

To clarify the significant genomic events of the tribe Parideae, we analysed the complete chloroplast genome sequences of two Trillium species representing two subgenera: Trillium and Phyllantherum. The results showed that the cpDNAs of Parideae are highly conserved across genome structure, gene order and contents. However, the chloroplast genome of T. maculatum contained a 3.4kb inverted sequence between ndhC - rbcL in the LSC region. In addition, we found three different types of trnI-CAU duplication. These genomic features provide informative molecular markers for identifying the infrageneric taxa of Trillium and improve our understanding of the evolution patterns of Parideae.
Keywords: Chloroplast genome; comparative genomics; gene duplication; single inversion; Trillium maculatum; Trillium tschonoskii, trnI-CAU
Abstract
Genomic events including gene loss, duplication, pseudogenization and rearrangement in plant genomes are valuable sources for exploring and understanding the process of evolution in angiosperms. The family Melanthiaceae is distributed in temperate regions of the Northern Hemisphere and divided into five tribes (Heloniadeae, Chionographideae, Xerophylleae, Melanthieae and Parideae) based on the molecular phylogenetic analyses. At present, complete chloroplast genomes of the Melanthiaceae have been reported from three species. In the previous genomic study of Liliales, a trnI-CAU gene duplication event was reported from Paris verticillata, a member of Parideae. To clarify the significant genomic events of the tribe Parideae, we analysed the complete chloroplast genome sequences of two Trillium species representing two subgenera: Trillium and Phyllantherum. In Trillium tschonoskii (subgenus Trillium), the circular double-stranded cpDNA sequence of 156 852 bp consists of two inverted repeat (IR) regions of 26 501 bp each, a large single-copy (LSC) region of 83 981 bp and a small single-copy (SSC) region of 19 869 bp. The chloroplast genome sequence of T. maculatum (subgenus Phyllantherum) is 157 359 bp in length, consisting of two IRs (25 535 bp), one SSC (19 949 bp) and one LSC (86 340 bp), and is longer than that of T. tschonoskii. The results showed that the cpDNAs of Parideae are highly conserved across genome structure, gene order and contents. However, the chloroplast genome of T. maculatum contained a 3.4-kb inverted sequence between ndhC and rbcL in the LSC region, and it was a unique feature for subgenera Phyllantherum. In addition, we found three different types of gene duplication in the intergenic spacer between rpl23 and ycf2 containing trnI-CAU, which were in agreement with the circumscription of subgenera and sections in Parideae excluding T. govanianum. These genomic features provide informative molecular markers for identifying the infrageneric taxa of Trillium and improve our understanding of the evolution patterns of Parideae in Melanthiaceae.
Introduction
The chloroplast that characterizes all green plants (Viridiplantae) originated from an endosymbiotic event between independent living cyanobacteria and a non-photosynthetic host (Dyall et al. 2004). Chloroplast genomes of flowering plants are typically circular double-stranded DNA molecules, and usually contain two inverted repeat (IR) regions (IRA and IRB) separated by a large single-copy and a small single-copy (LSC and SSC, respectively) regions (Ravi et al. 2008). The plastid genome is mostly stable in structure, gene content and gene order across land plant lineages (Jansen et al. 2005). Due to this stability, it demonstrated great utility for developing phylogenetic hypotheses across the plant tree of life (Jansen et al. 2007; Zhang et al. 2011; Li et al. 2013). Within seed plants, plastid genomes usually contain 101–118 unique genes with the majority of those 66–82 coding for proteins involved in photosynthesis and gene expression, 29–32 of these genes code for transfer RNAs and 4 code for the ribosomal RNA genes (Jansen and Ruhlman 2012). The advance of next-generation sequencing has facilitated rapid growth of complete chloroplast genomes due to time-saving and low-cost advantages (Shendure and Ji 2008). To date, ∼500 complete chloroplast DNA genome sequence data have been released in GenBank's Organelle Genome Resources (http://www.ncbi.nlm.nih.gov/genome).
Melanthiaceae, a member of Liliales, comprises 17 genera and ∼178 species of perennial herbs that are mostly distributed in the temperate regions of the Northern Hemisphere (Zomlefer et al. 2001). Species of this family are characterized by their extrorse anthers and carpels bearing three distinct styles (Rudall et al. 2000). The family has been divided into five tribes: Heloniadeae, Chionographideae, Xerophylleae, Melanthieae and Parideae (The Angiosperm Phylogeny Group 2009). Prior to any molecular systematic analyses, Melanthiaceae were divided into several taxonomically independent families by Takhtajan (1997) due to their unique autapomorphies. Trilliaceae, which is now recognized as tribe Parideae (Trillieae), are unique in having solitary flowers, berries, membranous nectary and large chromosomes with five chromosomes as the base number. The phylogeny of species within the Trilliaceae (now Parideae) was highly debated by many researchers using molecular and morphological data (Kato et al. 1995; Osaloo et al. 1999; Osaloo and Kawano 1999; Farmer and Schilling 2002; Farmer 2006). Tribe Parideae includes three genera: Paris, Trillium and Pseudotrillium. Paris has 4–15 leaves in a whorl, flowers 4-merous or more and inner perianth segments that are much narrower than outer ones, while Trillium has only 3 leaves in a whorl, flowers 3-merous and inner perianth segments that are a little narrower than the outer ones. Pseudotrillium has thick, tough, heart-shaped leaves, spotted petals and flower stalks that extend until the ripe fruit touches the ground. Trillium has been divided into two subgenera differing in the presence of pedicel: subgenus Trillium (with pedicels) and Phyllantherum (without pedicels) (Freeman 1969, 1975). The monophyly of subgenus Phyllantherum was strongly supported in many previous studies (Osaloo et al. 1999; Osaloo and Kawano 1999; Farmer and Schilling 2002; Farmer 2006). On the other hand, subgenus Trillium is rendered a paraphyletic group by the inclusion of Phyllantherum.
Currently, complete chloroplast genomes of the Melanthiaceae have been reported from Paris verticillata (KJ433485; Do et al. 2014), Veratrum patulum (KF437397; Do et al. 2013) and Chionographis japonica (KF951065; Bodin et al. 2013), which represent three tribes of Parideae, Melanthieae and Chionographideae, respectively. In this study, we analysed complete chloroplast genome sequences of subgenera Trillium and Phyllanthrum of Trillium to better understand the evolution of the chloroplast genomes in tribe Parideae and across the Melanthiaceae. We analysed the sequence variation between two subgenera and proposed novel molecular markers for phylogenetic studies by comparing the two newly generated genome sequences. In addition, we characterized the trnI-CAU duplication event in Parideae, detected in P. verticillata chloroplast genome (KJ433485), to determine the origin of the repeating unit. Consequently, these results provide additional knowledge about the patterns of the chloroplast genome evolution within tribe Parideae.
Methods
DNA extraction, sequencing and annotation
We collected Trillium tschonoskii from Ulleung Island, South Korea. The voucher specimen and plant materials were deposited at the herbarium (GCU) and Medicinal Plant Resources Bank (MPRB) of Gachon University. Trillium maculatum was obtained from the Abraham Baldwin Agricultural College, USA (voucher No. Susan Farmer 19990006). We used silica gel-dried leaves from each species to extract total genomic DNA using the DNeasy Plant Mini Kit (Qiagen, Seoul, South Korea).
The Hiseq 2000 system was employed to sequence chloroplast genomes of T. tschonoskii and T. maculatum. Raw data were assembled using Geneious ver. 7. 1 (Biomatters Ltd, New Zealand) with default settings. After trimming the sequences, we mapped pair-end reads to the reference sequence of P. verticillata (KJ433485). Aligned contigs were ordered according to the reference genome and the gaps were filled via direct sequencing of polymerase chain reaction (PCR) products with newly designed primers. In addition, the ambiguous sequences including low assembly coverage regions and the borders of the four junctions between LSC, SSC and IR regions were confirmed using the Sanger method.
Complete chloroplast genomes of both species were annotated by Geneious ver. 7. 1 (Biomatters Ltd), with manual corrections for putative start and stop codons. The exon positions of protein-coding genes and intron were determined using released Liliales chloroplast genome sequences as references. All tRNA sequences were confirmed utilizing the web-based online tool of tRNAScan-SE (Schattner et al. 2005) with default settings to corroborate tRNA boundaries identified by Geneious. The genome maps were generated using OGDraw (OrganellarGenomeDRAW; Lohse et al. 2007) followed by manual modification.
Comparison of the chloroplast genome sequences of two subgenera
The simple sequence repeats (SSRs) were analysed using Phobos Version 3.3.12 (Kraemer et al. 2009), with thresholds of eight repeat units for mononucleotide SSRs, four repeat units for dinucleotide, trinucleotide SSRs and three repeat units for tetranucleotide, pentanucleotide and hexanucleotide SSRs. All the detected repeats were manually verified, and the redundant results were removed. We aligned the plastid genome sequences of two Trillium using MAFFT (Katoh et al. 2002). The identified insertion/deletion mutations (indels) from the results were confirmed by reassembling the whole reads generated by HiSeq 2000. The single nucleotide polymorphisms (SNPs) were analysed using Geneious 7.1 (Kearse et al. 2012), and each indel and SNP were separated based on the position excluding one of IR regions. Since we are comparing only two genomes, we quantified the sequence divergence as the ratio of aligned nucleotide sites within specifically different regions (p-distance). Sanger sequences and assembled genomes were calculated using mean p-distance in MEGA 6.0 (Tamura et al. 2013).
Twenty-nine species, representing the two subgenera of Trillium in Parideae, were selected for comparative sequencing of inversion. The PCR amplification primers were designed based on the sequence comparisons among three chloroplast genome sequences of two Trillium species (in this study), and P. verticillata (KJ433485). Presence and absence PCR amplifications were carried out using various combinations of the three primers (I1F: 5′-CCC TAG GTT TTT TTC TTC AAG-3′, I1R: 5′-TTA TGT AGC TTA TCC TTT AGA CC-3′ and I2R: 5′-AGA AGG TCT ACG GTT CGA G-3′).
trnI-CAU duplication pattern in the tribe Parideae
To clarify the trnI-CAU duplication pattern in the tribe Parideae, we designed two primers (Primer 1: 5′-GAA GAG TTC GAC CCA ATG CT-3′, Primer 2: 5′-TTA TGA AAC TCT TTG ACC CC-3′) for amplifying the intergenic spacer (IGS) region of rpl23-ycf2 based on the identical sequence among the three species (P. verticillata, T. maculatum and T. tschonoskii). The PCR condition for IGS region of rpl23-ycf2 was at initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 50 °C for 1 min and extension at 72 °C for 2 min, with a final extension at 72 °C for 5 min. We obtained variously sized PCR products ranging from 500 to 1200 bp, and compared the sequences of this region from 33 species covering the infrageneric classification of the tribe. Sequence editing and assembly were performed using Sequencher (ver. 5.1). The sequence alignment was initially performed using MAFFT (Katoh et al. 2002) and was adjusted manually.
Results
Comparison of the complete chloroplast genomes of subgenera Trillium and Phyllantherum
We sequenced the complete chloroplast genome sequence of two Trillium species, T. tschonoskii (subgenera Trillium; GenBank accession number KR780076) and T. maculatum (subgenera Phyllantherum; GenBank accession number KR780075) (Fig. 1). In total, 4 292 702 (T. tschonoskii) and 18 348 134 (T. maculatum) paired-end reads were generated. Out of those, 60 805 and 246 240 reads were identified as the chloroplast genome sequences for T. tschonoskii and T. maculatum, respectively. The chloroplast genome of T. tschonoskii was composed of 156 852 bp in length (AT content 62.5 %), and it comprised a LSC region (83 981 bp), a SSC region (19 869 bp) and two IR regions (26 501 bp), while T. maculatum was 157 359 bp in length (AT content 62.5 %, 86 340 bp of LSC, 19 949 bp of SSC and 25 535 bp of IRs).
Figure 1.

Gene maps and summary of the T. tschonoskii Maxim. and T. maculatum Raf. chloroplast genomes. IR, inverted repeat; LCS, large single-copy region; SSC, small single-copy region.
The gene content and order were slightly different between both species because of the rpl22 position in the IR-LSC boundary and trnI-CAU duplication in IR. While the rpl22 gene remained in the LSC region of the T. maculatum plastid genome, this gene was present in the IR region of T. tschonoskii plastid genome (Fig. 2). In total, 116 genes of T. maculatum were identified and consisted of 78 coding genes, 4 rRNA genes, 31 tRNA genes and 3 pseudogenes, while those of T. tschonoskii were 115 genes without tRNA gene duplication [see Supporting Information—Table S1]. In addition, T. tschonoskii has 7 coding genes, 4 rRNA genes, 9 tRNA genes, 2 pseudogenes, whereas T. maculatum has 8 coding genes, 4 rRNA genes, 8 tRNA genes, 2 pseudogenes, duplicated in the IR region, making a total of 138 genes and 137 genes presented in the T. tschonoskii and T. maculatum chloroplast genome, respectively. Among these genes, 22 intron-containing genes were found including 15 protein-coding genes and 7 tRNA genes. Among them, ycf3 and clpP gene contained two introns. The trnK-UUU has the largest intron (T. tschonoskii: 2614 bp, T. maculatum: 2640 bp) including the matK gene. Ycf15 and ycf68 in the IR region were pseudogenized because of the presence of several internal stop codons. Furthermore, the cemA gene located in the LSC of both genomes was also pseudogenized.
Figure 2.

Comparison of the IR boundaries among five species within Melanthiaceae.
Characterization of single inversion in subgenus Phyllantherum
Based on comparison of T. maculatum, T. tschonoskii and P. verticillata, a single inversion of 3.4 kb is characterized in the chloroplast genome of T. maculatum. This inversion is located between the ndhC and rbcL genes. We designed three different primers including I1F (5′-CCC TAG GTT TTT TTC TTC AAG-3′), I1R (5′-TTA TGT AGC TTA TCC TTT AGA CC-3′) and I2R (5′-AGA AGG TCT ACG GTT CGA G-3′) to confirm and clarify the distribution of this inversion throughout the genus Trillium. Specifically, the primer pairs of I1F and I1R worked only in the normal type, while I2R and I1R primer pairs were utilized for the recognized inversion type among examined species. The results showed that the inversion occurred in all examined species of the subgenera Phyllantherum (Fig. 3A and B).
Figure 3.

Confirmation of inversion (3492 bp) between ndhC and rbcL in the genus Trillium. (A) Design of primer to amplify junction regions between atpB and rbcL regions. The positions of atpB and rbcL genes in LSC regions are drawn based on the sequence assembly results of T. tschonoskii, T. maculatum in this study (red text). *The data downloaded from the NCBI. The forward primer I1F contains the sequence in atpB region. The sequence of the reverse primer (I1R) is located in the rbcL gene. Polymerase chain reaction amplification of IGS between atpB and rbcL. Relationships of Parideae lineages followed the phylogenetic trees of S. C. Kim, J. S. Kim, W. C. Mark, F. F. Michael and J. H. Kim (unpublished data). (B) Primers were designed to amplify junction regions between trnV-UAC and rbcL regions. The positions of trnV-UAC and rbcL genes in LSC regions are drawn based on the sequence assembly results of T. tschonoskii, T. maculatum in this study (red text). *The data downloaded from the NCBI. The forward primer I2R contains the sequence in trnV-UAC region. The sequence of the reverse primer I1R is located in the rbcL gene. Polymerase chain reaction amplification of IGS between trnV-UAC and rbcL. Relationships of Parideae lineages followed the phylogenetic trees of S. C. Kim, J. S. Kim, W. C. Mark, F. F. Michael and J. H. Kim (unpublished data).
Indels, SNPs and SSR between two subgenera of Trillium
A total of 402 indels were detected between T. maculatum and T. tschonoskii, and most indels were located in the IGS regions (78.2 %). 66.2 % of the total number of indels were found in the LSC, while 22.1 and 11.7 % were present in the SSC and IR regions, respectively [Table 1, see Supporting Information—Table S2]. The average length of indels was 74.8 bp, and the largest indel was located in ycf1 and ycf2. The frequency of 1 bp indels was 10.6 %, while 79.3 % of all indels were over 20 bp in length. In rRNA sequences, one indel of 3 bp and four indels of 5 bp were found in 16S rRNA and 23S rRNA. In addition, indel events were identified in 20 coding genes of both species (accD, atpB, ccsA, cemA, clpP, infA, matK, ndhF, rpl2, rpl20, rpl22, rpl32, rpoC1, rpoC2, rps11, rps15, rps18, rps19, ycf1 and ycf2).
Table 1.
The number and total length of insertion–deletion mutations between the chloroplast genomes of T. tschonoskii and T. maculatum in Parideae.
| Region | Number of indels | Total length of indels |
|---|---|---|
| IGS | 262 | 5139 |
| Intron | 51 | 528 |
| Coding gene | 86 | 3005 |
A total of 2861 SNPs were detected between T. maculatum and T. tschonoskii (Table 2), and 1620 SNPs were transversions. In total, 1707 (59.7 %) SNPs were located in the coding regions, and 1154 (40.3 %) were within IGS regions or within introns.
Table 2.
Single nucleotide polymorphisms found between the plastid genomes of T. tschonoskii and T. maculatum. (A) Single nucleotide polymorphisms in coding gene. (B) Single nucleotide polymorphisms in intron. (C) Single nucleotide polymorphisms in IGS regions. Bold values represent p-distance >0.08.
| Gene | Aligned length (bp) | No. SNP | p-Distance | Gene | Aligned length (bp) | No. SNP | p-Distance | Gene | Aligned length (bp) | No. SNP | p-Distance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | |||||||||||
| psbD | 1062 | 2 | 0.002 | ycf4 | 555 | 4 | 0.007 | rpl2 | 828 | 14 | 0.017 |
| psaB | 2205 | 5 | 0.002 | cemA | 695 | 5 | 0.007 | rrn5S | 121 | 2 | 0.017 |
| psaA | 2253 | 4 | 0.002 | rrn23S | 2814 | 19 | 0.007 | rpl36 | 114 | 2 | 0.018 |
| atpE | 405 | 1 | 0.002 | ndhD | 1503 | 10 | 0.007 | rrn4.5S | 103 | 2 | 0.019 |
| psbB | 1527 | 3 | 0.002 | ndhE | 306 | 2 | 0.007 | rrn16S | 1494 | 30 | 0.02 |
| petB | 648 | 1 | 0.002 | ndhI | 543 | 4 | 0.007 | rpl14 | 369 | 9 | 0.024 |
| psbA | 1062 | 3 | 0.003 | rps15 | 276 | 3 | 0.007 | ycf2 | 7209 | 146 | 0.024 |
| ndhK | 768 | 2 | 0.003 | rpoB | 3213 | 25 | 0.008 | rps7 | 468 | 11 | 0.024 |
| ndhC | 363 | 1 | 0.003 | psbF | 120 | 1 | 0.008 | rpl20 | 387 | 9 | 0.025 |
| rbcL | 1434 | 5 | 0.003 | rps8 | 399 | 3 | 0.008 | rps18 | 363 | 8 | 0.026 |
| petA | 963 | 3 | 0.003 | psbN | 132 | 1 | 0.008 | rps3 | 657 | 18 | 0.027 |
| ndhB | 1533 | 5 | 0.003 | ycf15 | 234 | 2 | 0.009 | trnT-UGU | 73 | 2 | 0.027 |
| ccsA | 969 | 3 | 0.003 | pebT | 108 | 1 | 0.009 | trnE-UUC | 73 | 2 | 0.027 |
| rps16 | 252 | 1 | 0.004 | rps14 | 303 | 3 | 0.01 | trnC-GCA | 71 | 2 | 0.028 |
| atpA | 1524 | 6 | 0.004 | rps4 | 606 | 6 | 0.01 | trnQ-UUG | 72 | 2 | 0.028 |
| atpH | 246 | 1 | 0.004 | rpoA | 1023 | 10 | 0.01 | rps12 | 372 | 11 | 0.03 |
| psbC | 1422 | 6 | 0.004 | infA | 243 | 2 | 0.01 | rpl33 | 201 | 7 | 0.035 |
| psbE | 252 | 1 | 0.004 | rpoC1 | 2097 | 22 | 0.011 | rpl22 | 387 | 13 | 0.035 |
| petD | 483 | 2 | 0.004 | trnS-GCU | 88 | 1 | 0.011 | rpl32 | 156 | 6 | 0.04 |
| atpB | 1521 | 7 | 0.005 | rpl23 | 282 | 3 | 0.011 | trnP-UGG | 74 | 3 | 0.041 |
| ycf68 | 376 | 2 | 0.005 | ndhF | 2232 | 26 | 0.012 | trnI-CAU | 74 | 3 | 0.041 |
| ndhG | 531 | 3 | 0.006 | matK | 1554 | 22 | 0.014 | rps11 | 405 | 19 | 0.048 |
| ndhA | 1083 | 7 | 0.006 | trnH-GUG | 74 | 1 | 0.014 | rps19 | 351 | 24 | 0.084 |
| ndhH | 1182 | 7 | 0.006 | trnW-CCA | 74 | 1 | 0.014 | clpP | 639 | 65 | 0.111 |
| atpF | 555 | 4 | 0.007 | rpl16 | 411 | 6 | 0.015 | ycf1 | 6778 | 664 | 0.121 |
| rpoC2 | 4140 | 27 | 0.007 | rps2 | 711 | 12 | 0.017 | accD | 1566 | 323 | 0.23 |
| Gene | Aligned length (bp) | No. SNP | p-Distance | Gene | Aligned length (bp) | No. SNP | p-Distance | ||||
| (B) | |||||||||||
| atpF intron | 812 | 14 | 0.018 | rpoC1 intron | 714 | 9 | 0.013 | ||||
| clpP intron 1 | 709 | 12 | 0.034 | rps16 intron | 783 | 11 | 0.015 | ||||
| clpP intron 2 | 983 | 29 | 0.012 | trnI-GAU intron | 936 | 2 | 0.002 | ||||
| ndhA intron | 1077 | 6 | 0.006 | trnK-UUU intron | 1109 | 34 | 0.012 | ||||
| ndhB intron | 695 | 1 | 0.001 | trnL-UAA intron | 538 | 2 | 0.004 | ||||
| petB intron | 823 | 4 | 0.005 | trnV-UAC | 595 | 4 | 0.007 | ||||
| petD intron | 747 | 5 | 0.007 | ycf3 intron1 | 737 | 7 | 0.006 | ||||
| rpl16 intron | 1075 | 26 | 0.026 | ycf3 intron2 | 738 | 8 | 0.01 | ||||
| rpl2 intron | 664 | 3 | 0.005 | ||||||||
| IGS | Aligned length (bp) | No. SNP | p-Distance | IGS | Aligned length (bp) | No. SNP | p-Distance | ||||
| (C) | |||||||||||
| trnL-CAA_ndhB | 578 | 1 | 0.002 | rrn23S_rrn4.5S | 102 | 2 | 0.02 | ||||
| ndhB_rps7 | 323 | 1 | 0.003 | rpl32_trnL-UAG | 938 | 16 | 0.02 | ||||
| atpI_rps2 | 242 | 1 | 0.004 | ndhH_rps15 | 110 | 2 | 0.02 | ||||
| rps12_trnV-GAC | 1905 | 8 | 0.004 | rpoB_trnC-GCA | 859 | 16 | 0.022 | ||||
| psaI_ycf4 | 376 | 2 | 0.005 | rps19_trnH-GUG | 147 | 3 | 0.022 | ||||
| petB_petD | 205 | 1 | 0.005 | atpA_atpF | 92 | 2 | 0.023 | ||||
| psbE_petL | 952 | 6 | 0.006 | trnG-GCC_trnfM-CAU | 132 | 3 | 0.023 | ||||
| psbB_psbT | 168 | 1 | 0.006 | petA_psbJ | 1139 | 26 | 0.023 | ||||
| ycf4_cemA | 785 | 5 | 0.007 | petG_trnW-CCA | 138 | 3 | 0.023 | ||||
| rrn16S_trnI-GAU | 296 | 2 | 0.007 | rps8_rpl14 | 179 | 4 | 0.023 | ||||
| trnA-UGC_rrn23S | 144 | 1 | 0.007 | ndhE_ndhG | 301 | 4 | 0.023 | ||||
| trnR-ACG_trnN-GUU | 572 | 4 | 0.007 | rps15_ycf1 | 429 | 9 | 0.023 | ||||
| rps14_psaB | 132 | 1 | 0.008 | trnK-UUU_rps16 | 783 | 23 | 0.024 | ||||
| psbN_psbH | 124 | 1 | 0.008 | rpl20_clpP | 1208 | 25 | 0.024 | ||||
| infA_rps8 | 296 | 2 | 0.008 | trnL-UAG_ccsA | 82 | 2 | 0.025 | ||||
| trnC-GCA_petN | 831 | 8 | 0.01 | ndhD_psaC | 119 | 3 | 0.025 | ||||
| trnD-GUC_trnY-GUA | 412 | 4 | 0.01 | cemA_petA | 242 | 6 | 0.026 | ||||
| trnS-GGA_rps4 | 307 | 3 | 0.01 | trnQ-UUG_psbK | 360 | 9 | 0.027 | ||||
| ycf15_trnL-CAA | 674 | 2 | 0.01 | trnG-UCC_trnR-UCU | 150 | 4 | 0.027 | ||||
| psbM_trnD-GUC | 1031 | 11 | 0.011 | trnT-UGU_trnL-UAA | 739 | 21 | 0.029 | ||||
| trnT-GGU_psbD | 1016 | 12 | 0.011 | rpoA_rps11 | 68 | 2 | 0.029 | ||||
| ndhJ_ndhK | 89 | 1 | 0.011 | atpH_atpI | 655 | 19 | 0.031 | ||||
| psaJ_rpl33 | 482 | 3 | 0.011 | rps16_trnQ-UUG | 1204 | 37 | 0.037 | ||||
| petD_rpoA | 179 | 2 | 0.011 | trnW-CCA_trnP-UGG | 167 | 6 | 0.037 | ||||
| ndhG_ndhI | 283 | 3 | 0.011 | ndhF_rpl32 | 778 | 28 | 0.038 | ||||
| trnF-GAA_ndhJ | 686 | 8 | 0.012 | psbK_psbI | 396 | 15 | 0.039 | ||||
| rps2_rpoC2 | 246 | 3 | 0.013 | psaC_ndhE | 380 | 14 | 0.04 | ||||
| ycf3_trnS-GGA | 759 | 8 | 0.013 | psbH_petB | 134 | 5 | 0.042 | ||||
| rps4_trnT-UGU | 320 | 4 | 0.013 | trnE-UUC_trnT-GGU | 724 | 26 | 0.044 | ||||
| trnL-UAA_trnF-GAA | 386 | 5 | 0.013 | rpl33_rps18 | 200 | 8 | 0.049 | ||||
| atpF_atpH | 474 | 7 | 0.015 | clpP_psbB | 507 | 23 | 0.049 | ||||
| psbZ_trnG-GCC | 296 | 4 | 0.015 | rps11_rpl36 | 151 | 7 | 0.051 | ||||
| trnM_CAU- atpE | 206 | 3 | 0.015 | trnS-GCU_trnG-UCC | 1178 | 57 | 0.052 | ||||
| psaA_ycf3 | 642 | 10 | 0.016 | psbI_trnS-GCU | 124 | 6 | 0.054 | ||||
| petL_petG | 183 | 3 | 0.016 | psbC__trnS-UGA | 140 | 8 | 0.057 | ||||
| rpl36_infA | 154 | 2 | 0.016 | rpl23_trnI-CAU | 210 | 11 | 0.065 | ||||
| rpl14_rpl16 | 126 | 2 | 0.016 | rpoC1_rpoB | 37 | 2 | 0.077 | ||||
| psbA_trnK-UUU | 243 | 10 | 0.017 | trnN-GUU_ndhF | 782 | 31 | 0.086 | ||||
| petN_psbM | 712 | 12 | 0.017 | rpl22_rps19 | 115 | 4 | 0.091 | ||||
| trnY-GUA_trnE-UUC | 59 | 1 | 0.017 | rps3_rpl22 | 80 | 5 | 0.098 | ||||
| ccsA_ndhD | 242 | 4 | 0.018 | accD_psaI | 285 | 20 | 0.099 | ||||
| rpoC2_rpoC1 | 154 | 3 | 0.019 | psbT_psbN | 65 | 10 | 0.154 | ||||
| trnP-UGG_psaJ | 388 | 7 | 0.019 | trnH-GUG_rpl2 | 44 | 8 | 0.186 | ||||
| rpl16_rps3 | 166 | 3 | 0.019 | trnI-CAU_ycf2 | 210 | 20 | 0.238 | ||||
| rps7_rps12 | 54 | 1 | 0.019 | rbcL_accD | 2090 | 236 | 0.291 | ||||
In our result of SNPs, p-distance values in coding regions range from 0.002 to 0.23 and the average value was 0.02. On the other hand, the average p-distance value in non-coding regions was 0.034. Figure 4 shows the average p-distance for five classes of genomic regions: protein-coding genes, tRNAs, rRNAs, IGSs and introns. The IGS divergence is almost double that of the next highest class (genes). Introns hold the lowest sequence divergence, at an average of 0.011%.
Figure 4.
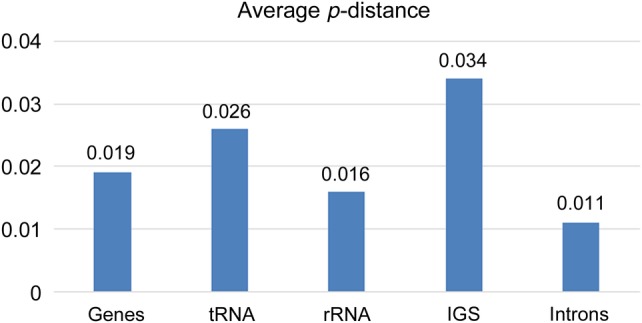
Average p-distance across five classes of genomic regions between two Trillium.
We detected SSRs longer than 8 bp in T. maculatum, T. tschonoskii and P. verticillata chloroplast genomes by the method of Qian et al. (2013). According to Qian et al., the threshold was set because 8 bp or longer SSRs are prone to slipstrand mispairing, which is thought to be the primary mutational mechanism causing their high level of polymorphism. In this analysis, the total number of SSRs was 204 in P. verticillata, 205 in T. maculatulatum and 213 in T. tschonoskii (Table 3). The most abundant type of SSR in Parideae was a mononucleotide, with 138 in P. verticillata, 121 in T. maculatulatum and 133 in T. tschonoskii. In addition to mononucleotide SSRs, there are 52 dinucleotide SSRs in P. verticillata, 57 in T. maculatulatum and 53 in T. tschonoskii. Trinucleotide SSRs were less frequent with 6, 14 and 7 in P. verticillata, T. maculatum and T. tschonoskii, respectively. The hexanucleotide SSRs were found only in Trillium species. The majority of mononucleotide repeats were A-T rich (Table 3).
Table 3.
Number of SSRs present in the three Parideae chloroplast genomes.
| Taxon | Paris verticillata | Trillium maculatum | Trillium tschonoskii |
|---|---|---|---|
| Genome size | 157 379 | 157 359 | 156 852 |
| No. of SSRs | |||
| A/T | 133 | 117 | 127 |
| C/G | 5 | 4 | 6 |
| AC/GT | 3 | 3 | 3 |
| AG/CT | 17 | 19 | 18 |
| AT/TA | 32 | 35 | 32 |
| AAG/CTT | 2 | 2 | |
| AAT/ATT | 6 | 10 | 3 |
| ACT/AGT | 2 | 1 | |
| ATC/GAT | 1 | ||
| AAAG/CTTT | – | – | 1 |
| AAAT/ATTT | 3 | 4 | 4 |
| AAGG/CCTT | 1 | 1 | 1 |
| AATC/GATT | 1 | 1 | 1 |
| AATG/CATT | 1 | 1 | 3 |
| AGAT/ATCT | 1 | 1 | 1 |
| ACTAT/ATAGT | 1 | – | 1 |
| AAAAT/ATTTT | – | 1 | 1 |
| AATAT/ATATT | – | 2 | 1 |
| AATATG/CATATT | – | 1 | – |
| AAAATC/GATTTT | – | 1 | – |
| ATATCC/GGATAT | – | – | 1 |
| AAAAAT/ATTTTT | – | – | 2 |
| AAGACT/AGTCTT | – | – | 1 |
| AACTAC/GTAGTT | – | – | 1 |
| AAAGAG/CTCTTT | – | – | 1 |
| Total | 204 | 205 | 213 |
Type of trnI-CAU of Parideae
We compared the sequences of the IGS region between rpl23 and ycf2 using 33 species including Xerophyllum to understand the evolutionary implication of trnI-CAU duplication, which was reported from the Paris chloroplast genome (Do et al. 2014). Based on the results, we found that this region is of highly variable length among the species, and we distinguished three major types based on the number of copies of trnI-CAU (Fig. 5). Type A was composed of a single trnI-CAU and was found in Xerophyllum, Pseudotrillium rivale and T. undulatum. It was also identified in several Trillium and Paris species, but with variable lengths: in subgenus Trillium species, the sequences ranged from 207 to 445 bp, in which there are two tandem repeats of ‘CAG GTA TTA TCA TAC TGA AA’ (20 bp) and ‘CAT ATT ATC ATA CTG AAA’ (18 bp). Similarly, in subgenus Daiswa of Paris, there were 24 bp random tandem repeats of TAT AAC TTA ACA GGA ATC ATC GTA. Type B contained two copies of trnI-CAU. This type is found in subgenus Phyllantherum of Trillium and section Kinugasa of subgenus Paris. The lengths of tandem repeat sequences were 180 bp (subgenera Phyllantherum) and 155 bp (section Kinugasa of subgenus Paris), which included 74 bp of trnI-CAU. Remarkably, section Kinugasa (Paris japonica) has the longest length of IGS between rpl23 and ycf2 among the tribe Parideae. Type C, possessing three copies of trnI-CAU genes in the sequenced region, was detected in T. govanianum and section Paris of subgenus Paris. They included three fully repeated units including trnI-CAU, and the lengths were 155 and 139 bp, respectively.
Figure 5.

Summary of three types of trnI-CAU gene duplication in the tribe Parideae. *Including tandem repeats.
Discussion
Comparison of complete plastid genomes of subgenera Trillium and Phyllantherum
The plastid genome structure of the two Trillium species, T. maculatum and T. tschonoskii, have a typical form found in most angiosperms (Zhang et al. 2011; Kim and Kim 2013; Li et al. 2013; Qian et al. 2013). The T. tschonoskii chloroplast genome was 507 bp shorter than T.maculatum, and we confirmed that the length variation among Parideae chloroplast genomes including Paris verticillata occurred by gene deletion and duplication as well as its IR expansion.
Although chloroplast genomes are considered highly conserved among land plants, sequence polymorphisms were often observed among closely related species. From the T. tschonoskii and T. maculatum chloroplast genome sequences, we confirmed that 402 indels and 2861 SNPs were present between the two species.
In addition, we found that SSRs (i.e. microsatellites), composed of 1–6 bp in length per unit, are distributed throughout both genomes. The SSRs have been accepted as one of the major molecular markers for genome variation between species or within populations due to their high polymorphism within the species and have been widely practiced for analysing plant population structure, diversity, differentiation and maternity analysis (Liu et al. 2013). Simple sequence repeats have successfully been applied to the study of Poaceae, Brassicaceae and Solanaceae (Provan et al. 1997, 1999; Bryan et al. 1999; Flannery et al. 2006). Simple sequence repeats detected in the present study will provide basic information for the further analysis of genetic diversity in Parideae.
Based on our results, the IR/LSC boundary and the IR/SSC boundary differed between the two subgenera of Trillium. Inverted repeat/large single-copy junction was expanded to a part of rps3 in T. tschonoskii, whereas that of T. maculatum was found at rps19. The ycf1 was completely located in SSC of T. tschonoskii, but a part of the ycf1 gene was duplicated in IR of T. maculatum. Within the Parideae, the IR boundary pattern of T. tschonoskii was more similar to P. verticillata than T. maculatum (Fig. 2).
Inversion events in Melanthiaceae
Inversions caused by the recombination between repeated sequences are considered to be a main mechanism for changes in gene order among plastid genomes (Jansen and Ruhlman 2012). Most of the reported inversions in plastid genomes are in the LSC region (Kim et al. 2005). In subtribe Phaseolinae of Fabaceae, there is a 78 kb inversion between trnH/rpl14 and rps19/rps8 in the chloroplast genome (Tangphatsornruang et al. 2010). Additionally, Kim et al. (2005) reported that the inversion occurred in the spacer between tRNAGly and tRNASer genes of Lactuca sativa. Also, they defined two inversions that characterize Asteraceae. The two inversions were identical across all members of Asteraceae, suggesting that the inversion events are likely to occur simultaneously or within a short period of time following the origin of the family. In Campanulaceae, >50 large inversions occurred during diversification of the family, in which at least 20 occurred in Cyphia, and a minimum of 53 are now known in Lobelia (Knox 2014). Fabaceae are known to exhibit a number of unusual phenomena in their chloroplast genome: Trifolium subterraneum has undergone extensive genomic reconfiguration, including the loss of six genes and two introns and numerous gene order changes, attributable to 14–18 inversions (Cai et al. 2008).
Our results confirmed a single inversion in Melanthiaceae. It was remarkable that a single inversion of 3492 bp embedded four genes between ndhC and rbcL genes, which specifically occurred in the monophyletic subgenus Phyllantherum (Fig. 3). This event is thought to have occurred after the evolutionary divergence between subgenus Phyllantherum and subgenus Trillium. This new finding may be an effective molecular marker for classifying subgenera of the genus Trillium.
Diverse patterns of trnI-CAU duplication in Parideae
Gene duplication is an important process in organellar genome evolution. Most duplicated genes occur within the IR regions due to the mechanisms underlying IR expansion and contraction (Xiong et al. 2009). Gene duplication in plastid genome has been reported in tRNA genes (Hipkins et al. 1995; Vijverberg and Bachmann 1999; Schmickl et al. 2009) and in some protein-coding genes. Most of the duplications can be detected only in rearranged chloroplast genomes, as in grasses, legumes and conifers. Hipkins et al. (1995) compared the number of direct repeats between partially duplicated trnY-GUA and the complete trnY-GUA gene in Pseudotsuga. They found that the length-variable region in Pseudotsuga comprised imperfect tandem direct repeats based on the trnY gene sequence. Schmickl et al. (2009) used the 5′-trnL-UAA_trnF-GAA region for phylogeographic reconstructions, gene diversity calculations and phylogenetic analyses among the genera Arabidopsis and Boechera. The Cruciferous taxa are characterized by these pseudogenes in at least four independent phylogenetic lineages. In addition, the tRNA gene as well as the coding gene could be confirmed by duplication events in Jasminum and Menodora, which have the duplicated rbcL_psaI region. Most chloroplast gene duplications outside of the IR involve tRNAs, as in the case of Oleaceae (Lee et al. 2007).
A total of 30–32 tRNA genes are present within the chloroplast genome of land plants (Tsudzuki et al. 1994; Vijverberg and Bachmann 1999), and they may be involved in chloroplast genome rearrangements through their secondary structure (Howe et al. 1988). These genes are dispersed throughout the genome, but five to eight genes are located in the IR (Maréchal-Drouard et al. 1993). We found that three major types of trnI-CAU gene duplication are located between rpl23 and ycf2 at the IR of tribe Parideae (Fig. 5). Traditionally, Parideae included two genera, Paris and Trillium; however, Trillium was separated into two genera Trillium and Pesudotrillium in recent classifications (Farmer and Schilling 2002). Using the various duplication patterns of trnI-CAU in the IR region, the infrageneric circumscription of Parideae member was strongly supported. The type of trnI-CAU that had been discovered in Xerophyllum, Pesudotrillium and T. undulatum with one trnI-CAU between rpl23 and ycf2 was seen to be similar to the ancestor of Parideae (Type A, Fig. 5). This type was found also in most chloroplast genomes of Liliales (Liu et al. 2012; Bodin et al. 2013; Do et al. 2013; Kim and Kim 2013). It was modified in subgenus Trillium of Trillium and subgenus Daiswa of Paris to be extended by the tandem repeat between trnI-CAU and ycf2. Type B was found in subgenus Phyllantherum of Trillium and section Kinugasa of subgenus Paris although section Kinugasa possessed the additional tandem repeat between trnI-CAU units. Type C, which was found in T. govanianum and section Paris of subgenus Paris, has three copies of the trnI-CAU gene. From the results, we suggested that duplicate events of trnI-CAU have occurred independently in the tribe Parideae of Melanthiaceae, and it provided useful information for determining the infrageneric circumscription. However, T. govanianum, which was classified into another genus Trillidium by Farmer and Schilling (2002) based on morphological characters and geographical distribution, was more similar to Paris than Trillium. Also, this result showed that the trnI-CAU gene duplication pattern of T. govanianum was more similar to Paris than Trillium. Interestingly, it was positioned at the same clade together with the North American species T. undulatum in the molecular phylogenetic tree (Farmer and Schilling 2002; S. C. Kim, J. S. Kim, W. C. Mark, F. F. Michael, J. H. Kim, unpublished data). Further studies are necessary to clarify the relationship between both species.
Conclusions
We analysed the complete chloroplast genomes of two species of T. tschonoskii (subgenus Trillium) and T. maculatum (subgenus Phyllantherum) to verify the specific feature in the genome level. As a result, we found a 3.4 kb inverted sequence between ndhC and rbcL in the LSC region in the chloroplast genome of T. maculatum, which was unique to subgenus Phyllantherum. In addition, three different gene duplication patterns of trnI-CAU gene were found and they were the informative molecular markers for identifying the infrageneric taxa of Trillium.
Sources of Funding
This work was supported by the National Research Foundation of Korea (NRF) Grant Foundation (MEST 2010-0029131) and Korea National Arboretum (KNA1-2-13,14-2).
Contributions by the Authors
J.-H.K conceived and designed the experiments, S.-C.K. performed the experiments, S.-C.K. and J.S.K. analysed the data and S.-C.K. and J.-H.K. wrote the paper.
Conflict of Interest Statement
None declared.
Supporting Information
The following additional information is available in the online version of this article –
Table S1. Genes found in Trillium tschonoskii and T. maculatum chloroplast genomes.
Table S2. The detailed list of insertion–deletion mutations between the chloroplast genomes of T. tschonoskii and T. maculatum in Parideae.
Table S3. Sequences of rpl23_ycf2 IGS among Parideae species.
Acknowledgements
The authors thank Dr Susan Farmer of Abraham Baldwin Agricultural College who kindly provided the Trillium maculatum for this study. Also we thank the editors and two reviewers for valuable comments and suggestions.
Literature Cited
- Bodin SS, Kim JS, Kim J-H. 2013. Complete chloroplast genome of Chionographis japonica (Willd.) Maxim. (Melanthiaceae): comparative genomics and evaluation of universal primers for Liliales. Plant Molecular Biology Reporter 31:1407–1421. 10.1007/s11105-013-0616-x [DOI] [Google Scholar]
- Bryan GJ, McNicoll J, Ramsay G, Meyer RC, De Jong WS. 1999. Polymorphic simple sequence repeat markers in chloroplast genomes of Solanaceous plants. Theoretical and Applied Genetics 99:859–867. 10.1007/s001220051306 [DOI] [Google Scholar]
- Cai Z, Guisinger M, Kim H-G, Ruck E, Blazier JC, Mcmurtry V, Kuehl JV, Boore J, Jansen RK. 2008. Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. Journal of Molecular Evolution 67:696–704. 10.1007/s00239-008-9180-7 [DOI] [PubMed] [Google Scholar]
- Do HD, Kim JS, Kim JH. 2013. Comparative genomics of four Liliales families inferred from the complete chloroplast genome sequence of Veratrum patulum O. Loes. (Melanthiaceae). Gene 530:229–235. 10.1016/j.gene.2013.07.100 [DOI] [PubMed] [Google Scholar]
- Do HDK, Kim JS, Kim J-H. 2014. A trnI_CAU triplication event in the complete chloroplast genome of Paris verticillata M.Bieb. (Melanthiaceae, Liliales). Genome Biology and Evolution 6:1699–1706. 10.1093/gbe/evu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyall SD, Brown MT, Johnson PJ. 2004. Ancient invasions: from endosymbionts to organelles. Science 304:253–257. 10.1126/science.1094884 [DOI] [PubMed] [Google Scholar]
- Farmer SB. 2006. Phylogenetic analyses and biogeography of Trilliaceae. Aliso 22:579–592. [Google Scholar]
- Farmer SB, Schilling EE. 2002. Phylogenetic analyses of Trilliaceae based on morphological and molecular data. Systematic Botany 27:674–692. [Google Scholar]
- Flannery ML, Mitchell FJG, Coyne S, Kavanagh TA, Burke JI, Salamin N, Dowding P, Hodkinson TR. 2006. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theoretical and Applied Genetics 113:1221–1231. 10.1007/s00122-006-0377-0 [DOI] [PubMed] [Google Scholar]
- Freeman JD. 1969. A revisionary study of sessile-flowered Trillium L. (Liliaceae). PhD Dissertation, Vanderbilt University, Nashville, USA. [Google Scholar]
- Freeman JD. 1975. Revision of Trillium subgenus Phyllantherum (Liliaceae). Brittonia 27:1–62. 10.2307/2805646 [DOI] [Google Scholar]
- Gene Codes Corporation. 2012. Sequencher (version 5.1). Ann Arbor, MI: Gene Codes Corporation. [Google Scholar]
- Hipkins VD, Marshall KA, Neale DB, Rottmann WH, Strauss SH. 1995. A mutation hotspot in the chloroplast genome of a conifer (Douglas-fir: Pseudotsuga) is caused by variability in the number of direct repeats derived from a partially duplicated tRNA gene. Current Genetics 27:572–579. 10.1007/BF00314450 [DOI] [PubMed] [Google Scholar]
- Howe CJ, Barker RF, Bowman CM, Dyer TA. 1988. Common features of three inversions in wheat chloroplast DNA. Current Genetics 13:343–349. 10.1007/BF00424430 [DOI] [PubMed] [Google Scholar]
- Jansen RK, Ruhlman TA. 2012. Plastid genomes of seed plants. In: Bock R, Knoop V, eds. Genomics of chloroplasts and mitochondria. Dordrecht, The Netherlands: Springer, 103–126. [Google Scholar]
- Jansen RK, Raubeson LA, Boore JL, Depamphilis CW, Chumley TW, Haberle RC, Wyman SK, Alverson AJ, Peery R, Herman SJ, Fourcade HM, Kuehl JV, Mcneal JR, Leebens-Mack J, Cui L. 2005. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods in Enzymology 395:348–384. 10.1016/S0076-6879(05)95020-9 [DOI] [PubMed] [Google Scholar]
- Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, Müller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, Chumley TW, Lee SB, Peery R, Mcneal JR, Kuehl JV, Boore JL. 2007. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proceedings of the National Academy of Sciences of the USA 104:19369–19374. 10.1073/pnas.0709121104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Terauchi R, Utech FH, Kawano S. 1995. Molecular systematics of the Trilliaceae sensu lato as inferred from rbcL sequence data. Molecular Phylogenetics and Evolution 4:184–193. 10.1006/mpev.1995.1018 [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 30:3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Kim J-H. 2013. Comparative genome analysis and phylogenetic relationship of order Liliales insight from the complete plastid genome sequences of two Lilies (Lilium longiflorum and Alstroemeria aurea). PLoS ONE 8:e68180 10.1371/journal.pone.0068180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-J, Choi K-S, Jansen RK. 2005. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Molecular Biology and Evolution 22:1783–1792. 10.1093/molbev/msi174 [DOI] [PubMed] [Google Scholar]
- Knox EB. 2014. The dynamic history of plastid genomes in the Campanulaceae sensu lato is unique among angiosperms. Proceedings of the National Academy of Sciences of the USA 111:11097–11102. 10.1073/pnas.1403363111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer L, Beszteri B, Gäebler-Schwarz S, Held C, Leese F, Mayer C, Pöehlmann K, Frickenhaus S. 2009. STAMP: extensions to the STADEN sequence analysis package for high throughput interactive microsatellite marker design. BMC Bioinformatics 10:41 10.1186/1471-2105-10-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-L, Jansen RK, Chumley TW, Kim K-J. 2007. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Molecular Biology and Evolution 24:1161–1180. 10.1093/molbev/msm036 [DOI] [PubMed] [Google Scholar]
- Li R, Ma P-F, Wen J, Yi T-S. 2013. Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications. PLoS ONE 8:e78568 10.1371/journal.pone.0078568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Qi ZC, Zhao YP, Fu CX, Jenny Xiang QY. 2012. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales—influences of gene partitions and taxon sampling. Molecular Phylogenetics and Evolution 64:545–562. 10.1016/j.ympev.2012.05.010 [DOI] [PubMed] [Google Scholar]
- Liu Y, Huo N, Dong L, Wang Y, Zhang S, Young HA, Gu YQ. 2013. Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS ONE 8:e57533 10.1371/journal.pone.0057533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Drechsel O, Bock R. 2007. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Current Genetics 52:267–274. 10.1007/s00294-007-0161-y [DOI] [PubMed] [Google Scholar]
- Maréchal-Drouard L, Weil JH, Dietrich A. 1993. Transfer RNAs and transfer RNA genes in plants. Annual Review of Plant Biology 44:13–32. 10.1146/annurev.pp.44.060193.000305 [DOI] [PubMed] [Google Scholar]
- Osaloo SK, Kawano S. 1999. Molecular systematics of Trilliaceae II. Phylogenetic analyses of Trillium and its allies using sequences of rbcL and matK genes of cpDNA and internal transcribed spacers of 18S–26S nrDNA. Plant Species Biology 14:75–94. 10.1046/j.1442-1984.1999.00009.x [DOI] [Google Scholar]
- Osaloo SK, Utech FH, Ohara M, Kawano S. 1999. Molecular systematics of Trilliaceae I. Phylogenetic analyses of Trillium using matK gene sequences. Journal of Plant Research 112:35–49. 10.1007/PL00013853 [DOI] [Google Scholar]
- Provan J, Corbett G, McNicol JW, Powell W. 1997. Chloroplast DNA variability in wild and cultivated rice (Oryza spp.) revealed by polymorphic chloroplast simple sequence repeats. Genome 40:104–110. 10.1139/g97-014 [DOI] [PubMed] [Google Scholar]
- Provan J, Russell JR, Booth A, Powell W. 1999. Polymorphic chloroplast simple sequence repeat primers for systematic and population studies in the genus Hordeum. Molecular Ecology 8:505–511. 10.1046/j.1365-294X.1999.00545.x [DOI] [PubMed] [Google Scholar]
- Qian J, Song J, Gao H, Zhu Y, Xu J, Pang X, Yao H, Sun C, Li X, Li C, Liu J, Xu H, Chen S. 2013. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8:e57607 10.1371/journal.pone.0057607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi V, Khurana JP, Tyagi AK, Khurana P. 2008. An update on chloroplast genome. Plant Systematics and Evolution 271:101–122. 10.1007/s00606-007-0608-0 [DOI] [Google Scholar]
- Rudall PJ, Stobart KL, Hong W-P, Conran JG, Furness CA, Kite GC, Chase MW. 2000. Consider the lilies: systematics of Liliales. In: Wilson KL, Morrison D, eds. Monocots: systematics and evolution. Collingwood: CSIRO Publishing, 347–359. [Google Scholar]
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research 33:W686–W689. 10.1093/nar/gki366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmickl R, Kiefer C, Dobeš C, Koch MA. 2009. Evolution of trnF(GAA) pseudogenes in cruciferous plants. Plant Systematics and Evolution 282:229–240. 10.1007/s00606-008-0030-2 [DOI] [Google Scholar]
- Shendure J, Ji H. 2008. Next-generation DNA sequencing. Nature Biotechnology 26:1135–1145. 10.1038/nbt1486 [DOI] [PubMed] [Google Scholar]
- Takhtajan A. 1997. Diversity and classification of flowering plants. New York: Columbia University Press. [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30:2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangphatsornruang S, Sangsrakru D, Chanprasert J, Uthaipaisanwong P, Yoocha T, Jomchai N, Tragoonrung S. 2010. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Research 17:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Angiosperm Phylogeny Group. 2009. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Botanical Journal of the Linnean Society 161:105–121. 10.1111/j.1095-8339.2009.00996.x [DOI] [Google Scholar]
- Tsudzuki J, Ito S, Tsudzuki T, Wakasugi T, Sugiura M. 1994. A new gene encoding tRNAPro (GGG) is present in the chloroplast genome of black pine: a compilation of 32 tRNA genes from black pine chloroplasts. Current Genetics 26:153–158. 10.1007/BF00313804 [DOI] [PubMed] [Google Scholar]
- Vijverberg K, Bachmann K. 1999. Molecular evolution of a tandemly repeated trnF(GAA) gene in the chloroplast genomes of Microseris (Asteraceae) and the use of structural mutations in phylogenetic analyses. Molecular Biology and Evolution 16:1329–1340. 10.1093/oxfordjournals.molbev.a026043 [DOI] [PubMed] [Google Scholar]
- Xiong AS, Peng RH, Zhuang J, Gao F, Zhu B, Fu XY, Yao QH. 2009. Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnology Advances 27:340–347. 10.1016/j.biotechadv.2009.01.012 [DOI] [PubMed] [Google Scholar]
- Zhang Y-J, Ma P-F, Li D-Z. 2011. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6:e20596 10.1371/journal.pone.0020596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomlefer WB, Williams NH, Whitten WM, Judd WS. 2001. Generic circumscription and relationships in the tribe Melanthieae (Liliales, Melanthiaceae), with emphasis on Zigadenus: evidence from ITS and trnL-F sequence data. American Journal of Botany 88:1657–1669. 10.2307/3558411 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.