

Abstract
Objectives
Antigen-specific CD4+ T cells epigenetically modified with DNA methylation inhibitors overexpress genes normally suppressed by this mechanism, including CD11a, CD70, CD40L and the KIR gene family. The altered cells become autoreactive, losing restriction for nominal antigen and responding to self-class II major histocompatibility complex (MHC) molecules without added antigen, and are sufficient to cause a lupus-like disease in syngeneic mice. T cells overexpressing the same genes are found in patients with active lupus. Whether these genes are co-overexpressed on the same or different cells is unknown. The goal of this study was to determine whether these genes are overexpressed on the same or different T cells and whether this subset of CD4+ T cells is also present in patients with lupus and other rheumatic diseases.
Methods
Multicolour flow cytometry was used to compare CD11a, CD70, CD40L and KIR expression on CD3+CD4+CD28+ T cells to their expression on experimentally demethylated CD3+CD4+CD28+ T cells and CD3+CD4+CD28+ T cells from patients with active lupus and other autoimmune diseases.
Results
Experimentally demethylated CD4+ T cells and T cells from patients with active lupus have a CD3+CD4+CD28+CD11ahiCD70+CD40LhiKIR+ subset, and the subset size is proportional to lupus flare severity. A similar subset is found in patients with other rheumatic diseases including rheumatoid arthritis, systemic sclerosis and Sjögren's syndrome but not retroperitoneal fibrosis.
Conclusions
Patients with active autoimmune rheumatic diseases have a previously undescribed CD3+CD4+CD28+CD11ahiCD70+CD40LhiKIR+ T cell subset. This subset may play an important role in flares of lupus and related autoimmune rheumatic diseases, provide a biomarker for disease activity and serve as a novel therapeutic target for the treatment of lupus flares.
Keywords: Systemic Lupus Erythematosus, T Cells, Kir, Autoimmune Diseases, DNA Methylation
Introduction
Epigenetically altered CD4+ T cells play a crucial role in human lupus flares. Reports that lupus goes into remission as CD4+ T cell numbers decline in patients with AIDS,1 2 and that anti-CD4 antibodies treat lupus in NZB/W and MRL/lpr mice,3 4 indicate that CD4+ T cells are necessary for lupus disease activity. Our group reported that treating human or mouse CD4+ T cells with the DNA methylation inhibitor 5-azacytidine (5-azaC) alters gene expression and makes the cells autoreactive,5 and that the epigenetically modified murine T cells are sufficient to cause a lupus-like disease with anti-DNA antibodies and an immune complex glomerulonephritis when injected into syngeneic mice.6
Genes such as the KIR gene family, CD11a, CD70 and CD40L, overexpressed by experimentally demethylated CD4+ T cells are also demethylated and overexpressed by CD4+ T cells from patients with active lupus.7 The number of T cells overexpressing these genes is directly related to lupus disease activity as measured by the systemic lupus erythematosus disease activity index (SLEDAI).7 8 However, whether KIR, CD11a, CD70 and CD40L are aberrantly overexpressed on the same CD4+ T cell, or on different T cells, is unknown. This is important to determine because coexpression on the same cell would lead to the development of new and safer treatments directed at eliminating this pathogenic subset.
A growing body of evidence has established that two or more of the related autoimmune rheumatic diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), systemic sclerosis (SSc) and Sjögren's syndrome can develop both within the same person and within families at higher rates than expected by chance.9–11 Characterisation of commonalities across these autoimmune diseases may yield important insights into their pathogenesis and treatment, and elucidation of the pleiotropic genetic and environmental risk factors for autoimmunity is an active area of investigation.12–14 Thus, to determine whether the epigenetically altered T cell subset is unique to SLE or has broader implications for these related forms of autoimmunity, we extended our studies to include RA, Sjögren's syndrome and SSc, as well as retroperitoneal fibrosis (RPF), a fibroinflammatory disease that can exist as an idiopathic process, as part of an IgG4-related (IgG4-RD) inflammatory disease, or in association with underlying disease such as malignancy or connective tissue disease including SLE and anti-neutrophil cytoplasmic antibody-associated vasculitis.15 16 RPF is not associated with antinuclear antibody production.
The present study uses multicolour flow cytometry to determine whether CD11a, CD40L, CD70 and the KIR genes are co-overexpressed on the same or different CD3+CD4+CD28+ T cells using T cells experimentally demethylated with 5-azaC and T cells from patients with active lupus. We also determined whether the size of the epigenetically altered T cell subset is related to lupus disease activity and whether the subset is present in patients with these related autoimmune rheumatic diseases.
Methods
Subjects
Patients with SLE, RA, SSc, Sjögren's syndrome and RPF were recruited from the rheumatology and nephrology outpatient clinics at the University of Michigan. Healthy controls, ages 23–64 years, were recruited by advertising. Study subjects' information is shown in online supplementary table S1. A total of 54 patients, 19 with SLE, 9 with RA, 12 with SSc, 8 with RPF and 6 with Sjögren's were recruited. In addition, five healthy control subjects were recruited for the present study. Patients with Sjögren's syndrome met criteria for the classification of primary Sjögren's syndrome,17 and disease activity was determined using the European League Against Rheumatism (EULAR) Sjögren's syndrome disease activity index.18 Patients with lupus met the American College of Rheumatology classification criteria for SLE,19 20 and disease activity was determined by SLEDAI.8 Patients with RA met the 2010 criteria for the classification of RA.21 RA disease activity was assessed on a qualitative 0–4 scale with 1 point given for each of the following criteria: (1) provider added an immunosuppressant or increased the dose of a currently prescribed immunosuppressant during the visit, (2) patient reported morning stiffness >30 min, (3) provider documented synovitis was present on exam, and (4) ESR or C-reactive protein (CRP) above normal or increased from prior visit when judged to be inactive.
Study Subjects
lupus-2016-000147supp_tableS1.pdf (208.4KB, pdf)
Patients receiving cyclophosphamide were excluded because of effects on T cell surface marker expression.22 Patients with SSc were further classified as sine scleroderma (no skin involvement), limited cutaneous SSc or diffuse cutaneous SSc based on skin examination and extent of involvement assessed using the modified Rodnan skin score (MRSS).23 Only subjects with idiopathic RPF were recruited. However, following the conclusion of the study, one patient was found to have IgG4-RD and a positive biopsy. The remaining seven patients were still deemed to have idiopathic RPF without IgG4-RD. The RPF diagnosis was confirmed by expert opinion of study rheumatologist (WM) and nephrologist (RS) as no standardised classification criteria for RPF currently exists. The diagnosis is suspected by clinical presentation with back pain and ureteral obstruction associated with characteristic imaging abnormalities, elevated in inflammatory markers, and tissue revealing fibrotic and non-specific lymphocytic infiltrates negative for malignancy.24 The majority of patients with RPF had characteristic CT imaging and ureteral obstruction.
T cell isolation and culture
Peripheral blood mononuclear cells (PBMCs) were isolated from the venous blood of patients or healthy controls by Ficoll density gradient separation (Histopaque-1077, Sigma Aldrich, St Louis, Missouri, USA). Where indicated, PBMCs from healthy female donors were stimulated with 1 µg/mL phytohemagglutinin (Remel, Lenexa, Kansas, USA) for 18–24 h at 37°C in a 5% CO2/balanced air environment, followed by treatment with 2.5 µM 5-azaC (Sigma Chemical Co, St Louis, Missouri, USA) for 3 days as previously described.25
Antibodies and flow cytometric analyses
The following antibodies were used: Phycoerytherin (PE)-anti-Kir2DL4/CD158D (clone 181703; R&D Systems, Minneapolis, Minnesota, USA), anti-CD40L-biotin (clone hCD40L-M91) and avidin-PECy7, PE-Cy7-anti-CD8 (clone RPA-T8), anti-CD11a-APC (clone HI111), Pacific Blue-anti-CD3 (clone UCHT1), PECy5-anti-CD28 (clone CD28.2), FITC-anti-CD70 (clone Ki-24) and APC-Cy7-anti-CD4 (clone RPA-T4) (Becton Dickinson, Franklin Lakes, New Jersey, USA). PE-anti-CD158b (clone CH-L), PE-anti-CD158i (clone FES172), PE-anti-CD158b1/b2, j (clone GL183) and PE-anti-CD158a,h (clone EB6B) were from Beckman Coulter (Brea, California, USA). All antibodies were titrated to determine their optimal concentrations prior to use.
PBMC from patients with autoimmune diseases or from cultures of 5-azaC-treated PBMC from healthy donors were incubated in phosphate-buffered saline (PBS)/0.001% azide containing 10% horse serum at 4°C for 30 min to block non-specific binding. All staining procedures were performed at 4°C in the dark. The cells were then washed, incubated for 60 min, with a single or mixture of fluorochrome-conjugated antibodies. Biotin-CD40L-stained cells were further washed and incubated for 30 min with a 1:1000 dilution of avidin-PE-Cy7, fixed with 4% paraformaldehyde and stored in PBS/0.001% azide in the dark at 4°C until analysed. Controls included isotype and fluorochrome matched antibodies, single antibodies and ‘full minus one’ (FMO) staining controls in which one of each of the fluorochrome-conjugated antibodies was serially omitted while retaining the rest.
Fluorescence-activated cell sorting (FACS) analyses were performed using a FACS ARIA IIIU flow cytometer and FACSDiva software V.6.1.3 (Becton Dickinson, New Jersey, USA) or an iCyte Synergy (Sony Biotechnology, San Jose California, USA) and WINLIST V.8 (Verty Software House, http://www.vsh.com) software. The fluorescent antibody conjugates, laser emission wavelengths and filter sets used are shown in online supplementary table S2.
Flow Cytometry Instrument Configuration
lupus-2016-000147supp_tableS2.pdf (15.7KB, pdf)
Statistical analysis
The difference between two means was evaluated with Student's t test, and between subset size and disease activity by regression analysis using GraphPad Prism V.6 software. The median and SD of populations identified by flow cytometry were calculated using the statistical functions within the FlowJo software (FlowJo, Ashland Oregon, USA).
Results
We first analysed the expression of methylation-sensitive genes in 5-azaC-treated cells from healthy subjects. PBMCs from a healthy woman were stimulated with phytohemagglutinin (PHA), then cultured with or without 5-azaC. Seventy-two hours later, the cells were washed, stained with fluorochrome-conjugated antibodies to CD3, CD4, CD28, CD11a, CD70 and CD40L with and without a ‘cocktail’ of PE conjugated antibodies to five distinct KIR proteins, then analysed by flow cytometry as outlined in figure 1. An example of the gating strategy used to select the T cell subsets and establish negative and positive staining for each antibody is shown in the online supplementary figure. Doublets were excluded based on forward and side scatter (panels A–C), thus assuring analysis of single cells. Greater than 95% of CD3+CD4+ T cells from healthy donors were CD28+ (panel E), a value typical for young healthy donors, although this per cent declines somewhat with age.26 Panel F shows CD11a and KIR expression on the CD3+CD4+CD28+ T cells selected in panel E. CD3+CD4+CD28+ KIR+ T cells were heterogeneous in their KIR expression. Since a cocktail of five antibodies directed to different KIR gene products was used to provide the broadest coverage of KIR expression, the heterogeneity observed may reflect different clones, different amounts of a given KIR isotype on the cells or heterogeneity in the assortment of KIR genes inherited by any given person.27
Figure 1.
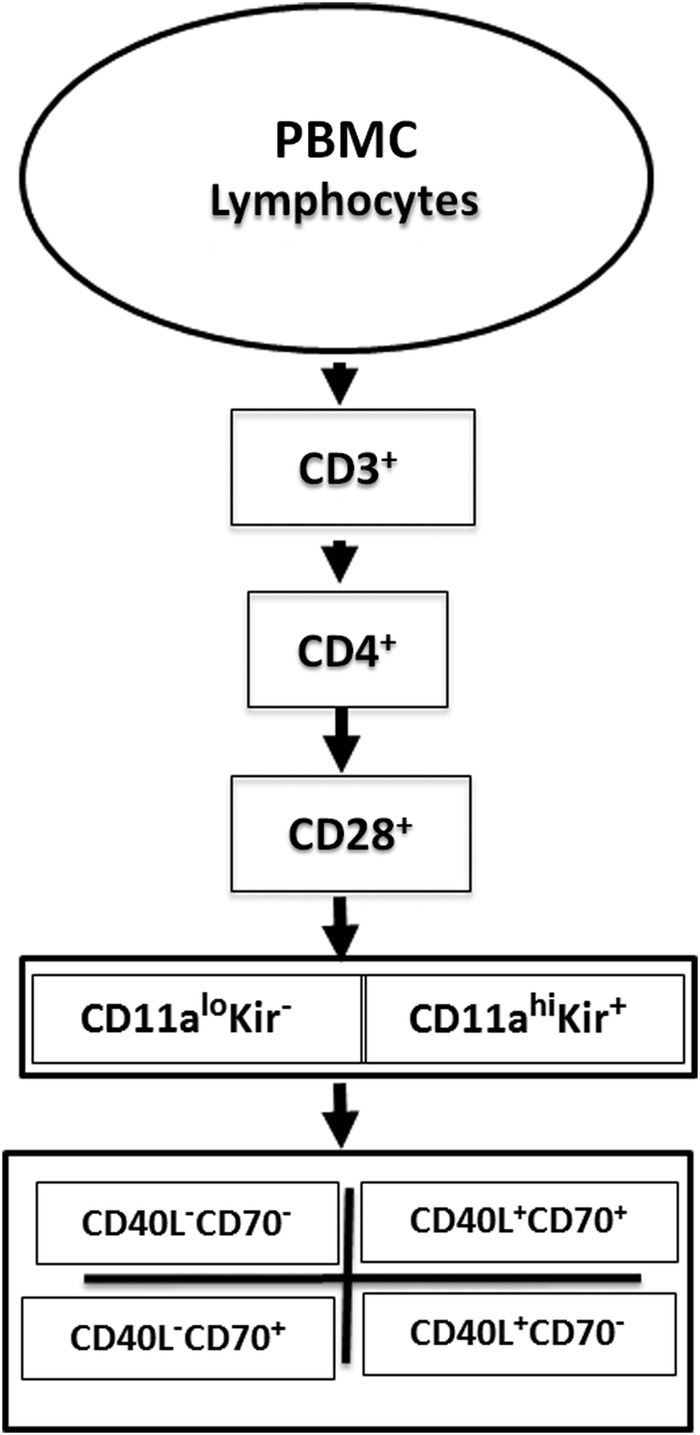
Flow cytometric analysis strategy. Peripheral blood mononuclear cell (PBMC) from patients with systemic autoimmune rheumatic diseases or PHA stimulated, 5-azacytidine-treated PBMC from healthy subjects were stained with fluorochrome-conjugated antibodies to CD3, CD4, CD28, CD11a, CD70, CD40L and the KIR gene family, then analysed by gating on CD3+CD4+CD28+ cells and comparing the levels of KIR and CD11a, then CD40L and CD70 on the cells.
lupus-2016-000147supp_figure.pdf (386KB, pdf)
Figure 2 shows an example of KIR and CD11a expression by PHA-stimulated untreated and 5-azaC-treated T cells from healthy subjects. In the absence of treatment with the demethylating agent 5-azaC, very few KIR+ cells were observed (figure 2A). The small number of KIR+ cells observed in the peripheral blood of healthy people varies and increases somewhat with age.28 Treatment with 5-azaC increases the numbers of KIR+ cells (figure 2B), as previously described by our group.29 Further, CD11a expression on the majority (>70%) of the KIR+CD3+CD4+CD28+ cells treated with 5-azaC (figure 2B, rectangle) was nearly twice that of the corresponding KIR− T cells (figure 2B, circle) from the same cultures (median fluorescence intensity 2298±748 SD vs 1394±572, respectively, n=9, analysed in seven different experiments, p<0.001). We therefore referred to the KIR+ subset as CD3+CD4+CD28+CD11ahiKIR+. While all CD4+ T cells express CD11a, overexpression of CD11a confers autoreactivity on CD4+ T cells.11 Single antibody (PE-KIRs) and FMO (full staining minus PE-KIRs) staining demonstrated that the KIR staining was specific (figure 2C, D respectively).
Figure 2.

Example of 5-azacytidine (5-azaC) treatment of PHA-stimulated peripheral blood mononuclear cell (PBMC) induces the expression of KIR proteins on a subset of CD4+CD28+CD11ahi T cells. PBMCs from a healthy female donor were stimulated for 18–24 h with PHA followed by treatment with 2.5 µM 5-azacytidine or saline for 72 h. The cells were then stained with fluorochrome-conjugated antibodies to methylation-sensitive gene products, fixed and analysed by flow cytometry as described in ‘Methods’. Gating was established to select CD3+CD4+CD28+ T cells. The CD3+CD4+CD28+ T cells were then analysed for CD11a and KIR using a cocktail of five PE-conjugated anti-KIR antibodies and imaged on a FACS Synergy flow cytometer. (A) Untreated T cells. (B) 5-azaC-treated T cells. (C) 5-azaC-treated PBMC stained only with the cocktail of anti-KIR antibodies. (D) 5-azaC-treated T cells stained with antibodies to CD3, CD4, CD28, CD11a, CD70 and CD40L but not KIR. The figure shown is representative of nine separate cultures.
Epigenetically altered T cells in autoimmune diseases
We next tested whether patients with active lupus had a similar CD3+CD4+CD28+CD11ahiKIR+ subset. Figure 3A shows an example of CD11a and KIR expression on CD3+CD4+CD28+ T cells from one of six patients with inactive SLE. Less than 1% of CD3+CD4+CD28+ cells were also KIR+ (rectangle). However, the per cent of CD3+CD4+CD28+KIR+ cells increased with SLE disease activity (figure 3B, C). The KIR+ T cells also overexpressed CD11a. Evidence for the CD3+CD4+CD28+CD11ahiKIR+ T cell subset in other systemic autoimmune rheumatic diseases was tested in nine patients with active and inactive RA and 12 patients with SSc. Controls included T cells from patients with RPF, an idiopathic inflammatory fibrosing disorder30 and healthy age-matched donors. Background levels of CD3+CD4+CD28+CD11ahiKIR+ T cells were observed in patients with RA and SSc with inactive RA or sine/limited SSc (figure 3D, F). As in the patients with SLE, the size of the KIR+ subset was larger in patients with active RA (figure 3E) and more extensive SSc (figure 3G) and appeared to express more CD11a protein than the KIR− cells. In contrast, the size of the KIR+ subset in patients with RPF was comparable to that of the CD3+CD4+CD28+KIR+ subset in healthy controls and did not increase with disease activity (figure 3H, I).
Figure 3.

CD3+CD4+CD28+CD11ahiKIR+ T cells in patients with rheumatic autoimmune diseases. The selected examples are representative of 4–7 patients for each level of disease activity. (A) Inactive systemic lupus erythematosus (systemic lupus erythematosus disease activity index (SLEDAI) 0–2, n=6). (B) Mildly active lupus (SLEDAI 4–5, n=7). (C) Active lupus (SLEDAI ≥6 , n=6). (D) Inactive rheumatoid arthritis (RA) (n=5). (E) Active RA (n=4). (F) Diffuse/limited systemic sclerosis (n=4). (G) Diffuse systemic sclerosis (n=8). Retroperitoneal fibrosis (n=8) served as a control. Disease activity was clinically determined as described in the ‘Methods’. Gating was established based on single antibody and full minus one controls for each experiment. The rectangles indicate KIR+ cells selected in quadrant 2. Numbers above the rectangles are the per cent of total cells. MRSS, modified Rodnan skin score.
The significance of the relationship between the size of the epigenetically altered T cell subset and disease activity was investigated. Figure 4 shows there was a direct relationship between the subset size and the SLEDAI (n=19, p=0.0004, R2=0.52 by linear regression, figure 4A). Similarly, the relationship between the size of the KIR+ subset and RA and SSc disease activity was significant (figure 4B, C). The subset was seen in two of the six patients with Sjögren's syndrome (not shown); however, four of these subjects had a EULAR Sjögren's syndrome disease activity index31 of 0 and two had a score of 1, with no relation of the score to subset size. Four of these subjects were positive for Sjogren's Syndrome antigen A (SSA) and Sjogren's Syndrome antigen B (SSB), and of the other two, one was positive for rheumatoid factor and the other for anti-DNA antibodies. Further studies of the KIR+ subset in Sjögren’s patients with more active disease are underway. The subset was not seen in eight patients with RPF, two of whom were judged to have active disease as determined by the clinical criteria described by Swartz.24
Figure 4.

Relationship between disease activity and size of the CD3+CD4+CD28+CD11ahiKIR+ T cell subset in patients with rheumatic diseases. (A–C) The size of the CD3+CD4+CD28+CD11ahiKIR+ T cell subset in patients is plotted against their disease activity/involvement scores. p Values shown were determined by linear regression. Demographic and disease information for patients and controls analysed is shown in online supplementary table S1. (D) Size of the CD3+CD4+CD28+CD11ahiKIR+ T cell subset from A to C. The numbers in the bars represent the number of subjects studied, and results are presented as the mean±SEM. p Values shown were calculated using Student's t test. Control, healthy subjects, retroperitoneal fibrosis (RPF), systemic lupus erythematosus (SLE) and systemic lupus erythematosus disease activity index (SLEDAI) scores 0–2, 4–5 and ≥6, inactive rheumatoid arthritis (RA(I)), active RA (RA(A)), sine/limited systemic sclerosis (SSc (modified Rodnan skin score (MRSS) 0–2), diffuse systemic sclerosis (SSc (MRSS 9–26)) and Sjögren's syndrome.
Coexpression of other methylation-sensitive genes
The data above show that the CD4+ T cells from patients with active lupus-related autoimmune diseases as well as patients with Sjögren's syndrome express elevated levels of CD11a and KIR and that the per cent of the KIR+ cells correlates with disease activity in patients with SLE, RA and SSc. We next asked whether the methylation-sensitive genes CD70 and CD40L were also expressed on the KIR+ T cells and to what level they were coexpressed on KIR− T cells. All studies of CD40L activation were confined to women because CD40L is encoded on the X chromosome, one of which is inactivated by DNA methylation in women, so female CD4+ T cells demethylated with 5-azaC or in women with active SLE overexpress CD40L while CD40L expression is not affected in male T cells.32 Examples of coexpression in 5azaC-treated T cells from a healthy woman and PBMC from a woman with active SLE are shown in figure 5. Nearly all CD4+ T cells also express CD40L (figure 5B, C, E, F, dotted line). However, with experimentally induced DNA demethylation and in patients with active SLE, CD40L expression is elevated in Kir+ T cells32 (figure 5B, E, G) compared with Kir− counterparts (figure 5C, F, G). In women with active SLE (SLEDAI 4–8), the majority of the CD4+CD28+CD11ahi KIR+ CD40Lhi cells were also CD70+ (figure 5G, geometric mean 71±8% SEM, range 45–95%, n=5, p=0.001 vs Kir−). In women with active RA, 69±8% SEM (n=5, range 55–94%, p=0.004) of their CD4+CD28+CD11ahiKIR+CD40Lhi T cells also overexpressed CD70. Similar results (64%±10%, n=5, p=0.007) were observed in women with diffuse/MRSS >9 SSc). In contrast, 3% or less of the CD4+CD28+CD11a+KIR− T cells from 5azaC-treated healthy controls or patients with SLE, RA or SSc overexpressed CD40L and only 10% of expressed CD70 protein. A small number (<3%) of CD4+CD28+CD11a+KIR− cells expressed CD70 but did not exhibit elevated levels of CD40L (figure 5F).
Figure 5.

CD70/CD40L coexpression on KIR+ versus KIR−CD4+CD28+ T cells. The dotted line demarks positive from negative CD40L staining. (A) Peripheral blood mononuclear cell (PBMC) from a healthy female donor were stimulated 18–24 h with PHA followed by treatment with 5-azacytidine (5-azaC) for an additional 72 h as described in ‘Methods’. CD3+CD4+CD28+CD11a+Kir+ (rectangle) or Kir− (circle) cells were gated and analysed for their expression of CD70 and CD40L. PBMCs from one of the female systemic lupus erythematosus (SLE) patients with active disease (systemic lupus erythematosus disease activity index 6) shown in figure 4 were similarly gated and analysed for their CD70 and CD70 coexpression (D–F). (G) CD70 and CD40L expression on Kir+ and Kir− cells as depicted in (B, C) and (E, F) above. Data are presented as the geometric mean± SEM per cent expression of groups of six 5azaC, five SLE, five rheumatoid arthritis (RA) and five systemic sclerosis (SSc) patients. CD40L overexpression (CD40Lhi) is calculated as the per cent values in (Q2+Q4) for Kir+ (B and E) and Kir− (C and F). Per cent CD40Lhi that also expresses CD70 is calculated as (Q2/(Q2+Q3)) (C and F). *p=0.03–0.01; **p<0.01 Kir+ vs Kir− by Student's t test.
Discussion
Previous work from our group demonstrates that inhibiting DNA methylation in human CD4+ T cells with the Dnmt inhibitor 5-azaC causes demethylation and overexpression of genes including CD11a, CD70, CD40L, perforin and the KIR gene family, making the cells autoreactive, cytotoxic and pro-inflammatory.6 33 We also reported that CD4+ murine T cells treated with 5-azaC overexpress similar genes and also become autoreactive, and that the epigenetically altered murine T cells are sufficient to cause a lupus-like disease when injected into syngeneic recipients.34 CD4+ T cells from patients with active lupus have hypomethylated DNA, overexpress the same genes as those activated by DNA methylation inhibitors and similarly become autoreactive,6 suggesting that the demethylated T cells may contribute to flares of human lupus as they do in the murine models.6 More recently, we found that oxidative stress, such as that caused by H2O2 and ONOO−, also causes T cell DNA demethylation in vitro,35 and that CD4+ T cells treated with these oxidisers also cause a lupus-like disease in mice.36 Since lupus flares are characterised by oxidative stress as evidenced by extensive protein nitration,37 our observations suggest that antioxidants may help treat lupus flares. This is supported by reports that the antioxidant N-acetylcysteine decreases human lupus flare severity.38
The present study extends these reports by demonstrating that the genes activated in experimentally demethylated T cells39 are all overexpressed on a previously undescribed CD4+CD28+ T cell subset found in patients with active lupus, and that the size of this subset is directly proportional to lupus disease activity as measured by the SLEDAI. Since experimentally demethylated murine CD4+ T cells are sufficient to cause a lupus-like disease in lupus-prone (SJL) mice,40 the epigenetically altered T cell subset described above may similarly contribute to human lupus flares, making the subset a therapeutic target for agents or antibodies directed at gene products uniquely expressed by this subset such as KIR proteins.
The human KIR gene locus encodes approximately 15 KIR genes. The KIR genes are expressed clonally on natural killer (NK) cells, and the non-expressed genes are silenced by DNA methylation. Treating NK cells with DNA methylation inhibitors like 5-azaC activates expression of the KIR family.41 Normal T cells do not express KIR genes. However, treating CD4+ T cells with 5-azaC also activates expression of the KIR gene family.42 Since KIR genes are expressed clonally on NK cells, but the KIR gene family is expressed on experimentally demethylated CD4+ T cells and CD4+ T cells from patients with active lupus,29 39 antibodies to a single KIR protein will deplete the epigenetically altered KIR+ subset in lupus patients, while only affecting a clone of NK cells.
KIR is also expressed on ‘senescent’ CD4+CD28− T cells that develop with ageing and in chronic inflammatory diseases, and are implicated in atherosclerotic plaque development and rupture.25 This suggests that anti-KIR antibodies may also prevent atherosclerotic plaque development in lupus patients as well as others with CD4+CD28− T cells. These studies are the subject of ongoing investigations (manuscript in preparation).
Interestingly, the same epigenetically altered CD4+CD28+ T cell subset is also found in patients with related autoimmune rheumatic diseases including RA, SSc and possibly Sjögren's syndrome, and the size of the subset is similarly related to disease activity in RA and to disease activity in SSc. The studies summarised above focused primarily on lupus, so it is possible that the subset may contribute to only the development of antinuclear antibodies in these other diseases,43 and that inflammation generated by flares of RA or other autoimmune diseases is responsible for the presence of the subset in these patients. However, this epigenetically altered autoreactive lupus T cell subset, which responds to self-class II MHC molecules without added antigen,5 resembles the semi-allogeneic T cells that respond to host class II MHC molecules in the murine chronic graft-versus-host disease model. In this model, semi-allogeneic T cells cause a spectrum of autoimmune diseases that resemble human RA, lupus, Sjögren's syndrome and SSc, depending on the genetic background of the mice.44 Similarly, patients receiving allogeneic bone transplants can develop graft-versus-host disease resembling SSc, Sjögren's syndrome or RA.45 Further, pristane, which stimulates oxidative stress when injected into mice,46 causes a lupus-like disease in lupus-prone SJL mice,47 but induces a T cell-dependent inflammatory arthritis resembling human RA in CBA mice48 and a disease with features of both RA and lupus in Balb/c mice.49 Myeloperoxidase suppresses the development of autoimmunity and renal disease in the pristane-induced lupus model,50 supporting a role for oxidative stress in this model. This suggests that the epigenetically altered T cell subset described above may contribute to the pathogenesis of the other autoimmune rheumatic diseases as well as overlap syndromes such as ‘rhupus’ (RA and lupus)51 and other overlapping connective tissue diseases52 depending on the genetic makeup of the host. This hypothesis is supported by a recent report describing co-aggregation of systemic autoimmune diseases including Sjögren's syndrome, lupus, RA, SSc and inflammatory myopathies, as well as others in affected families.53
In summary, these studies define a novel, epigenetically modified CD4+ T cell subset that is found in patients with active lupus and other autoimmune rheumatic diseases but not the idiopathic inflammatory disease RPF. This subset may participate in the development of these diseases and be a therapeutic target for the treatment of human lupus and perhaps the related autoimmune rheumatic diseases.
Acknowledgments
The authors thank MsSushma Yarlagadda and Ms Erin Grant for their expert technical assistance and Ms Stacy Fry for her excellent secretarial support. The authors also thank Dr Amr Sawalha for helpful discussions, Dr William J McCune and the Rheumatology Clinic Recruitment Team for recruiting patients.
Footnotes
Contributors: All authors have contributed to the production of data, writing and reviewing of the contents of the manuscript and meet the criteria for co-authorship.
Funding: The studies were supported by PHS grants AR42525 (BR), AI110502 (BR), ES017885 (BR), K01ES019909 (ES), K12HD001438 (WM), K24AR063120 (DK), a Merit grant from the Department of Veterans Affairs (BR), and the Lupus Insight Prize (BR) from the Lupus Foundation of America, the Lupus Research Institute and the Alliance for Lupus Research.
Competing interests: None declared.
Patient consent: Obtained.
Ethics approval: The University of Michigan Institutional Review Board.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Palacios R, Santos J, Valdivielso P et al. Human immunodeficiency virus infection and systemic lupus erythematosus. An unusual case and a review of the literature. Lupus 2002;11:60–3. doi:10.1191/0961203302lu141cr [DOI] [PubMed] [Google Scholar]
- 2.Fox RA, Isenberg DA. Human immunodeficiency virus infection in systemic lupus erythematosus. Arthritis Rheum 1997;40:1168–72. doi:10.1002/art.1780400623 [DOI] [PubMed] [Google Scholar]
- 3.Wofsy D, Seaman WE. Successful treatment of autoimmunity in NZB/NZW F1 mice with monoclonal antibody to L3T4. J Exp Med 1985;161:378–91. doi:10.1084/jem.161.2.378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jabs DA, Burek CL, Hu Q et al. Anti-CD4 monoclonal antibody therapy suppresses autoimmune disease in MRL/Mp-lpr/lpr mice. Cell Immunol 1992;141:496–507. doi:10.1016/0008-8749(92)90166-M [DOI] [PubMed] [Google Scholar]
- 5.Richardson B. Effect of an inhibitor of DNA methylation on T cells. II. 5-Azacytidine induces self-reactivity in antigen-specific T4+ cells. Human Immunol 1986;17:456–70. doi:10.1016/0198-8859(86)90304-6 [DOI] [PubMed] [Google Scholar]
- 6.Patel DR, Richardson BC. Dissecting complex epigenetic alterations in human lupus. Arthritis Res Ther 2013;15:201 doi:10.1186/ar4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol 2007;3:521–7. doi:10.1038/ncprheum0573 [DOI] [PubMed] [Google Scholar]
- 8.Bombardier C, Gladman DD, Urowitz MB et al. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992;35:630–40. doi:10.1002/art.1780350606 [DOI] [PubMed] [Google Scholar]
- 9.Somers EC, Thomas SL, Smeeth L et al. Autoimmune diseases co-occurring within individuals and within families: a systematic review. Epidemiology 2006;17:202–17. doi:10.1097/01.ede.0000193605.93416.df [DOI] [PubMed] [Google Scholar]
- 10.Somers EC, Thomas SL, Smeeth L et al. Are individuals with an autoimmune disease at higher risk of a second autoimmune disorder? Am J Epidemiol 2009;169:749–55. doi:10.1093/aje/kwn408 [DOI] [PubMed] [Google Scholar]
- 11.Cooper GS, Bynum ML, Somers EC. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun 2009;33:197–207. doi:10.1016/j.jaut.2009.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anaya JM, Gómez L, Castiblanco J. Is there a common genetic basis for autoimmune diseases? Clin Dev Immunol 2006;13:185–95. doi:10.1080/17402520600876762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramos PS, Criswell LA, Moser KL et al. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genet 2011;7:e1002406 doi:10.1371/journal.pgen.1002406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Somers EC, Ganser MA, Warren JS et al. Mercury Exposure and Antinuclear Antibodies among Females of Reproductive Age in the United States: NHANES. Environ Health Perspect 2015;123:792–8. doi:10.1289/ehp.1408751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubo K, Yamamoto K. IgG4-related disease. Int J Rheumatic Dis 2015:1–9. doi:10.1111/1756-185X.12586 [DOI] [PubMed] [Google Scholar]
- 16.Urban ML, Palmisano A, Nicastro M et al. Idiopathic and secondary forms of retroperitoneal fibrosis: a diagnostic approach. Rev Med Interne 2015;36:15–21. doi:10.1016/j.revmed.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 17.Vitali C, Bombardieri S, Jonsson R et al. Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61:554–8. doi:10.1136/ard.61.6.554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seror R, Ravaud P, Bowman SJ et al. EULAR Sjogren's syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren's syndrome. Ann Rheum Dis 2010;69:1103–9. doi:10.1136/ard.2009.110619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan EM, Cohen AS, Fries JF et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271–7. doi:10.1002/art.1780251101 [DOI] [PubMed] [Google Scholar]
- 20.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725 doi:10.1002/art.1780400928 [DOI] [PubMed] [Google Scholar]
- 21.Aletaha D, Neogi T, Silman AJ et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. doi:10.1002/art.27584 [DOI] [PubMed] [Google Scholar]
- 22.McCune WJ, Golbus J, Zeldes W et al. Clinical and immunologic effects of monthly administration of intravenous cyclophosphamide in severe systemic lupus erythematosus. N Engl J Med 1988;318:1423–31. doi:10.1056/NEJM198806023182203 [DOI] [PubMed] [Google Scholar]
- 23.Khanna D, Denton CP. Evidence-based management of rapidly progressing systemic sclerosis. Best Pract Res Clin Rheumatol 2010;24:387–400. doi:10.1016/j.berh.2009.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swartz RD. Idiopathic retroperitoneal fibrosis: a review of the pathogenesis and approaches to treatment. Am J Kidney Dis 2009;54:546–53. doi:10.1053/j.ajkd.2009.04.019 [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Chen Y, Richardson B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4(+)CD28(−) T cells. Clin Immunol 2009;132:257–65. doi:10.1016/j.clim.2009.03.529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weyand CM, Brandes JC, Schmidt D et al. Functional properties of CD4+ CD28− T cells in the aging immune system. Mech Ageing Dev 1998;102:131–47. doi:10.1016/S0047-6374(97)00161-9 [DOI] [PubMed] [Google Scholar]
- 27.Middleton D, Gonzelez F. The extensive polymorphism of KIR genes. Immunology 2010;129:8–19. doi:10.1111/j.1365-2567.2009.03208.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Liu Y, Strickland FM et al. Age-dependent decreases in DNA methyltransferase levels and low transmethylation micronutrient levels synergize to promote overexpression of genes implicated in autoimmunity and acute coronary syndromes. Exp Gerontol 2010;45:312–22. doi:10.1016/j.exger.2009.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Kuick R, Hanash S et al. DNA methylation inhibition increases T cell KIR expression through effects on both promoter methylation and transcription factors. Clin Immunol 2009;130:213–24. doi:10.1016/j.clim.2008.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brito-Zerón P, Ramos-Casals M, Bosch X et al. The clinical spectrum of IgG4-related disease. Autoimmun Rev 2014;13:1203–10. doi:10.1016/j.autrev.2014.08.013 [DOI] [PubMed] [Google Scholar]
- 31.Seror R, Bootsma H, Saraux A et al. Defining disease activity states and clinically meaningful improvement in primary Sjogren's syndrome with EULAR primary Sjogren's syndrome disease activity (ESSDAI) and patient-reported indexes (ESSPRI). Ann Rheum Dis 2016;75(2):382–9. doi:10.1136/annrheumdis-2014-206008 [DOI] [PubMed] [Google Scholar]
- 32.Lu Q, Wu A, Tesmer L et al. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol 2007;179:6352–8. doi:10.4049/jimmunol.179.9.6352 [DOI] [PubMed] [Google Scholar]
- 33.Farrar WL, Ruscetti FW, Young HA. 5-Azacytidine treatment of a murine cytotoxic T cell line alters interferon-gamma gene induction by interleukin 2. J Immunol 1985;135:1551–4. [PubMed] [Google Scholar]
- 34.Yung RL, Quddus J, Chrisp CE et al. Mechanism of drug-induced lupus. I. Cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol 1995;154:3025–35. [PubMed] [Google Scholar]
- 35.Li Y, Gorelik G, Strickland FM et al. Oxidative stress, T cell DNA methylation, and lupus. Arthritis Rheumatol 2014;66:1574–82. doi:10.1002/art.38427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strickland FM, Li Y, Johnson K et al. CD4(+) T cells epigenetically modified by oxidative stress cause lupus-like autoimmunity in mice. J Autoimmun 2015;62:75–80. doi:10.1016/j.jaut.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahsan H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum Immunol 2013;74:1392–9. doi:10.1016/j.humimm.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 38.Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol 2013;9:674–86. doi:10.1038/nrrheum.2013.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Driscoll T, Jacklyn G, Orchard J et al. The global burden of occupationally related low back pain: estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis 2014;73:975–81. doi:10.1136/annrheumdis-2013-204631 [DOI] [PubMed] [Google Scholar]
- 40.Gorelik G, Sawalha AH, Patel D et al. T cell PKCδ kinase inactivation induces lupus-like autoimmunity in mice. Clin Immunol 2015;158:193–203. doi:10.1016/j.clim.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santourlidis S, Trompeter HI, Weinhold S et al. Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. J Immunol 2002;169:4253–61. doi:10.4049/jimmunol.169.8.4253 [DOI] [PubMed] [Google Scholar]
- 42.Basu D, Liu Y, Wu A et al. Stimulatory and inhibitory killer Ig-like receptor molecules are expressed and functional on lupus T cells. J Immunol 2009;183:3481–7. doi:10.4049/jimmunol.0900034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson B, Epstein WV. Utility of the fluorescent antinuclear antibody test in a single patient. Ann Intern Med 1981;95:333–8. doi:10.7326/0003-4819-95-3-333 [DOI] [PubMed] [Google Scholar]
- 44.Pals ST, Radaszkiewicz T, Roozendaal L et al. Chronic progressive polyarthritis and other symptoms of collagen vascular disease induced by graft-vs-host reaction. J Immunol 1985;134:1475–82. [PubMed] [Google Scholar]
- 45.Daikeler T, Tyndall A. Autoimmunity following haematopoietic stem-cell transplantation. Best Pract Res Clin Haematol 2007;20:349–60. doi:10.1016/j.beha.2006.09.008 [DOI] [PubMed] [Google Scholar]
- 46.Minhas U, Das P, Bhatnagar A. Role of reactive intermediates in the immunopathogenesis of the pristane-induced Balb/c model of lupus. Lupus 2011;20:1421–5. doi:10.1177/0961203311418791 [DOI] [PubMed] [Google Scholar]
- 47.Satoh M, Hamilton KJ, Ajmani AK et al. Autoantibodies to ribosomal P antigens with immune complex glomerulonephritis in SJL mice treated with pristane. J Immunol 1996;157:3200–6. [PubMed] [Google Scholar]
- 48.Stasiuk LM, Ghoraishian M, Elson CJ et al. Pristane-induced arthritis is CD4+ T-cell dependent. Immunology 1997;90:81–6. doi:10.1046/j.1365-2567.1997.00121.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leiss H, Niederreiter B, Bandur T et al. Pristane-induced lupus as a model of human lupus arthritis: evolvement of autoantibodies, internal organ and joint inflammation. Lupus 2013;22:778–92. doi:10.1177/0961203313492869 [DOI] [PubMed] [Google Scholar]
- 50.Odobasic D, Muljadi RC, O'Sullivan KM, et al. Suppression of Autoimmunity and Renal Disease in Pristane-Induced Lupus by Myeloperoxidase. Arthritis Rheumatol 2015;67:1868–80. doi:10.1002/art.39109 [DOI] [PubMed] [Google Scholar]
- 51.Li J, Wu H, Huang X et al. Clinical analysis of 56 patients with rhupus syndrome: manifestations and comparisons with systemic lupus erythematosus: a retrospective case-control study. Medicine 2014;93:e49 doi:10.1097/MD.0000000000000049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosca M, Tani C, Vagnani S et al. The diagnosis and classification of undifferentiated connective tissue diseases. J Autoimmun 2014;48–49:50–2. doi:10.1016/j.jaut.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 53.Kuo CF, Grainge MJ, Valdes AM et al. Familial Aggregation of Systemic Lupus Erythematosus and Coaggregation of Autoimmune Diseases in Affected Families. JAMA Intern Med 2015;175:1518–26. doi:10.1001/jamainternmed.2015.3528 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Study Subjects
lupus-2016-000147supp_tableS1.pdf (208.4KB, pdf)
Flow Cytometry Instrument Configuration
lupus-2016-000147supp_tableS2.pdf (15.7KB, pdf)
lupus-2016-000147supp_figure.pdf (386KB, pdf)