work and potentially computational modeling to better understand and explore the complex parameter space that is DBS and the mechanisms behind it, if we are to optimize the potential of neurostimulation for clinical translation.
Conclusion
After years of research, there are few proven interventions that reduce injury-induced cellular cascades and ultimately, cell death and dysfunction following TBI. While the latest census estimates over 5.3 million patients live with chronic disability, it is clear that that number has grown and continues to grow. Therefore, there is a clear need for pre-clinical research expressly focused on the injured nervous system in the chronic stages of the disease. Oscillations are known to play a key role in physiological circuit function, whether it is the progression of oscillations through the sleep cycle or theta oscillations in the hippocampus. Initial evidence suggests that injury-induced disruption of these oscillations has a profound impact on neural connectivity and behavior. In fact, changes in EEG can be used as a biomarker to confirm mild and moderate TBI. Additionally, limited studies of DBS in brain injured rats demonstrate that the injured brain can be modulated by entraining or replacing oscillations, with improved outcomes. Future preclinical studies are needed to explore a very large parametric space that spans not only multiple stimulation targets and paradigms but also different injury mechanisms as well as a range of cognitive behavioral tasks and dependent measures, extending beyond spatial navigation. The potential for DBS is clear. We believe that further research into electrical neuromodulation of the injured brain will result in an exciting avenue to promote behavioral, cognitive and neurophysiological recovery following TBI.
Author Contributions
AP, KS, and GGG contributed to the conception, writing and editing of the manuscript. AI, DJL contributed significantly to the conception and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Almeida-Suhett C. P., Prager E. M., Pidoplichko V., Figueiredo T. H., Marini A. M., Li Z., et al. (2015). GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp. Neurol. 273, 11–23. 10.1016/j.expneurol.2015.07.028 [DOI] [PubMed] [Google Scholar]
- Alonso A., García-Austt E. (1987). Neuronal sources of theta rhythm in the entorhinal cortex of the rat. I. Laminar distribution of theta field potentials. Exp. Brain Res. 67, 493–501. 10.1007/bf00247282 [DOI] [PubMed] [Google Scholar]
- Alonso A., Klink R. (1993). Differential electroresponsiveness of stellate and pyramidal-like cells of medial entorhinal cortex layer II. J. Neurophysiol. 70, 128–143. [DOI] [PubMed] [Google Scholar]
- Alonso A., Llinas R. R. (1989). Subthreshold Na+-dependent theta-like rhythmicity in stellate cells of entorhinal cortex layer II. Nature 342, 175–177. 10.1038/342175a0 [DOI] [PubMed] [Google Scholar]
- Alvarez X. A., Sampedro C., Figueroa J., Tellado I., González A., García-Fantini M., et al. (2008). Reductions in qEEG slowing over 1 year and after treatment with Cerebrolysin in patients with moderate-severe traumatic brain injury. J. Neural Transm. (Vienna) 115, 683–692. 10.1007/s00702-008-0024-9 [DOI] [PubMed] [Google Scholar]
- Alvarez X. A., Sampedro C., Pérez P., Laredo M., Couceiro V., Hernández A., et al. (2003). Positive effects of cerebrolysin on electroencephalogram slowing, cognition and clinical outcome in patients with postacute traumatic brain injury: an exploratory study. Int. Clin. Psychopharmacol. 18, 271–278. 10.1097/00004850-200309000-00003 [DOI] [PubMed] [Google Scholar]
- Apartis E., Poindessous-Jazat F. R., Lamour Y. A., Bassant M. H. (1998). Loss of rhythmically bursting neurons in rat medial septum following selective lesion of septohippocampal cholinergic system. J. Neurophysiol. 79, 1633–1642. [DOI] [PubMed] [Google Scholar]
- Arciniegas D. B. (2011). Clinical electrophysiologic assessments and mild traumatic brain injury: state-of-the-science and implications for clinical practice. Int. J. Psychophysiol. 82, 41–52. 10.1016/j.ijpsycho.2011.03.004 [DOI] [PubMed] [Google Scholar]
- Avanzini G., de Curtis M., Panzica F., Spreafico R. (1989). Intrinsic properties of nucleus reticularis thalami neurones of the rat studied in vitro. J. Physiol. 416, 111–122. 10.1113/jphysiol.1989.sp017752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axmacher N., Mormann F., Fernández G., Elger C. E., Fell J. (2006). Memory formation by neuronal synchronization. Brain Res. Rev. 52, 170–182. 10.1016/j.brainresrev.2006.01.007 [DOI] [PubMed] [Google Scholar]
- Baker A. J., Phan N., Moulton R. J., Fehlings M. G., Yucel Y., Zhao M., et al. (2002). Attenuation of the electrophysiological function of the corpus callosum after fluid percussion injury in the rat. J. Neurotrauma 19, 587–599. 10.1089/089771502753754064 [DOI] [PubMed] [Google Scholar]
- Bal T., von Krosigk M., McCormick D. A. (1995a). Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J. Physiol. 483, 665–685. 10.1113/jphysiol.1995.sp020613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T., von Krosigk M., McCormick D. A. (1995b). Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro. J. Physiol. 483, 641–663. 10.1113/jphysiol.1995.sp020612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales J. W., Wagner A. K., Kline A. E., Dixon C. E. (2009). Persistent cognitive dysfunction after traumatic brain injury: a dopamine hypothesis. Neurosci. Biobehav. Rev. 33, 981–1003. 10.1016/j.neubiorev.2009.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball G. J., Gloor P., Schaul N. (1977). The cortical electromicrophysiology of pathological delta waves in the electroencephalogram of cats. Electroencephalogr. Clin. Neurophysiol. 43, 346–361. 10.1016/0013-4694(77)90258-9 [DOI] [PubMed] [Google Scholar]
- Başar E., Başar-Eroglu C., Karakaş S., Schürmann M. (2001). Gamma, alpha, delta and theta oscillations govern cognitive processes. Int. J. Psychophysiol. 39, 241–248. 10.1016/s0167-8760(00)00145-8 [DOI] [PubMed] [Google Scholar]
- Battaglia F. P., Sutherland G. R., McNaughton B. L. (2004). Hippocampal sharp wave bursts coincide with neocortical “up-state” transitions. Learn. Mem. 11, 697–704. 10.1101/lm.73504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluscio M. A., Mizuseki K., Schmidt R., Kempter R., Buzsáki G. (2012). Cross-frequency phase-phase coupling between theta and gamma oscillations in the hippocampus. J. Neurosci. 32, 423–435. 10.1523/JNEUROSCI.4122-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergey G. K., Morrell M. J., Mizrahi E. M., Goldman A., King-Stephens D., Nair D., et al. (2015). Long-term treatment with responsive brain stimulation in adults with refractory partial seizures. Neurology 84, 810–817. 10.1212/WNL.0000000000001280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. J., Rapp P. E. (1979). A comparative survey of the function, mechanism and control of cellular oscillators. J. Exp. Biol. 81, 217–279. [DOI] [PubMed] [Google Scholar]
- Berry S. D., Thompson R. F. (1978). Prediction of learning rate from the hippocampal electroencephalogram. Science 200, 1298–1300. 10.1126/science.663612 [DOI] [PubMed] [Google Scholar]
- Birdno M. J., Grill W. M. (2008). Mechanisms of deep brain stimulation in movement disorders as revealed by changes in stimulus frequency. Neurotherapeutics 5, 14–25. 10.1016/j.nurt.2007.10.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland B. H. (1986). The physiology and pharmacology of hippocampal formation theta rhythms. Prog. Neurobiol. 26, 1–54. 10.1016/0301-0082(86)90019-5 [DOI] [PubMed] [Google Scholar]
- Bonislawski D. P., Schwarzbach E. P., Cohen A. S. (2007). Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol. Dis. 25, 163–169. 10.1016/j.nbd.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootes K., Chapparo C. (2010). Difficulties with multitasking on return to work after TBI: a critical case study. Work 36, 207–216. 10.3233/WOR-2010-1021 [DOI] [PubMed] [Google Scholar]
- Borich M., Babul A. N., Yuan P. H., Boyd L., Virji-Babul N. (2015). Alterations in resting-state brain networks in concussed adolescent athletes. J. Neurotrauma 32, 265–271. 10.1089/neu.2013.3269 [DOI] [PubMed] [Google Scholar]
- Bragin A., Jandó G., Nádasdy Z., Hetke J., Wise K., Buzsáki G. (1995). Gamma (40–100 Hz) oscillation in the hippocampus of the behaving rat. J. Neurosci. 15, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. E., Basheer R., McKenna J. T., Strecker R. E., McCarley R. W. (2012). Control of sleep and wakefulness. Physiol. Rev. 92, 1087–1187. 10.1152/physrev.00032.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunoni A. R., Fregni F., Pagano R. L. (2011). Translational research in transcranial direct current stimulation (tDCS): a systematic review of studies in animals. Rev. Neurosci. 22, 471–481. 10.1515/RNS.2011.042 [DOI] [PubMed] [Google Scholar]
- Butson C. R., McIntyre C. C. (2007). Differences among implanted pulse generator waveforms cause variations in the neural response to deep brain stimulation. Clin. Neurophysiol. 118, 1889–1894. 10.1016/j.clinph.2007.05.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G. (1989). Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience 31, 551–570. 10.1016/0306-4522(89)90423-5 [DOI] [PubMed] [Google Scholar]
- Buzsáki G. (2005). Theta rhythm of navigation: link between path integration and landmark navigation, episodic and semantic memory. Hippocampus 15, 827–840. 10.1002/hipo.20113 [DOI] [PubMed] [Google Scholar]
- Buzsáki G. (2010). Neural syntax: cell assemblies, synapsembles and readers. Neuron 68, 362–385. 10.1016/j.neuron.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Anastassiou C. A., Koch C. (2012). The origin of extracellular fields and currents–EEG, ECoG, LFP and spikes. Nat. Rev. Neurosci. 13, 407–420. 10.1038/nrn3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Czopf J., Kondákor I., Kellényi L. (1986). Laminar distribution of hippocampal rhythmic slow activity (RSA) in the behaving rat: current-source density analysis, effects of urethane and atropine. Brain Res. 365, 125–137. 10.1016/0006-8993(86)90729-8 [DOI] [PubMed] [Google Scholar]
- Buzsáki G., Draguhn A. (2004). Neuronal oscillations in cortical networks. Science 304, 1926–1929. 10.1126/science.1099745 [DOI] [PubMed] [Google Scholar]
- Buzsáki G., Horváth Z., Urioste R., Hetke J., Wise K. (1992). High-frequency network oscillation in the hippocampus. Science 256, 1025–1027. 10.1126/science.1589772 [DOI] [PubMed] [Google Scholar]
- Buzsáki G., Leung L. W., Vanderwolf C. H. (1983). Cellular bases of hippocampal EEG in the behaving rat. Brain Res. 287, 139–171. 10.1016/0165-0173(83)90037-1 [DOI] [PubMed] [Google Scholar]
- Buzsáki G., Moser E. I. (2013). Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat. Neurosci. 16, 130–138. 10.1038/nn.3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Schomburg E. W. (2015). What does gamma coherence tell us about inter-regional neural communication? Nat. Neurosci. 18, 484–489. 10.1038/nn.3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Silva F. L. (2012). High frequency oscillations in the intact brain. Prog Neurobiol 98, 241–249. 10.1016/j.pneurobio.2012.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Watson B. O. (2012). Brain rhythms and neural syntax: implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues Clin. Neurosci. 14, 345–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballosa Gonzalez M. M., Blaya M. O., Alonso O. F., Bramlett H. M., Hentall I. D. (2013). Midbrain raphe stimulation improves behavioral and anatomical recovery from fluid-percussion brain injury. J. Neurotrauma 30, 119–130. 10.1089/neu.2012.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman C. A., Lacaille J. C. (1999). Intrinsic theta-frequency membrane potential oscillations in hippocampal CA1 interneurons of stratum lacunosum-moleculare. J. Neurophysiol. 81, 1296–1307. [DOI] [PubMed] [Google Scholar]
- Choi D. W. (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. 10.1016/0896-6273(88)90162-6 [DOI] [PubMed] [Google Scholar]
- Ciallella J. R., Yan H. Q., Ma X., Wolfson B. M., Marion D. W., DeKosky S. T., et al. (1998). Chronic effects of traumatic brain injury on hippocampal vesicular acetylcholine transporter and M2 muscarinic receptor protein in rats. Exp. Neurol. 152, 11–19. 10.1006/exnr.1998.6831 [DOI] [PubMed] [Google Scholar]
- Cole A. E., Nicoll R. A. (1984). Characterization of a slow cholinergic post-synaptic potential recorded in vitro from rat hippocampal pyramidal cells. J. Physiol. 352, 173–188. 10.1113/jphysiol.1984.sp015285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin L. L. (2013). Mechanisms and functions of theta rhythms. Annu. Rev. Neurosci. 36, 295–312. 10.1146/annurev-neuro-062012-170330 [DOI] [PubMed] [Google Scholar]
- Colgin L. L., Denninger T., Fyhn M., Hafting T., Bonnevie T., Jensen O., et al. (2009). Frequency of gamma oscillations routes flow of information in the hippocampus. Nature 462, 353–357. 10.1038/nature08573 [DOI] [PubMed] [Google Scholar]
- Colom L. V., Castaneda M. T., Reyna T., Hernandez S., Garrido-Sanabria E. (2005). Characterization of medial septal glutamatergic neurons and their projection to the hippocampus. Synapse 58, 151–164. 10.1002/syn.20184 [DOI] [PubMed] [Google Scholar]
- Colom L. V., García-Hernández A., Castañeda M. T., Perez-Cordova M. G., Garrido-Sanabria E. R. (2006). Septo-hippocampal networks in chronically epileptic rats: potential antiepileptic effects of theta rhythm generation. J. Neurophysiol. 95, 3645–3653. 10.1152/jn.00040.2006 [DOI] [PubMed] [Google Scholar]
- Coronado V. G., McGuire L. C., Sarmiento K., Bell J., Lionbarger M. R., Jones C. D., et al. (2012). Trends in traumatic brain injury in the USA and the public health response: 1995–2009. J. Safety Res 43, 299–307. 10.1016/j.jsr.2012.08.011 [DOI] [PubMed] [Google Scholar]
- Cowan R. L., Wilson C. J. (1994). Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J. Neurophysiol. 71, 17–32. [DOI] [PubMed] [Google Scholar]
- Crunelli V., Lightowler S., Pollard C. E. (1989). A T-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J. Physiol. 413, 543–561. 10.1113/jphysiol.1989.sp017668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R., Maris D. O., Grady M. S., Winn H. R., Janigro D. (1998). Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 786, 64–79. 10.1016/s0006-8993(97)01412-1 [DOI] [PubMed] [Google Scholar]
- De Gennaro L., Ferrara M. (2003). Sleep spindles: an overview. Sleep Med. Rev. 7, 423–440. 10.1053/smrv.2002.0252 [DOI] [PubMed] [Google Scholar]
- de Hemptinne C., Swann N. C., Ostrem J. L., Ryapolova-Webb E. S., San Luciano M., Galifianakis N. B., et al. (2015). Therapeutic deep brain stimulation reduces cortical phase-amplitude coupling in Parkinson’s disease. Nat. Neurosci. 18, 779–786. 10.1038/nn.3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky S. T., Ikonomovic M. D., Gandy S. (2010). Traumatic brain injury–football, warfare and long-term effects. N. Engl. J. Med. 363, 1293–1296. 10.1056/nejmp1007051 [DOI] [PubMed] [Google Scholar]
- Delahunty T. M. (1992). Mild traumatic brain injury enhances muscarinic receptor-linked inositol phosphate production in rat hippocampus. Brain Res. 594, 307–310. 10.1016/0006-8993(92)91140-a [DOI] [PubMed] [Google Scholar]
- Delahunty T. M., Jiang J. Y., Gong Q. Z., Black R. T., Lyeth B. G. (1995). Differential consequences of lateral and central fluid percussion brain injury on receptor coupling in rat hippocampus. J. Neurotrauma 12, 1045–1057. 10.1089/neu.1995.12.1045 [DOI] [PubMed] [Google Scholar]
- Deupree D., Coppock W., Willer H. (1982). Pretraining septal driving of hippocampal rhythmic slow activity facilitates acquisition of visual discrimination. J. Comp. Physiol. Psychol. 96, 557–562. 10.1037/h0077908 [DOI] [PubMed] [Google Scholar]
- Diamond D. M., Dunwiddie T. V., Rose G. M. (1988). Characteristics of hippocampal primed burst potentiation in vitro and in the awake rat. J. Neurosci. 8, 4079–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson C. T., Magistretti J., Shalinsky M. H., Fransen E., Hasselmo M. E., Alonso A. (2000). Properties and role of Ih in the pacing of subthreshold oscillations in entorhinal cortex layer II neurons. J. Neurophysiol. 83, 2562–2579. [DOI] [PubMed] [Google Scholar]
- Dinocourt C., Aungst S., Yang K., Thompson S. M. (2011). Homeostatic increase in excitability in area CA1 after Schaffer collateral transection in vivo. Epilepsia 52, 1656–1665. 10.1111/j.1528-1167.2011.03113.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon C. E., Bao J., Long D. A., Hayes R. L. (1996). Reduced evoked release of acetylcholine in the rodent hippocampus following traumatic brain injury. Pharmacol. Biochem. Behav. 53, 679–686. 10.1016/0091-3057(95)02069-1 [DOI] [PubMed] [Google Scholar]
- Dixon C. E., Lighthall J. W., Anderson T. E. (1988). Physiologic, histopathologic and cineradiographic characterization of a new fluid-percussion model of experimental brain injury in the rat. J. Neurotrauma 5, 91–104. 10.1089/neu.1988.5.91 [DOI] [PubMed] [Google Scholar]
- Dixon C. E., Lyeth B. G., Povlishock J. T., Findling R. L., Hamm R. J., Marmarou A., et al. (1987). A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 67, 110–119. 10.3171/jns.1987.67.1.0110 [DOI] [PubMed] [Google Scholar]
- Dixon C. E., Ma X., Marion D. W. (1997). Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury. Brain Res. 749, 127–130. 10.1016/s0006-8993(96)01310-8 [DOI] [PubMed] [Google Scholar]
- Donnemiller E., Brenneis C., Wissel J., Scherfler C., Poewe W., Riccabona G., et al. (2000). Impaired dopaminergic neurotransmission in patients with traumatic brain injury: a SPECT study using 123I-beta-CIT and 123I-IBZM. Eur. J. Nucl. Med. 27, 1410–1414. 10.1007/s002590000308 [DOI] [PubMed] [Google Scholar]
- Drexel M., Puhakka N., Kirchmair E., Hörtnagl H., Pitkänen A., Sperk G. (2015). Expression of GABA receptor subunits in the hippocampus and thalamus after experimental traumatic brain injury. Neuropharmacology 88, 122–133. 10.1016/j.neuropharm.2014.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel A. K., König P., Kreiter A. K., Singer W. (1991). Interhemispheric synchronization of oscillatory neuronal responses in cat visual cortex. Science 252, 1177–1179. 10.1126/science.252.5009.1177 [DOI] [PubMed] [Google Scholar]
- Faden A. I., Demediuk P., Panter S. S., Vink R. (1989). The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800. 10.1126/science.2567056 [DOI] [PubMed] [Google Scholar]
- Fedor M., Berman R. F., Muizelaar J. P., Lyeth B. G. (2010). Hippocampal θ dysfunction after lateral fluid percussion injury. J. Neurotrauma 27, 1605–1615. 10.1089/neu.2010.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell J., Axmacher N. (2011). The role of phase synchronization in memory processes. Nat. Rev. Neurosci. 12, 105–118. 10.1038/nrn2979 [DOI] [PubMed] [Google Scholar]
- Fineman I., Hovda D. A., Smith M., Yoshino A., Becker D. P. (1993). Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res. 624, 94–102. 10.1016/0006-8993(93)90064-t [DOI] [PubMed] [Google Scholar]
- Fisher R. S. (2015). Stimulation of the medial septum should benefit patients with temporal lobe epilepsy. Med. Hypotheses 84, 543–550. 10.1016/j.mehy.2015.02.016 [DOI] [PubMed] [Google Scholar]
- Fisher R., Salanova V., Witt T., Worth R., Henry T., Gross R., et al. (2010). Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 51, 899–908. 10.1111/j.1528-1167.2010.02536.x [DOI] [PubMed] [Google Scholar]
- Floyd C. L., Golden K. M., Black R. T., Hamm R. J., Lyeth B. G. (2002). Craniectomy position affects morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. J. Neurotrauma 19, 303–316. 10.1089/089771502753594873 [DOI] [PubMed] [Google Scholar]
- Folkerts M. M., Berman R. F., Muizelaar J. P., Rafols J. A. (1998). Disruption of MAP-2 immunostaining in rat hippocampus after traumatic brain injury. J. Neurotrauma 15, 349–363. 10.1089/neu.1998.15.349 [DOI] [PubMed] [Google Scholar]
- Fransén E., Alonso A. A., Dickson C. T., Magistretti J., Hasselmo M. E. (2004). Ionic mechanisms in the generation of subthreshold oscillations and action potential clustering in entorhinal layer II stellate neurons. Hippocampus 14, 368–384. 10.1002/hipo.10198 [DOI] [PubMed] [Google Scholar]
- Freund T. F., Antal M. (1988). GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature 336, 170–173. 10.1038/336170a0 [DOI] [PubMed] [Google Scholar]
- Frey L. C. (2003). Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia 44, 11–17. 10.1046/j.1528-1157.44.s10.4.x [DOI] [PubMed] [Google Scholar]
- Frieboes R. M., Müller U., Murck H., von Cramon D. Y., Holsboer F., Steiger A. (1999). Nocturnal hormone secretion and the sleep EEG in patients several months after traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 11, 354–360. 10.1176/jnp.11.3.354 [DOI] [PubMed] [Google Scholar]
- Fries P. (2005). A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn. Sci. 9, 474–480. 10.1016/j.tics.2005.08.011 [DOI] [PubMed] [Google Scholar]
- Fries P., Reynolds J. H., Rorie A. E., Desimone R. (2001). Modulation of oscillatory neuronal synchronization by selective visual attention. Science 291, 1560–1563. 10.1126/science.1055465 [DOI] [PubMed] [Google Scholar]
- Fuhrmann F., Justus D., Sosulina L., Kaneko H., Beutel T., Friedrichs D., et al. (2015). Locomotion, theta oscillations and the speed-correlated firing of hippocampal neurons are controlled by a medial septal glutamatergic circuit. Neuron 86, 1253–1264. 10.1016/j.neuron.2015.05.001 [DOI] [PubMed] [Google Scholar]
- Fujisawa S., Buzsáki G. (2011). A 4 Hz oscillation adaptively synchronizes prefrontal, VTA and hippocampal activities. Neuron 72, 153–165. 10.1016/j.neuron.2011.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaito J., Nobrega J. N., Gaito S. T. (1980). Interference effect of 3 Hz brain stimulation on kindling behavior induced by 60 Hz stimulation. Epilepsia 21, 73–84. 10.1111/j.1528-1157.1980.tb04046.x [DOI] [PubMed] [Google Scholar]
- García-Muñoz A., Barrio L. C., Buño W. (1993). Membrane potential oscillations in CA1 hippocampal pyramidal neurons in vitro: intrinsic rhythms and fluctuations entrained by sinusoidal injected current. Exp. Brain Res. 97, 325–333. 10.1007/bf00228702 [DOI] [PubMed] [Google Scholar]
- Garrido Sanabria E. R., Castañeda M. T., Banuelos C., Perez-Cordova M. G., Hernandez S., Colom L. V. (2006). Septal GABAergic neurons are selectively vulnerable to pilocarpine-induced status epilepticus and chronic spontaneous seizures. Neuroscience 142, 871–883. 10.1016/j.neuroscience.2006.06.057 [DOI] [PubMed] [Google Scholar]
- Gaykema R. P., van der Kuil J., Hersh L. B., Luiten P. G. (1991). Patterns of direct projections from the hippocampus to the medial septum-diagonal band complex: anterograde tracing with Phaseolus vulgaris leucoagglutinin combined with immunohistochemistry of choline acetyltransferase. Neuroscience 43, 349–360. 10.1016/0306-4522(91)90299-4 [DOI] [PubMed] [Google Scholar]
- Goforth P. B., Ren J., Schwartz B. S., Satin L. S. (2011). Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. J. Neurophysiol. 105, 2350–2363. 10.1152/jn.00467.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golarai G., Greenwood A. C., Feeney D. M., Connor J. A. (2001). Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J. Neurosci. 21, 8523–8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutagny R., Jackson J., Williams S. (2009). Self-generated theta oscillations in the hippocampus. Nat. Neurosci. 12, 1491–1493. 10.1038/nn.2440 [DOI] [PubMed] [Google Scholar]
- Graham D. I., Raghupathi R., Saatman K. E., Meaney D., McIntosh T. K. (2000). Tissue tears in the white matter after lateral fluid percussion brain injury in the rat: relevance to human brain injury. Acta Neuropathol. 99, 117–124. 10.1007/pl00007414 [DOI] [PubMed] [Google Scholar]
- Gray C. M., König P., Engel A. K., Singer W. (1989). Oscillatory responses in cat visual cortex exhibit inter-columnar synchronization which reflects global stimulus properties. Nature 338, 334–337. 10.1038/338334a0 [DOI] [PubMed] [Google Scholar]
- Green J. D., Arduini A. A. (1954). Hippocampal electrical activity in arousal. J. Neurophysiol. 17, 533–557. [DOI] [PubMed] [Google Scholar]
- Griesbach G. S., Gomez-Pinilla F., Hovda D. A. (2004). The upregulation of plasticity-related proteins following TBI is disrupted with acute voluntary exercise. Brain Res. 1016, 154–162. 10.1016/j.brainres.2004.04.079 [DOI] [PubMed] [Google Scholar]
- Gupta A., Elgammal F. S., Proddutur A., Shah S., Santhakumar V. (2012). Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J. Neurosci. 32, 2523–2537. 10.1523/JNEUROSCI.4141-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkoff G. G., Gahan J. D., Ghiasvand R. T., Hunsaker M. R., Van K., Feng J. F., et al. (2013). Evaluation of metric, topological and temporal ordering memory tasks after lateral fluid percussion injury. J. Neurotrauma 30, 292–300. 10.1089/neu.2012.2463 [DOI] [PubMed] [Google Scholar]
- Hajszan T., Alreja M., Leranth C. (2004). Intrinsic vesicular glutamate transporter 2-immunoreactive input to septohippocampal parvalbumin-containing neurons: novel glutamatergic local circuit cells. Hippocampus 14, 499–509. 10.1002/hipo.10195 [DOI] [PubMed] [Google Scholar]
- Hanks R. A., Rapport L. J., Millis S. R., Deshpande S. A. (1999). Measures of executive functioning as predictors of functional ability and social integration in a rehabilitation sample. Arch. Phys. Med. Rehabil. 80, 1030–1037. 10.1016/s0003-9993(99)90056-4 [DOI] [PubMed] [Google Scholar]
- Headley D. B., Paré D. (2013). In sync: gamma oscillations and emotional memory. Front. Behav. Neurosci. 7:170. 10.3389/fnbeh.2013.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentall I. D., Burns S. B. (2009). Restorative effects of stimulating medullary raphe after spinal cord injury. J. Rehabil. Res. Dev. 46, 109–122. 10.1682/JRRD.2008.04.0054 [DOI] [PubMed] [Google Scholar]
- Hentall I. D., Gonzalez M. M. (2012). Promotion of recovery from thoracic spinal cord contusion in rats by stimulation of medullary raphe or its midbrain input. Neurorehabil. Neural Repair 26, 374–384. 10.1177/1545968311425178 [DOI] [PubMed] [Google Scholar]
- Hicks R. R., Smith D. H., McIntosh T. K. (1995). Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury in rats. Brain Res. 678, 151–160. 10.1016/0006-8993(95)00179-t [DOI] [PubMed] [Google Scholar]
- Hicks R. R., Smith D. H., Lowenstein D. H., Saint Marie R., McIntosh T. K. (1993). Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J. Neurotrauma 10, 405–414. 10.1089/neu.1993.10.405 [DOI] [PubMed] [Google Scholar]
- Hu H., Vervaeke K., Storm J. F. (2002). Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J. Physiol. 545, 783–805. 10.1113/jphysiol.2002.029249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta P. T., Lisman J. E. (1995). Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron 15, 1053–1063. 10.1016/0896-6273(95)90094-2 [DOI] [PubMed] [Google Scholar]
- Hunt R. F., Scheff S. W., Smith B. N. (2011). Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J. Neurosci. 31, 6880–6890. 10.1523/jneurosci.0032-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko N., Römer C., Ndode-Ekane X. E., Lukasiuk K., Pitkänen A. (2015). Loss of hippocampal interneurons and epileptogenesis: a comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 220, 153–191. 10.1007/s00429-013-0644-1 [DOI] [PubMed] [Google Scholar]
- Hyman J. M., Zilli E. A., Paley A. M., Hasselmo M. E. (2005). Medial prefrontal cortex cells show dynamic modulation with the hippocampal theta rhythm dependent on behavior. Hippocampus 15, 739–749. 10.1002/hipo.20106 [DOI] [PubMed] [Google Scholar]
- Ip E. Y., Giza C. C., Griesbach G. S., Hovda D. A. (2002). Effects of enriched environment and fluid percussion injury on dendritic arborization within the cerebral cortex of the developing rat. J. Neurotrauma 19, 573–585. 10.1089/089771502753754055 [DOI] [PubMed] [Google Scholar]
- Ishige N., Pitts L. H., Hashimoto T., Nishimura M. C., Bartkowski H. M. (1987). Effect of hypoxia on traumatic brain injury in rats: part (1). Changes in neurological function, electroencephalograms and histopathology. Neurosurgery 20, 848–853. 10.1097/00006123-198706000-00005 [DOI] [PubMed] [Google Scholar]
- Isomura Y., Sirota A., Ozen S., Montgomery S., Mizuseki K., Henze D. A., et al. (2006). Integration and segregation of activity in entorhinal-hippocampal subregions by neocortical slow oscillations. Neuron 52, 871–882. 10.1016/j.neuron.2006.10.023 [DOI] [PubMed] [Google Scholar]
- James D. T. D., McNaughton N., Rawlins J. N. P., Feldon J., Gray J. A. (1977). Septal driving of hippocampal theta rhythm as a function of frequency in free-moving male rat. Neuroscience 2, 1007–1017. 10.1016/0306-4522(77)90123-3 [DOI] [Google Scholar]
- Jennett B., Snoek J., Bond M. R., Brooks N. (1981). Disability after severe head injury: observations on the use of the Glasgow Outcome Scale. J. Neurol. Neurosurg. Psychiatry 44, 285–293. 10.1136/jnnp.44.4.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O., Colgin L. L. (2007). Cross-frequency coupling between neuronal oscillations. Trends Cogn. Sci. 11, 267–269. 10.1016/j.tics.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Ji D., Wilson M. A. (2007). Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat. Neurosci. 10, 100–107. 10.1038/nn1825 [DOI] [PubMed] [Google Scholar]
- Jiang J. Y., Lyeth B. G., Delahunty T. M., Phillips L. L., Hamm R. J. (1994). Muscarinic cholinergic receptor binding in rat brain at 15 days following traumatic brain injury. Brain Res. 651, 123–128. 10.1016/0006-8993(94)90687-4 [DOI] [PubMed] [Google Scholar]
- Johnson B., Neuberger T., Gay M., Hallett M., Slobounov S. (2014). Effects of subconcussive head trauma on the default mode network of the brain. J. Neurotrauma 31, 1907–1913. 10.1089/neu.2014.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson V. E., Stewart W., Smith D. H. (2013). Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. 10.1016/j.expneurol.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. W., Wilson M. A. (2005). Phase precession of medial prefrontal cortical activity relative to the hippocampal theta rhythm. Hippocampus 15, 867–873. 10.1002/hipo.20119 [DOI] [PubMed] [Google Scholar]
- Kamondi A., Acsády L., Wang X. J., Buzsáki G. (1998). Theta oscillations in somata and dendrites of hippocampal pyramidal cells in vivo: activity-dependent phase-precession of action potentials. Hippocampus 8, 244–261. [DOI] [PubMed] [Google Scholar]
- Kampfl A., Posmantur R. M., Zhao X., Schmutzhard E., Clifton G. L., Hayes R. L. (1997). Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J. Neurotrauma 14, 121–134. 10.1089/neu.1997.14.121 [DOI] [PubMed] [Google Scholar]
- Kao C. Q., Goforth P. B., Ellis E. F., Satin L. S. (2004). Potentiation of GABAA currents after mechanical injury of cortical neurons. J. Neurotrauma 21, 259–270. 10.1089/089771504322972059 [DOI] [PubMed] [Google Scholar]
- Katayama Y., Becker D. P., Tamura T., Hovda D. A. (1990). Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 73, 889–900. 10.3171/jns.1990.73.6.0889 [DOI] [PubMed] [Google Scholar]
- Kato T., Nakayama N., Yasokawa Y., Okumura A., Shinoda J., Iwama T. (2007). Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J. Neurotrauma 24, 919–926. 10.1089/neu.2006.0203 [DOI] [PubMed] [Google Scholar]
- Kempf J., Werth E., Kaiser P. R., Bassetti C. L., Baumann C. R. (2010). Sleep-wake disturbances 3 years after traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 81, 1402–1405. 10.1136/jnnp.2009.201913 [DOI] [PubMed] [Google Scholar]
- Kenney C., Simpson R., Hunter C., Ondo W., Almaguer M., Davidson A., et al. (2007). Short-term and long-term safety of deep brain stimulation in the treatment of movement disorders. J. Neurosurg. 106, 621–625. 10.3171/jns.2007.106.4.621 [DOI] [PubMed] [Google Scholar]
- Khorchid A., Ikura M. (2002). How calpain is activated by calcium. Nat. Struct. Biol. 9, 239–241. 10.1038/nsb0402-239 [DOI] [PubMed] [Google Scholar]
- Kile K. B., Tian N., Durand D. M. (2010). Low frequency stimulation decreases seizure activity in a mutation model of epilepsy. Epilepsia 51, 1745–1753. 10.1111/j.1528-1167.2010.02679.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk I. J. (1998). Frequency modulation of hippocampal theta by the supramammillary nucleus and other hypothalamo-hippocampal interactions: mechanisms and functional implications. Neurosci. Biobehav. Rev. 22, 291–302. 10.1016/s0149-7634(97)00015-8 [DOI] [PubMed] [Google Scholar]
- Kirkwood A., Dudek S. M., Gold J. T., Aizenman C. D., Bear M. F. (1993). Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science 260, 1518–1521. 10.1126/science.8502997 [DOI] [PubMed] [Google Scholar]
- Kita T., Tanaka T., Tanaka N., Kinoshita Y. (2000). The role of tumor necrosis factor-alpha in diffuse axonal injury following fluid-percussive brain injury in rats. Int. J. Legal. Med. 113, 221–228. 10.1007/s004149900095 [DOI] [PubMed] [Google Scholar]
- Kitchigina V., Popova I., Sinelnikova V., Malkov A., Astasheva E., Shubina L., et al. (2013). Disturbances of septohippocampal theta oscillations in the epileptic brain: reasons and consequences. Exp. Neurol. 247, 314–327. 10.1016/j.expneurol.2013.01.029 [DOI] [PubMed] [Google Scholar]
- Klausberger T., Magill P. J., Márton L. F., Roberts J. D., Cobden P. M., Buzsáki G., et al. (2003). Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421, 844–848. 10.1038/nature01374 [DOI] [PubMed] [Google Scholar]
- Klink R., Alonso A. (1993). Ionic mechanisms for the subthreshold oscillations and differential electroresponsiveness of medial entorhinal cortex layer II neurons. J. Neurophysiol. 70, 144–157. [DOI] [PubMed] [Google Scholar]
- Knyazev G. G. (2012). EEG delta oscillations as a correlate of basic homeostatic and motivational processes. Neurosci. Biobehav. Rev. 36, 677–695. 10.1016/j.neubiorev.2011.10.002 [DOI] [PubMed] [Google Scholar]
- Kocsis B., Bragin A., Buzsáki G. (1999). Interdependence of multiple theta generators in the hippocampus: a partial coherence analysis. J. Neurosci. 19, 6200–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopacki J., MacIver M. B., Bland B. H., Roth S. H. (1987). Carbachol-induced EEG ’theta’ activity in hippocampal brain slices. Brain Res. 405, 196–198. 10.1016/0006-8993(87)91009-2 [DOI] [PubMed] [Google Scholar]
- Koubeissi M. Z., Kahriman E., Syed T. U., Miller J., Durand D. M. (2013). Low-frequency electrical stimulation of a fiber tract in temporal lobe epilepsy. Ann. Neurol. 74, 223–231. 10.1002/ana.23915 [DOI] [PubMed] [Google Scholar]
- Kramis R., Vanderwolf C. H., Bland B. H. (1975). Two types of hippocampal rhythmical slow activity in both the rabbit and the rat: relations to behavior and effects of atropine, diethyl ether, urethane and pentobarbital. Exp. Neurol. 49, 58–85. 10.1016/0014-4886(75)90195-8 [DOI] [PubMed] [Google Scholar]
- Kringelbach M. L., Jenkinson N., Owen S. L., Aziz T. Z. (2007). Translational principles of deep brain stimulation. Nat. Rev. Neurosci. 8, 623–635. 10.1038/nrn2196 [DOI] [PubMed] [Google Scholar]
- Kuncel A. M., Grill W. M. (2004). Selection of stimulus parameters for deep brain stimulation. Clin. Neurophysiol. 115, 2431–2441. 10.1016/j.clinph.2004.05.031 [DOI] [PubMed] [Google Scholar]
- Lakatos P., Karmos G., Mehta A. D., Ulbert I., Schroeder C. E. (2008). Entrainment of neuronal oscillations as a mechanism of attentional selection. Science 320, 110–113. 10.1126/science.1154735 [DOI] [PubMed] [Google Scholar]
- Landfield P. W. (1977). Different effects of posttrial driving or blocking of the theta rhythm on avoidance learning in rats. Physiol. Behav. 18, 439–445. 10.1016/0031-9384(77)90257-8 [DOI] [Google Scholar]
- Landfield P. W., McGaugh J. L., Tusa R. J. (1972). Theta rhythm: a temporal correlate of memory storage processes in the rat. Science 175, 87–89. 10.1126/science.175.4017.87 [DOI] [PubMed] [Google Scholar]
- Langlois J. A., Rutland-Brown W., Wald M. M. (2006). The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378. 10.1097/00001199-200609000-00001 [DOI] [PubMed] [Google Scholar]
- Lee D. J., Gurkoff G. G., Izadi A., Berman R. F., Ekstrom A. D., Muizelaar J. P., et al. (2013). Medial septal nucleus theta frequency deep brain stimulation improves spatial working memory after traumatic brain injury. J. Neurotrauma 30, 131–139. 10.1089/neu.2012.2646 [DOI] [PubMed] [Google Scholar]
- Lee D. J., Gurkoff G. G., Izadi A., Seidl S. E., Echeverri A., Melnik M., et al. (2015). Septohippocampal neuromodulation improves cognition after traumatic brain injury. J. Neurotrauma 32, 1822–1832. 10.1089/neu.2014.3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leininger B. E., Gramling S. E., Farrell A. D., Kreutzer J. S., Peck E. A., III (1990). Neuropsychological deficits in symptomatic minor head injury patients after concussion and mild concussion. J. Neurol. Neurosurg. Psychiatry 53, 293–296. 10.1136/jnnp.53.4.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J. R., Grady M. S., Lee M. E., Paz J. C., Westrum L. E. (1997). Fluid percussion injury causes disruption of the septohippocampal pathway in the rat. Exp. Neurol. 143, 177–187. 10.1006/exnr.1996.6366 [DOI] [PubMed] [Google Scholar]
- Leonard J. R., Maris D. O., Grady M. S. (1994). Fluid percussion injury causes loss of forebrain choline acetyltransferase and nerve growth factor receptor immunoreactive cells in the rat. J. Neurotrauma 11, 379–392. 10.1089/neu.1994.11.379 [DOI] [PubMed] [Google Scholar]
- Leung L. W., Yim C. Y. (1991). Intrinsic membrane potential oscillations in hippocampal neurons in vitro. Brain Res. 553, 261–274. 10.1016/0006-8993(91)90834-i [DOI] [PubMed] [Google Scholar]
- Levin H. S., Goldstein F. C., High W. M., Jr., Williams D. (1988a). Automatic and effortful processing after severe closed head injury. Brain Cogn. 7, 283–297. 10.1016/0278-2626(88)90003-6 [DOI] [PubMed] [Google Scholar]
- Levin H. S., High W. M., Jr., Ewing-Cobbs L., Fletcher J. M., Eisenberg H. M., Miner M. E., et al. (1988b). Memory functioning during the first year after closed head injury in children and adolescents. Neurosurgery 22, 1043–1052. 10.1097/00006123-198806010-00012 [DOI] [PubMed] [Google Scholar]
- Li N., Yang Y., Glover D. P., Zhang J., Saraswati M., Robertson C., et al. (2014). Evidence for impaired plasticity after traumatic brain injury in the developing brain. J. Neurotrauma 31, 395–403. 10.1089/neu.2013.3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J., Buzsáki G. (2008). A neural coding scheme formed by the combined function of gamma and theta oscillations. Schizophr. Bull. 34, 974–980. 10.1093/schbul/sbn060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. E., Jensen O. (2013). The θ-γ neural code. Neuron 77, 1002–1016. 10.1016/j.neuron.2013.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein D. H., Thomas M. J., Smith D. H., McIntosh T. K. (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 12, 4846–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano A. M., Lipsman N. (2013). Probing and regulating dysfunctional circuits using deep brain stimulation. Neuron 77, 406–424. 10.1016/j.neuron.2013.01.020 [DOI] [PubMed] [Google Scholar]
- Lyeth B. G., Jiang J. Y., Delahunty T. M., Phillips L. L., Hamm R. J. (1994). Muscarinic cholinergic receptor binding in rat brain following traumatic brain injury. Brain Res. 640, 240–245. 10.1016/0006-8993(94)91879-1 [DOI] [PubMed] [Google Scholar]
- Lyeth B. G., Jiang J. Y., Robinson S. E., Guo H., Jenkins L. W. (1993). Hypothermia blunts acetylcholine increase in CSF of traumatically brain injured rats. Mol. Chem. Neuropathol. 18, 247–256. 10.1007/bf03160117 [DOI] [PubMed] [Google Scholar]
- Lyeth B. G., Ray M., Hamm R. J., Schnabel J., Saady J. J., Poklis A., et al. (1992). Postinjury scopolamine administration in experimental traumatic brain injury. Brain Res. 569, 281–286. 10.1016/0006-8993(92)90640-u [DOI] [PubMed] [Google Scholar]
- Maccaferri G., McBain C. J. (1996). The hyperpolarization-activated current Ih and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurones. J. Physiol. 497, 119–130. 10.1113/jphysiol.1996.sp021754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäki-Marttunen V., Kuusinen V., Brause M., Peräkylä J., Polvivaara M., dos Santos Ribeiro R., et al. (2015). Enhanced attention capture by emotional stimuli in mild traumatic brain injury. J. Neurotrauma 32, 272–279. 10.1089/neu.2014.3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandile P., Giuditta A., Romano F., Montagnese P., Piscopo S., Cotugno M., et al. (2003). Waking EEG power spectra in the rat: correlations with training performance. Brain Res. Cogn. Brain Res. 17, 94–105. 10.1016/s0926-6410(03)00084-3 [DOI] [PubMed] [Google Scholar]
- Manseau F., Goutagny R., Danik M., Williams S. (2008). The hippocamposeptal pathway generates rhythmic firing of GABAergic neurons in the medial septum and diagonal bands: an investigation using a complete septohippocampal preparation in vitro. J. Neurosci. 28, 4096–4107. 10.1523/JNEUROSCI.0247-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J., McArthur D. A., Miller C., Glenn T. C., Villablanca P., Martin N. A., et al. (2008). Persistent metabolic crisis as measured by elevated cerebral microdialysis lactate-pyruvate ratio predicts chronic frontal lobe brain atrophy after traumatic brain injury. Crit. Care Med. 36, 2871–2877. 10.1097/CCM.0b013e318186a4a0 [DOI] [PubMed] [Google Scholar]
- Marshall L., Henze D. A., Hirase H., Leinekugel X., Dragoi G., Buzs’aki G. (2002). Hippocampal pyramidal cell-interneuron spike transmission is frequency dependent and responsible for place modulation of interneuron discharge. J. Neurosci. 22:RC197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massucci J. L., Kline A. E., Ma X., Zafonte R. D., Dixon C. E. (2004). Time dependent alterations in dopamine tissue levels and metabolism after experimental traumatic brain injury in rats. Neurosci. Lett. 372, 127–131. 10.1016/j.neulet.2004.09.026 [DOI] [PubMed] [Google Scholar]
- Mathias J. L., Alvaro P. K. (2012). Prevalence of sleep disturbances, disorders and problems following traumatic brain injury: a meta-analysis. Sleep Med. 13, 898–905. 10.1016/j.sleep.2012.04.006 [DOI] [PubMed] [Google Scholar]
- Mattis J., Brill J., Evans S., Lerner T. N., Davidson T. J., Hyun M., et al. (2014). Frequency-dependent, cell type-divergent signaling in the hippocamposeptal projection. J. Neurosci. 34, 11769–11780. 10.1523/JNEUROSCI.5188-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell W. L., Povlishock J. T., Graham D. L. (1997). A mechanistic analysis of nondisruptive axonal injury: a review. J. Neurotrauma 14, 419–440. 10.1089/neu.1997.14.419 [DOI] [PubMed] [Google Scholar]
- McCauley S. R., Wilde E. A., Barnes A., Hanten G., Hunter J. V., Levin H. S., et al. (2014). Patterns of early emotional and neuropsychological sequelae after mild traumatic brain injury. J. Neurotrauma 31, 914–925. 10.1089/neu.2012.2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick D. A., Bal T. (1997). Sleep and arousal: thalamocortical mechanisms. Annu. Rev. Neurosci. 20, 185–215. 10.1146/annurev.neuro.20.1.185 [DOI] [PubMed] [Google Scholar]
- McDowell S., Whyte J., D’Esposito M. (1997). Working memory impairments in traumatic brain injury: evidence from a dual-task paradigm. Neuropsychologia 35, 1341–1353. 10.1016/s0028-3932(97)00082-1 [DOI] [PubMed] [Google Scholar]
- McIntosh T. K., Noble L., Andrews B., Faden A. I. (1987). Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma 4, 119–134. [DOI] [PubMed] [Google Scholar]
- McIntosh T. K., Vink R., Noble L., Yamakami I., Fernyak S., Soares H., et al. (1989). Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience 28, 233–244. 10.1016/0306-4522(89)90247-9 [DOI] [PubMed] [Google Scholar]
- McNaughton N., Ruan M., Woodnorth M. A. (2006). Restoring theta-like rhythmicity in rats restores initial learning in the Morris water maze. Hippocampus 16, 1102–1110. 10.1002/hipo.20235 [DOI] [PubMed] [Google Scholar]
- Miller L. P., Lyeth B. G., Jenkins L. W., Oleniak L., Panchision D., Hamm R. J., et al. (1990). Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Res. 526, 103–107. 10.1016/0006-8993(90)90254-9 [DOI] [PubMed] [Google Scholar]
- Millis S. R., Rosenthal M., Novack T. A., Sherer M., Nick T. G., Kreutzer J. S., et al. (2001). Long-term neuropsychological outcome after traumatic brain injury. J. Head Trauma Rehabil. 16, 343–355. 10.1097/00001199-200108000-00005 [DOI] [PubMed] [Google Scholar]
- Mitchell D. J., McNaughton N., Flanagan D., Kirk I. J. (2008). Frontal-midline theta from the perspective of hippocampal “θ”. Prog. Neurobiol. 86, 156–185. 10.1016/j.pneurobio.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Miyazaki S., Katayama Y., Lyeth B. G., Jenkins L. W., Dewitt D. S., Goldberg S. J., et al. (1992). Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 585, 335–339. 10.1016/0006-8993(92)91232-4 [DOI] [PubMed] [Google Scholar]
- Mölle M., Yeshenko O., Marshall L., Sara S. J., Born J. (2006). Hippocampal sharp wave-ripples linked to slow oscillations in rat slow-wave sleep. J. Neurophysiol. 96, 62–70. 10.1152/jn.00014.2006 [DOI] [PubMed] [Google Scholar]
- Montgomery S. M., Betancur M. I., Buzsáki G. (2009). Behavior-dependent coordination of multiple theta dipoles in the hippocampus. J. Neurosci. 29, 1381–1394. 10.1523/JNEUROSCI.4339-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S. M., Buzsáki G. (2007). Gamma oscillations dynamically couple hippocampal CA3 and CA1 regions during memory task performance. Proc. Natl. Acad. Sci. U S A 104, 14495–14500. 10.1073/pnas.0701826104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S. M., Sirota A., Buzsáki G. (2008). θ and γ coordination of hippocampal networks during waking and rapid eye movement sleep. J. Neurosci. 28, 6731–6741. 10.1523/JNEUROSCI.1227-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormann F., Osterhage H., Andrzejak R. G., Weber B., Fernández G., Fell J., et al. (2008). Independent delta/theta rhythms in the human hippocampus and entorhinal cortex. Front. Hum. Neurosci. 2:3. 10.3389/neuro.09.003.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtchedlishvili Z., Lepsveridze E., Xu H., Kharlamov E. A., Lu B., Kelly K. M. (2010). Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiol. Dis. 38, 464–475. 10.1016/j.nbd.2010.03.012 [DOI] [PubMed] [Google Scholar]
- Nilsson P., Hillered L., Olsson Y., Sheardown M. J., Hansen A. J. (1993). Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J. Cereb. Blood Flow Metab. 13, 183–192. 10.1038/jcbfm.1993.22 [DOI] [PubMed] [Google Scholar]
- Nilsson P., Hillered L., Pontén U., Ungerstedt U. (1990). Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J. Cereb. Blood Flow Metab. 10, 631–637. 10.1038/jcbfm.1990.115 [DOI] [PubMed] [Google Scholar]
- O’Dell D. M., Gibson C. J., Wilson M. S., DeFord S. M., Hamm R. J. (2000). Positive and negative modulation of the GABAA receptor and outcome after traumatic brain injury in rats. Brain Res. 861, 325–332. 10.1016/s0006-8993(00)02055-2 [DOI] [PubMed] [Google Scholar]
- O’Keefe J., Recce M. L. (1993). Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus 3, 317–330. 10.1002/hipo.450030307 [DOI] [PubMed] [Google Scholar]
- Parcell D. L., Ponsford J. L., Rajaratnam S. M., Redman J. R. (2006). Self-reported changes to nighttime sleep after traumatic brain injury. Arch. Phys. Med. Rehabil. 87, 278–285. 10.1016/j.apmr.2005.10.024 [DOI] [PubMed] [Google Scholar]
- Parsons L. C., Crosby L. J., Perlis M., Britt T., Jones P. (1997). Longitudinal sleep EEG power spectral analysis studies in adolescents with minor head injury. J. Neurotrauma 14, 549–559. 10.1089/neu.1997.14.549 [DOI] [PubMed] [Google Scholar]
- Patel T., Gores G. J., Kaufmann S. H. (1996). The role of proteases during apoptosis. FASEB J. 10, 587–597. [DOI] [PubMed] [Google Scholar]
- Paterno R., Metheny H., Xiong G., Elkind J., Cohen A. S. (2016). Mild traumatic brain injury decreases broadband power in area CA1. J. Neurotrauma [Epub ahead of print]. 10.1089/neu.2015.4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penttonen M., Buzsáki G. (2003). Natural logarithmic relationship between brain oscillators. Thalamus Relat. Syst. 2, 145–152. 10.1017/s1472928803000074 [DOI] [Google Scholar]
- Petsche H., Stumpf C., Gogolak G. (1962). [The significance of the rabbit’s septum as a relay station between the midbrain and the hippocampus I. The control of hippocampus arousal activity by the septum cells]. Electroencephalogr. Clin. Neurophysiol. 14, 202–211. 10.1016/0013-4694(62)90030-5 [DOI] [PubMed] [Google Scholar]
- Pike F. G., Goddard R. S., Suckling J. M., Ganter P., Kasthuri N., Paulsen O. (2000). Distinct frequency preferences of different types of rat hippocampal neurones in response to oscillatory input currents. J. Physiol. 529, 205–213. 10.1111/j.1469-7793.2000.00205.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A., McIntosh T. K. (2006). Animal models of post-traumatic epilepsy. J. Neurotrauma 23, 241–261. 10.1089/neu.2006.23.241 [DOI] [PubMed] [Google Scholar]
- Ponsford J. L., Downing M. G., Olver J., Ponsford M., Acher R., Carty M., et al. (2014). Longitudinal follow-up of patients with traumatic brain injury: outcome at two, five and ten years post-injury. J. Neurotrauma 31, 64–77. 10.1089/neu.2013.2997 [DOI] [PubMed] [Google Scholar]
- Povlishock J. T., Katz D. I. (2005). Update of neuropathology and neurological recovery after traumatic brain injury. J. Head Trauma Rehabil. 20, 76–94. 10.1097/00001199-200501000-00008 [DOI] [PubMed] [Google Scholar]
- Quilichini P., Sirota A., Buzsáki G. (2010). Intrinsic circuit organization and theta-gamma oscillation dynamics in the entorhinal cortex of the rat. J. Neurosci. 30, 11128–11142. 10.1523/JNEUROSCI.1327-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp P. E., Keyser D. O., Albano A., Hernandez R., Gibson D. B., Zambon R. A., et al. (2015). Traumatic brain injury detection using electrophysiological methods. Front. Hum. Neurosci. 9:11. 10.3389/fnhum.2015.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves T. M., Lyeth B. G., Phillips L. L., Hamm R. J., Povlishock J. T. (1997). The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 757, 119–132. 10.1016/s0006-8993(97)00170-4 [DOI] [PubMed] [Google Scholar]
- Reeves T. M., Lyeth B. G., Povlishock J. T. (1995). Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp. Brain Res. 106, 248–256. 10.1007/bf00241120 [DOI] [PubMed] [Google Scholar]
- Rimel R. W., Giordani B., Barth J. T., Boll T. J., Jane J. A. (1981). Disability caused by minor head injury. Neurosurgery 9, 221–228. 10.1097/00006123-198109000-00001 [DOI] [PubMed] [Google Scholar]
- Robinson S. E., Foxx S. D., Posner M. G., Martin R. M., Davis T. R., Guo H. Z., et al. (1990). The effect of M1 muscarinic blockade on behavior and physiological responses following traumatic brain injury in the rat. Brain Res. 511, 141–148. 10.1016/0006-8993(90)90233-2 [DOI] [PubMed] [Google Scholar]
- Roozenbeek B., Maas A. I., Menon D. K. (2013). Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 9, 231–236. 10.1038/nrneurol.2013.22 [DOI] [PubMed] [Google Scholar]
- Rose G. M., Dunwiddie T. V. (1986). Induction of hippocampal long-term potentiation using physiologically patterned stimulation. Neurosci. Lett. 69, 244–248. 10.1016/0304-3940(86)90487-8 [DOI] [PubMed] [Google Scholar]
- Rosin B., Slovik M., Mitelman R., Rivlin-Etzion M., Haber S. N., Israel Z., et al. (2011). Closed-loop deep brain stimulation is superior in ameliorating parkinsonism. Neuron 72, 370–384. 10.1016/j.neuron.2011.08.023 [DOI] [PubMed] [Google Scholar]
- Ross S. T., Soltesz I. (2000). Selective depolarization of interneurons in the early posttraumatic dentate gyrus: involvement of the Na+/K+-ATPase. J. Neurophysiol. 83, 2916–2930. [DOI] [PubMed] [Google Scholar]
- Saatman K. E., Graham D. I., McIntosh T. K. (1998). The neuronal cytoskeleton is at risk after mild and moderate brain injury. J. Neurotrauma 15, 1047–1058. 10.1089/neu.1998.15.1047 [DOI] [PubMed] [Google Scholar]
- Saija A., Hayes R. L., Lyeth B. G., Dixon C. E., Yamamoto T., Robinson S. E. (1988). The effect of concussive head injury on central cholinergic neurons. Brain Res. 452, 303–311. 10.1016/0006-8993(88)90034-0 [DOI] [PubMed] [Google Scholar]
- Salanova V., Witt T., Worth R., Henry T. R., Gross R. E., Nazzaro J. M., et al. (2015). Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology 84, 1017–1025. 10.1212/WNL.0000000000001334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders M. J., Sick T. J., Perez-Pinzon M. A., Dietrich W. D., Green E. J. (2000). Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 861, 69–76. 10.1016/s0006-8993(00)01986-7 [DOI] [PubMed] [Google Scholar]
- Santhakumar V., Ratzliff A. D., Jeng J., Toth Z., Soltesz I. (2001). Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann. Neurol. 50, 708–717. 10.1002/ana.1230 [DOI] [PubMed] [Google Scholar]
- Schmidt R. H., Grady M. S. (1995). Loss of forebrain cholinergic neurons following fluid-percussion injury: implications for cognitive impairment in closed head injury. J. Neurosurg. 83, 496–502. 10.3171/jns.1995.83.3.0496 [DOI] [PubMed] [Google Scholar]
- Schomburg E. W., Fernández-Ruiz A., Mizuseki K., Berényi A., Anastassiou C. A., Koch C., et al. (2014). Theta phase segregation of input-specific gamma patterns in entorhinal-hippocampal networks. Neuron 84, 470–485. 10.1016/j.neuron.2014.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbach E., Bonislawski D. P., Xiong G., Cohen A. S. (2006). Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus 16, 541–550. 10.1002/hipo.20183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seager M. A., Johnson L. D., Chabot E. S., Asaka Y., Berry S. D. (2002). Oscillatory brain states and learning: impact of hippocampal theta-contingent training. Proc. Natl. Acad. Sci. U S A 99, 1616–1620. 10.1073/pnas.032662099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior T. J., Huxter J. R., Allen K., O’Neill J., Csicsvari J. (2008). Gamma oscillatory firing reveals distinct populations of pyramidal cells in the CA1 region of the hippocampus. J. Neurosci. 28, 2274–2286. 10.1523/JNEUROSCI.4669-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay N., Yeates K. O., Walz N. C., Stancin T., Taylor H. G., Beebe D. W., et al. (2014). Sleep problems and their relationship to cognitive and behavioral outcomes in young children with traumatic brain injury. J. Neurotrauma 31, 1305–1312. 10.1089/neu.2013.3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S. S., Bray E. R., Zhang C. Q., Dixon C. E. (2011). Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. 1369, 208–215. 10.1016/j.brainres.2010.10.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S. S., Dixon C. E. (2015). Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J. Neurotrauma 32, 1429–1440. 10.1089/neu.2014.3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S. S., Dixon C. E., Okonkwo D. O., Richardson R. M. (2014). Neurostimulation for traumatic brain injury. J. Neurosurg. 121, 1219–1231. 10.3171/2014.7.JNS131826 [DOI] [PubMed] [Google Scholar]
- Siapas A. G., Lubenov E. V., Wilson M. A. (2005). Prefrontal phase locking to hippocampal theta oscillations. Neuron 46, 141–151. 10.1016/j.neuron.2005.02.028 [DOI] [PubMed] [Google Scholar]
- Siapas A. G., Wilson M. A. (1998). Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron 21, 1123–1128. 10.1016/s0896-6273(00)80629-7 [DOI] [PubMed] [Google Scholar]
- Sick T. J., Pérez-Pinzón M. A., Feng Z. Z. (1998). Impaired expression of long-term potentiation in hippocampal slices 4 and 48 h following mild fluid-percussion brain injury in vivo. Brain Res. 785, 287–292. 10.1016/s0006-8993(97)01418-2 [DOI] [PubMed] [Google Scholar]
- Sirota A., Csicsvari J., Buhl D., Buzsáki G. (2003). Communication between neocortex and hippocampus during sleep in rodents. Proc. Natl. Acad. Sci. U S A 100, 2065–2069. 10.1073/pnas.0437938100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaggs W. E., McNaughton B. L., Wilson M. A., Barnes C. A. (1996). Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus 6, 149–172. [DOI] [PubMed] [Google Scholar]
- Skopin M. D., Kabadi S. V., Viechweg S. S., Mong J. A., Faden A. I. (2015). Chronic decrease in wakefulness and disruption of sleep-wake behavior after experimental traumatic brain injury. J. Neurotrauma 32, 289–296. 10.1089/neu.2014.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobounov S., Sebastianelli W., Hallett M. (2012). Residual brain dysfunction observed one year post-mild traumatic brain injury: combined EEG and balance study. Clin. Neurophysiol. 123, 1755–1761. 10.1016/j.clinph.2011.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. H., Chen X. H., Pierce J. E., Wolf J. A., Trojanowski J. Q., Graham D. I., et al. (1997). Progressive atrophy and neuron death for one year following brain trauma in the rat. J. Neurotrauma 14, 715–727. 10.1089/neu.1997.14.715 [DOI] [PubMed] [Google Scholar]
- Smith D. H., Okiyama K., Thomas M. J., McIntosh T. K. (1993). Effects of the excitatory amino acid receptor antagonists kynurenate and indole-2-carboxylic acid on behavioral and neurochemical outcome following experimental brain injury. J. Neurosci. 13, 5383–5392. 10.1016/0006-8993(93)90885-q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe J. W., Colom L. V., Bland B. H. (1992). The extrinsic modulation of hippocampal theta depends on the coactivation of cholinergic and GABA-ergic medial septal inputs. Neurosci. Biobehav. Rev. 16, 289–308. 10.1016/s0149-7634(05)80203-9 [DOI] [PubMed] [Google Scholar]
- Soares H. D., Thomas M., Cloherty K., McIntosh T. K. (1992). Development of prolonged focal cerebral edema and regional cation changes following experimental brain injury in the rat. J. Neurochem. 58, 1845–1852. 10.1111/j.1471-4159.1992.tb10061.x [DOI] [PubMed] [Google Scholar]
- Spain A., Daumas S., Lifshitz J., Rhodes J., Andrews P. J., Horsburgh K., et al. (2010). Mild fluid percussion injury in mice produces evolving selective axonal pathology and cognitive deficits relevant to human brain injury. J. Neurotrauma 27, 1429–1438. 10.1089/neu.2010.1288 [DOI] [PubMed] [Google Scholar]
- Staubli U., Lynch G. (1987). Stable hippocampal long-term potentiation elicited by ‘theta’ pattern stimulation. Brain Res. 435, 227–234. 10.1016/0006-8993(87)91605-2 [DOI] [PubMed] [Google Scholar]
- Steriade M., Deschenes M. (1984). The thalamus as a neuronal oscillator. Brain Res. 320, 1–63. 10.1016/0165-0173(84)90017-1 [DOI] [PubMed] [Google Scholar]
- Steriade M., McCormick D. A., Sejnowski T. J. (1993a). Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685. 10.1126/science.8235588 [DOI] [PubMed] [Google Scholar]
- Steriade M., Nuñez A., Amzica F. (1993b). Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J. Neurosci. 13, 3266–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan H. G., Martinez J., Becker D. P., Miller J. D., Griffith R., Wist A. O. (1976). Fluid-percussion model of mechanical brain injury in the cat. J. Neurosurg. 45, 521–534. 10.3171/jns.1976.45.5.0520 [DOI] [PubMed] [Google Scholar]
- Sun Y., Nguyen A. Q., Nguyen J. P., Le L., Saur D., Choi J., et al. (2014). Cell-type-specific circuit connectivity of hippocampal CA1 revealed through Cre-dependent rabies tracing. Cell Rep. 7, 269–280. 10.1016/j.celrep.2014.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthana N., Fried I. (2014). Deep brain stimulation for enhancement of learning and memory. Neuroimage 85, 996–1002. 10.1016/j.neuroimage.2013.07.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet J. A., Eakin K. C., Munyon C. N., Miller J. P. (2014). Improved learning and memory with theta-burst stimulation of the fornix in rat model of traumatic brain injury. Hippocampus 24, 1592–1600. 10.1002/hipo.22338 [DOI] [PubMed] [Google Scholar]
- Tekriwal A., Baltuch G. (2015). Deep brain stimulation: expanding applications. Neurol. Med. Chir. 55, 861–877. 10.2176/nmc.ra.2015-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher R. W., Cantor D. S., McAlaster R., Geisler F., Krause P. (1991). Comprehensive predictions of outcome in closed head-injured patients. The development of prognostic equations. Ann. N Y Acad. Sci. 620, 82–101. 10.1111/j.1749-6632.1991.tb51576.x [DOI] [PubMed] [Google Scholar]
- Thatcher R. W., North D. M., Curtin R. T., Walker R. A., Biver C. J., Gomez J. F., et al. (2001). An EEG severity index of traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 13, 77–87. 10.1176/appi.neuropsych.13.1.77 [DOI] [PubMed] [Google Scholar]
- Thatcher R. W., Walker R. A., Gerson I., Geisler F. H. (1989). EEG discriminant analyses of mild head trauma. Electroencephalogr. Clin. Neurophysiol. 73, 94–106. 10.1016/0013-4694(89)90188-0 [DOI] [PubMed] [Google Scholar]
- Timofeev I., Grenier F., Steriade M. (2001). Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc. Natl. Acad. Sci. U S A 98, 1924–1929. 10.1073/pnas.98.4.1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O., Feintuch A., Benifla M., Cohen A., Friedman A., Shelef I. (2011). Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc. Psychiatry Neurol. 2011:765923. 10.1155/2011/765923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth K., Borhegyi Z., Freund T. F. (1993). Postsynaptic targets of GABAergic hippocampal neurons in the medial septum-diagonal band of broca complex. J. Neurosci. 13, 3712–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth K., Freund T. F. (1992). Calbindin D28k-containing nonpyramidal cells in the rat hippocampus: their immunoreactivity for GABA and projection to the medial septum. Neuroscience 49, 793–805. 10.1016/0306-4522(92)90357-8 [DOI] [PubMed] [Google Scholar]
- Tóth K., Freund T. F., Miles R. (1997). Disinhibition of rat hippocampal pyramidal cells by GABAergic afferents from the septum. J. Physiol. 500, 463–474. 10.1113/jphysiol.1997.sp022033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth Z., Hollrigel G. S., Gorcs T., Soltesz I. (1997b). Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J. Neurosci. 17, 8106–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukker J. J., Fuentealba P., Hartwich K., Somogyi P., Klausberger T. (2007). Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J. Neurosci. 27, 8184–8189. 10.1523/jneurosci.1685-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandecasteele M., Varga V., Berényi A., Papp E., Barthó P., Venance L., et al. (2014). Optogenetic activation of septal cholinergic neurons suppresses sharp wave ripples and enhances theta oscillations in the hippocampus. Proc. Natl. Acad. Sci. U S A 111, 13535–13540. 10.1073/pnas.1411233111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol A. N., Obrietan K., Chen G. (1996). Excitatory actions of GABA after neuronal trauma. J. Neurosci. 16, 4283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderwolf C. H. (1969). Hippocampal electrical activity and voluntary movement in the rat. Electroencephalogr. Clin. Neurophysiol. 26, 407–418. 10.1016/0013-4694(69)90092-3 [DOI] [PubMed] [Google Scholar]
- Vertes R. P., Hoover W. B., Viana Di Prisco G. (2004). Theta rhythm of the hippocampus: subcortical control and functional significance. Behav. Cogn. Neurosci. Rev. 3, 173–200. 10.1177/1534582304273594 [DOI] [PubMed] [Google Scholar]
- Vink R., McIntosh T. K., Demediuk P., Weiner M. W., Faden A. I. (1988). Decline in intracellular free Mg2+ is associated with irreversible tissue injury after brain trauma. J. Biol. Chem. 263, 757–761. [PubMed] [Google Scholar]
- Vinogradova O. S., Brazhnik E. S., Karanov A. M., Zhadina S. D. (1980). Neuronal activity of the septum following various types of deafferentation. Brain Res. 187, 353–368. 10.1016/0006-8993(80)90208-5 [DOI] [PubMed] [Google Scholar]
- Virji-Babul N., Hilderman C. G., Makan N., Liu A., Smith-Forrester J., Franks C., et al. (2014). Changes in functional brain networks following sports-related concussion in adolescents. J. Neurotrauma 31, 1914–1919. 10.1089/neu.2014.3450 [DOI] [PubMed] [Google Scholar]
- Wallace B. E., Wagner A. K., Wagner E. P., McDeavitt J. T. (2001). A history and review of quantitative electroencephalography in traumatic brain injury. J. Head Trauma Rehabil. 16, 165–190. 10.1097/00001199-200104000-00006 [DOI] [PubMed] [Google Scholar]
- Wang X. J. (2002). Pacemaker neurons for the theta rhythm and their synchronization in the septohippocampal reciprocal loop. J. Neurophysiol. 87, 889–900. [DOI] [PubMed] [Google Scholar]
- Wang X. J. (2010). Neurophysiological and computational principles of cortical rhythms in cognition. Physiol. Rev. 90, 1195–1268. 10.1152/physrev.00035.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous A. J., Lee D. J., Izadi A., Gurkoff G. G., Shahlaie K., Ekstrom A. D. (2013). A comparative study of human and rat hippocampal low-frequency oscillations during spatial navigation. Hippocampus 23, 656–661. 10.1002/hipo.22124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel W., Ott T., H A. M. (1977). Post-training hippocampal rhythmic slow activity (“theta”) elicited by septal stimulation improves memory consolidation in rats. Behav. Biol. 21, 32–40. 10.1016/s0091-6773(77)92231-3 [DOI] [PubMed] [Google Scholar]
- Whiteneck G., Brooks C. A., Mellick D., Harrison-Felix C., Terrill M. S., Noble K. (2004). Population-based estimates of outcomes after hospitalization for traumatic brain injury in Colorado. Arch. Phys. Med. Rehabil. 85, S73–S81. 10.1016/j.apmr.2003.08.107 [DOI] [PubMed] [Google Scholar]
- Wierzynski C. M., Lubenov E. V., Gu M., Siapas A. G. (2009). State-dependent spike-timing relationships between hippocampal and prefrontal circuits during sleep. Neuron 61, 587–596. 10.1016/j.neuron.2009.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. A., McNaughton B. L. (1994). Reactivation of hippocampal ensemble memories during sleep. Science 265, 676–679. 10.1126/science.8036517 [DOI] [PubMed] [Google Scholar]
- Witgen B. M., Lifshitz J., Smith M. L., Schwarzbach E., Liang S. L., Grady M. S., et al. (2005). Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience 133, 1–15. 10.1016/j.neuroscience.2005.01.052 [DOI] [PubMed] [Google Scholar]
- Yakovlev A. G., Knoblach S. M., Fan L., Fox G. B., Goodnight R., Faden A. I. (1997). Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 17, 7415–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T., Ozawa Y., Suzuki M., Yamamoto M., Nakamura T., Yamaura A. (1996). Indeloxazine hydrochloride improves impairment of passive avoidance performance after fluid percussion brain injury in rats. Neuropharmacology 35, 329–336. 10.1016/0028-3908(95)00171-9 [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Ooyama M., Ozawa Y., Okada M., Tada S., Yamaguchi T., et al. (1993). Effects of indeloxazine hydrochloride, a cerebral activator, on passive avoidance learning impaired by disruption of cholinergic transmission in rats. Neuropharmacology 32, 695–701. 10.1016/0028-3908(93)90083-f [DOI] [PubMed] [Google Scholar]
- Ylinen A., Soltész I., Bragin A., Penttonen M., Sik A., Buzsáki G. (1995). Intracellular correlates of hippocampal theta rhythm in identified pyramidal cells, granule cells and basket cells. Hippocampus 5, 78–90. 10.1002/hipo.450050110 [DOI] [PubMed] [Google Scholar]
- Zhadina S. D., Vinogradova O. S. (1983). Effects of acetylcholine, norepinephrine and serotonin on neurons of the septum in vitro. Neurosci. Behav. Physiol. 13, 405–411. 10.1007/bf01182682 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Saraswati M., Koehler R. C., Robertson C., Kannan S. (2015). A new rabbit model of pediatric traumatic brain injury. J. Neurotrauma 32, 1369–1379. 10.1089/neu.2014.3701 [DOI] [PMC free article] [PubMed] [Google Scholar]