

Abstract
CHARGE syndrome is a rare autosomal dominant developmental disorder involving multiple organs. CHD7 is a major causative gene of CHARGE syndrome. We performed targeted-exome sequencing using a next-generation sequencer for molecular diagnosis of a 4-month-old male patient who was clinically suspected to have CHARGE syndrome, and report a novel monoallelic mutation in CHD7, NM_017780.3(CHD7_v001):c.2966del causing a reading frameshift [p.(Cys989Serfs*3)].
CHARGE syndrome (OMIM 214800) is an autosomal dominant congenital multi-system disorder occurring in an estimated 1:10,000 births worldwide and displaying broad clinical variability.1,2 Hall3 and Hittner et al.4 were the first to independently describe a non-random cluster of malformations that was later referred to as CHARGE association, named by the acronym summarizing the six major clinical features: ocular coloboma, heart malformations, atresia of the choanae, retardation of growth, genital hypoplasia and ear abnormalities.5 The clinical diagnosis of CHARGE syndrome with major and minor criteria was then proposed by Blake et al.6 and updated by Verloes.7 As CHARGE syndrome can closely resemble several other genetic and teratogenic conditions, genetic testing should be emphasized as a useful tool in patients with clinical suspicion.8 Monoallelic mutations or deletions of CHD7 (OMIM 608892), encoding the chromodomain helicase DNA-bindingprotein 7 at chromosome 8q12, are present in 65–70% of patients clinically diagnosed with CHARGE syndrome.9 CHD7 mutations in typical CHARGE syndrome patients occur de novo in the vast majority of the cases.8 There is also evidence that CHARGE syndrome can be caused by mutations in SEMA3E (OMIM 608166) at chromosome 7q21, although only one case has been reported.10
The difficulty in clinical diagnosis of CHARGE syndrome lies in differentiating it from various other diseases sharing its clinical features, such as 22q deletion syndrome, Kabuki syndrome, velo-cardio-facial syndrome, cat eye syndrome, retinoic acid embryopathy, VACTERL association and PAX2 abnormalities.8 Molecular diagnosis is the most accurate and definitive method for differential diagnosis.11,12 Conventional Sanger sequencing has routinely been used to identify disease-causing mutations for genetic diseases, but is laborious, expensive and time-consuming for large genes with numerous exons, such as CHD7 (37 exons covering 8,994 bp of the coding sequence), as well as for multiple candidate genes. Recently, targeted-exome sequencing for candidate or known disease-associated genes using next-generation sequencing (NGS) has been used to diagnose patients with various types of disorders including CHARGE syndrome.13 Here, we report a novel monoallelic frameshift mutation of CDH7, NM_017780.3(CHD7 v001):c.2966delG, detected in a targeted-exome panel for the coding regions of 4,813 clinical phenotype-associated genes including CHARGE syndrome-associated genes in a Japanese patient with CHARGE syndrome.
A 4-month-old male infant was the second child born to non-consanguineous Japanese healthy parents with an unremarkable family history, after uncomplicated pregnancy and delivery at a local hospital. He was born at 38 weeks and 2 days of gestation with a birth weight of 3,086 g (−0.1 s.d.), a length of 47 cm (−1.1 s.d.) and an occipitofrontal circumference of 33.4 cm (+0.1 s.d.). Apgar scores were 9 and 9 at 1 and 5 min, respectively. At birth, he showed cleft lip and palate resulting in poor feeding, and required tube feeding as well as oxygen; he was transferred to our hospital at the age of 11 days. Results of repeatedly performed auditory brainstem response test showed bilateral profound deafness with auditory brainstem response thresholds >95 dB. Computed tomography revealed bilateral hypoplastic semicircular canals and filled tympanic cavities. Two-dimensional color-Doppler echocardiography showed incomplete atrioventricular septal defect, patent ductus arteriosus, aortic valve stenosis and bicuspid aortic valve. As the child had ventilation dysfunction due to glossoptosis and upper airway obstruction detected by multi-detector row computed tomography were observed, he required artificial ventilation with endotracheal intubation at 14 days, and was subjected to a tracheotomy owing to difficulties of extubation at 81 days. He still required nasogastric tube feeding owing to swallowing difficulty. Symptoms of heart failure due to the progression of aortic valve stenosis developed at 90 days. He showed dysmorphic features, including choanal stenosis, bilateral low-set ears, micropenis and cryptorchidism. His motor development, such as head control and rolling over, was almost normal. Karyotype analysis revealed a normal male karyotype of 46,XY. Biochemical and gas chromatography-mass spectrometry metabolic disorder tests were negative, with no evidence of metabolic disease. No clinical characteristics of CHARGE syndrome were detected in the patient’s parents. Although coloboma was not observed, the patient was suspected to have CHARGE syndrome because two of four major characteristics (choanal atresia and characteristic outer and inner ears, and deafness) and three minor characteristics (cardiovascular malformation, genital hypoplasia and cleft lip and palate) of CHARGE syndrome were observed (Table 1).8
Table 1. Symptoms of the patient compared with major and minor diagnostic criteria in CHARGE patients8 .
| Diagnostic criteria | Symptoms of the patient | Frequency in CHARGE patients8 |
|---|---|---|
|
Major
| ||
| Ocular coloboma | − | 80–90% |
| Choanal atresia or stenosis | + | 50–60% |
| Cranial nerve dysfunction | Unclear (VIII, IX/X) | Frequent (VIII) or 70–90% (IX/X) |
| Characteristic CHARGE syndrome ear | + (Characteristic outer and inner ears and deafness) | 80–100% |
|
Minor
| ||
| Genital hypoplasia due to congenital hypogonadotropic hypogonadism | + (Micropenis, cryptorchidism) | 50–60% (male; micropenis, cryptorchidism) |
| Developmental delay | − | ⩽100% |
| Cardiovascular malformations | + | 75–85% |
| Growth deficiency | − | 70–80% |
| Orofacial cleft | + | 15–20% |
| Tracheoesophageal fistula | − | 15–20% |
| Distinctive facial feature | − | 70–80% |
Molecular diagnosis was performed using genomic DNA extracted from a blood sample, after informed consent was obtained from the parents. The study was approved by the ethics committees of Tokushima University. To screen known disease-associated genes including CHD7, we first used a MiSeq bench-top sequencer (Illumina, San Diego, CA, USA) to perform NGS with a TruSight One Sequencing Panel (Illumina), and our pipeline for NGS data analysis as described elsewhere.14,15 To identify presumably pathogenic single-nucleotide variants (SNVs), we excluded sequence variants with minor allele frequency >0.05 from the 1000 Genomes Project database (http://www.1000genomes.org/), NHLBI Grand Opportunity Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS/) and the Human Genetic Variation Database (HGVD, http://www.genome.med.kyoto-u.ac.jp/SnpDB/). Copy number aberration (CNA) analysis was also performed as described elsewhere.16 These analyses identified a monoallelic, one-base (guanine) deletion of CHD7, NM_017780.3(CHD7_v001):c.2966del (NM_017780.3:n.3458del), which was confirmed by Sanger sequencing (Figure 1a). Primer sequences are available upon request. This mutation causes a reading frameshift starting from residue Cys989, with the new reading frame ending with a premature stop codon two codons downstream, NM_017780.3(CHD7_i001):p.(Cys989Serfs*3). As shown in Figure 1, the mutation is located in exon 12, encoding the SNF2 family N-terminal (SNF2 N) and DEAD-like helicase superfamily including an ATP-binding (DEXDc) domains.9,17 This mutation is not present in dbSNP138, 1000 Genomes Project, ESP6500 and HGVD. The mutation is also not present in the Human Gene Mutation Database professional 2015.2 (HGMD, http://www.hgmd.org/) or ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), suggesting it to be novel. No other potentially pathogenic SNVs and CNAs were detected in genes possibly responsible for CHARGE syndrome or other related diseases.17 The mutation was not confirmed as de novo because of the unavailability of parental DNA.
Figure 1.
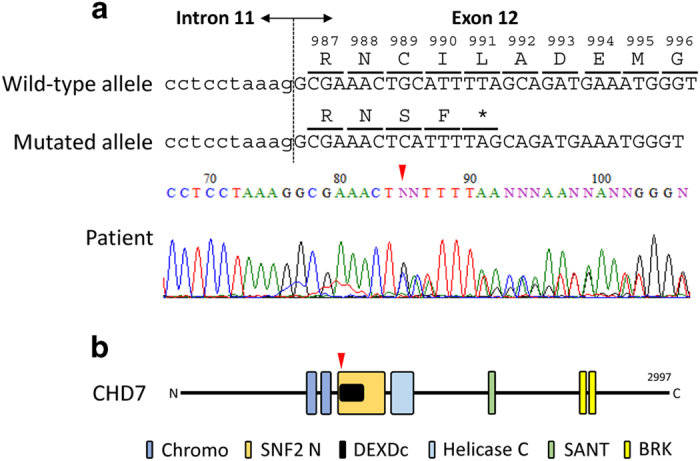
(a) Electropherogram of CHD7 (NM_017780.3) exon 12 and flanking intron 11 sequences showing the monoallelic germline mutation NM_017780.3:c.2966del (arrowhead) in the patient’s DNA. The DNA and corresponding amino acid sequences of the wild-type and mutant CHD7 alleles are also shown. (b) A schematic representation of the CHD7 protein. Domains are depicted approximately to the scale adapted from human reference sequence (NM_017780.3(CHD7_i001)) and reference 17. The arrowhead shows the position corresponding to the mutation. BRK, Brahma and Kismet domain; Chromo, chromodomain; DEXDC, DEAD-like helicase superfamily including an ATP-binding domain; Helicase C, helicase superfamily C-terminal domain; SANT, switching-defective protein 3, adaptor 2, nuclear receptor corepressor, transcription factor IIIB domain; SNF2 N, SNF2 family N-terminal domain.
In HGMD professional 2015.2, 615 CHD7 mutations were reported in CHARGE syndrome. The majority of these mutations are nonsense, frameshift or splice site mutations, which are predicted to be loss-of-function mutations, likely leading to an aberrant messenger RNA targeted for degradation via nonsense-mediated decay (NMD).18 Therefore, haploinsufficiency for CHD7 is the most likely pathogenic mechanism underlying CHARGE syndrome.18 Even if the mutated transcript escapes NMD, the truncated CHD7 protein predicted in this patient contains only the N-terminal tandem chromodomains, which are incapable of performing the protein’s functions owing to the loss of chromatin remodeling activity (Figure 1b).
NGS emergence for the use in diagnostic laboratories will facilitate the characterization of new sequence variants in patients with genetic disorders. In a diagnostic setting for CHARGE syndrome, CHD7 mutation analysis using Sanger sequencing may miss genetic alterations causing CHARGE syndrome or phenocopies of CHARGE syndrome, such as gross deletions or deep intronic mutations of CHD7, alterations in SEMA3E, unique chromosomal imbalances and 22q11.2 deletion syndrome.17 Given that targeted-exome or whole-exome sequencing can detect both exonic SNVs and CNAs in disease-causing genes,15,16 those platforms can facilitate differential diagnosis, appropriate therapy and genetic counseling for patients with this syndrome and related diseases in a cost-effective manner. Coverage of all genetic alterations causing CHARGE and CHARGE-like diseases will be extended with whole-genome sequencing when the cost of sequencing is lowered.
Acknowledgments
Authors would like to thank the patient and his family for participation in this study. This work was supported by JSPS KAKENHI Grant Numbers 26293304 (II) and 15K19620 (TN) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
The authors declare no conflict of interest.
References
- Issekutz KA , Graham JM Jr , Prasad C , Smith IM , Blake KD . An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A 2005; 133: 309–317. [DOI] [PubMed] [Google Scholar]
- Sanlaville D , Verloes A . CHARGE syndrome: an update. Eur J Hum Genet 2007; 15: 389–399. [DOI] [PubMed] [Google Scholar]
- Hall BD . Choanal atresia and associated multiple anomalies. J Pediatr 1979; 95: 395–398. [DOI] [PubMed] [Google Scholar]
- Hittner HM , Hirsch NJ , Kreh GM , Rudolph AJ . Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation—a syndrome. J Pediatr Ophthalmol Strabismus 1979; 16: 122–128. [DOI] [PubMed] [Google Scholar]
- Pagon RA , Graham JM Jr , Zonana J , Yong SL . Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 1981; 99: 223–227. [DOI] [PubMed] [Google Scholar]
- Blake KD , Davenport SL , Hall BD , Hefner MA , Pagon RA , Williams MS et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1998; 37: 159–173. [DOI] [PubMed] [Google Scholar]
- Verloes A . Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 2005; 133A: 306–308. [DOI] [PubMed] [Google Scholar]
- Lalani SR , Hefner MA , Belmont JW , Davenport SLH . CHARGE syndrome. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJH (eds). GeneReviews [Internet]. Seattle, WA, USA: University of Washington, 2006. Oct 2 [updated 2012 Feb 2]. [Google Scholar]
- Vissers LE , van Ravenswaaij CM , Admiraal R , Hurst JA , de Vries BB , Janssen IM et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004; 36: 955–957. [DOI] [PubMed] [Google Scholar]
- Lalani SR , Safiullah AM , Molinari LM , Fernbach SD , Martin DM , Belmont JW . SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet 2004; 41: e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake KD , Prasad C . CHARGE syndrome. Orphanet J Rare Dis 2006; 1: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsten-Janssen N , Saitta SC , Hoefsloot LH , McDonald-McGinn DM , Driscoll DA , Derks R et al. More clinical overlap between 22q11.2 deletion syndrome and CHARGE syndrome than often anticipated. Mol Syndromol 2013; 4: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J , Ma D , Wu Y , Luo C , Huang C , Hu P et al. Identification of one novel CHD7 mutation in a patient from China with atypical CHARGE syndrome. Gene 2015; 571: 298–302. [DOI] [PubMed] [Google Scholar]
- Okamoto N , Naruto T , Kohmoto T , Komori T , Imoto I . A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum Genome Variation 2014; 1: 14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruto T , Okamoto N , Masuda K , Endo T , Hatsukawa Y , Kohmoto T et al. Deep intronic GPR143 mutation in a Japanese family with ocular albinism. Sci Rep 2015; 5: 11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K , Naruto T , Tanimoto K , Yasukawa C , Oikawa Y , Masuda K et al. Simultaneous detection of both single nucleotide variations and copy number alterations by next-generation sequencing in Gorlin syndrome. PLoS ONE 2015; 10: e0140480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen N , Bergman JE , Swertz MA , Tranebjaerg L , Lodahl M , Schoots J et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 2012; 33: 1149–1160. [DOI] [PubMed] [Google Scholar]
- Zentner GE , Layman WS , Martin DM , Scacherin PC . Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A 2010; 152A: 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Citations
- Imoto Issei.HGV Database. 2016. 10.6084/m9.figshare.hgv.777. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Imoto Issei.HGV Database. 2016. 10.6084/m9.figshare.hgv.777. [DOI]