

Abstract
Heat shock response (HSR) generally plays a major role in sustaining protein homeostasis. In Escherichia coli, the activity and amount of the dedicated transcription factor σ32 transiently increase upon heat shock. The initial induction is followed by chaperone-mediated negative feedback to inactivate and degrade σ32. Previous work reported that signal recognition particle (SRP)-dependent targeting of σ32 to the membrane is essential for feedback control, though how SRP recognizes σ32 remained unknown. Extensive photo- and disulfide cross-linking studies in vivo now reveal that the highly conserved regulatory region of σ32 that lacks a consecutive hydrophobic stretch interacts with the signal peptide-binding site of Ffh (the protein subunit of SRP). Importantly, the σ32–Ffh interaction observed was significantly affected by mutations in this region that compromise the feedback regulation, but not by deleting the DnaK/DnaJ chaperones. Homeostatic regulation of HSR thus requires a novel type of SRP recognition mechanism.
The heat shock response (HSR) is a ubiquitous cellular strategy for coping with damaged proteins and maintaining homeostasis by ensuring appropriate expression levels of heat shock proteins (HSPs)1. Upon exposure to heat or other stressors, cells undergo rapid and transient induction of HSPs, such as chaperones and proteases, which aid in protein folding or degradation, thereby protecting cells from the stress. Not surprisingly, the HSR requires complex regulatory circuits to meet the needs of various cell types, organisms, and environments. In Escherichia coli and other bacteria, σ32, the rpoH gene product, directs RNA polymerase to promote transcription of a set of HSP genes2,3,4,5,6. σ32 is extremely unstable, and it is normally present at very low levels. When the cell is exposed to heat stress, the activity and level of σ32 rapidly increase, due to both elevated translation of rpoH mRNA7,8,9,10 and transient stabilization of the σ32 protein11,12,13. This induction phase is soon followed by the recovery (adaptive) phase, in which the activity/level of σ32 gradually decreases to reach a new steady-state. The latter mode of regulation, known as negative feedback control, is mediated by a set of conserved chaperones, including DnaK/DnaJ and proteases that accumulate during induction phase; however, the detailed mechanisms remain unknown, even in E. coli, the best-studied bacterial system.
Degradation of σ32 is primarily mediated by the essential membrane-localized protease FtsH14,15. DnaK/DnaJ and GroEL/GroES chaperones can bind σ32 directly and promote its degradation in vivo16,17,18,19,20, but this has not been recapitulated in vitro21. Extensive work on a class of σ32 mutants with altered stability and feedback control (dysregulation mutants), identified the homeostatic control region (a segment of Leu-47 to Leu-55 in region 2.1) of σ32 that is important for regulating both stability and activity of σ32 22,23,24, although its actual role is not known. Moreover, contrary to the expectations based on in vivo results, purified σ32 with a strong dysregulation mutation (I54N) exhibits almost normal binding to the chaperones and wild-type sensitivity to inhibition by the chaperones when tested in an in vitro transcription system, suggesting that additional factors are involved in σ32 regulation24. A subsequent search for the missing link led to the finding that signal recognition particle (SRP), which consists of the Ffh protein and 4.5 S RNA, SRP receptor (SR: FtsY), and the SecYEG translocon play essential roles in both chaperone-mediated inactivation and FtsH-mediated degradation of σ32 25. This unexpected finding not only revealed a new regulatory pathway for σ32-mediated HSR, but also explained how damage to the SRP pathway, as well as cytoplasmic protein damage, can induce HSR. Moreover, this observation suggested that protein-folding states in the cytoplasm and those in the inner membrane (IM) are integrated and/or coordinated.
In protein transportation to the inner membrane (IM) by the SRP pathway, the M domain of Ffh binds a hydrophobic signal peptide (SP) or a transmembrane segment of the membrane proteins during (or just after) translation on the ribosome (Fig. 1, left)26. The nascent chain is targeted to the SecYEG translocon through the SRP–SR interaction, and inserted into the lipid bilayer via the SecY polypeptide–conducting channel and subsequently its lateral gate27. Recent crystal structures of an Ffh homolog in complex with an SP have revealed diverse modes of Ffh–SP interactions28,29,30.
Figure 1. Roles of SRP in membrane protein biogenesis and feedback control of σ32.

(left) Membrane targeting of inner membrane (IM) proteins. The Ffh M domain of SRP interacts with a transmembrane segment (or a signal peptide) of a membrane protein emerging from the ribosomal tunnel. SRP targets the ribosome–nascent chain complex (RNC) to the SecYEG translocon through the interaction between SRP and SR (FtsY). (right) Negative feedback control of σ32. The homoeostatic control region of σ32 (red star) is recognized by the Ffh M domain, and σ32 is targeted to SecYEG through the SRP–SR interaction, and degraded by FtsH protease in a chaperone-dependent manner.
Full-length σ32 can bind to the M domain of Ffh in vitro25. Also, analysis of σ32–PhoA fusion proteins suggested that the N-terminal part of σ32 carries a sequence that can bring the PhoA mature sequence to the membrane-proximal region of the cell in an SRP-dependent manner25. Nonetheless, σ32 does not contain a stretch that is sufficiently hydrophobic to serve as a typical signal sequence or transmembrane segment, and it was unclear whether the SP-binding site of Ffh is actually involved in the binding of σ32. Thus, the mechanism by which Ffh recognizes σ32 remained as an intriguing problem to be addressed from the standpoints of both SRP function and heat shock regulation.
In this study, we used in vivo cross-linking approaches31,32,33,34 to demonstrate that the homeostatic control region of σ32 directly interacts with the SP-binding site in the M domain of Ffh and that this interaction is intimately involved in feedback control of σ32 (Fig. 1, right). Although the region 2.1 of σ32 also interacts with DnaK/DnaJ and other chaperones, the Ffh–σ32 interaction revealed by this work does not depend on these chaperones.
Results
Ffh binds to the homeostatic control region of σ32 in vivo
To search for proteins that interact with the homeostatic control region of σ32, we employed the in vivo photo-cross-linking approach. For this purpose, we introduced a non-natural, photo-reactive amino acid, p-benzoyl-L-phenylalanine (pBPA), into each of the 17 positions (Arg-35 to Asn-67; see Fig. 2A) in and around the homeostatic control region of N-terminally His6-tagged σ32 (His6-σ32); we accomplished these substitutions by amber suppression using the laboratory evolved Methanocaldococcus jannaschii aminoacyl-tRNA synthetase/suppressor tRNA pair35. While most of the His6-σ32 variants containing pBPA (His6-σ32pBPA) exhibited accumulation levels comparable to that of WT His6-σ32, some (A38, A49 and I64pBPA variants) accumulated at much lower levels presumably due to instability, although they all showed similar σ32 activities, as determined by LacZ expression from the reporter (PhtpG-lacZ) (Supplementary Fig. S1A,B).
Figure 2. The homeostatic control region of σ32 binds to Ffh in vivo.

(A) The region of σ32 examined (top) and a summary of the cross-linking studies (below). The homeostatic control region of σ32 is located within the regulatory region, region 2.1, conserved among all σ factors. The residues individually mutated to an amber codon are shown in bold letters, and those involved in the known dysregulation mutations are shown in red. Positions where cross-linking with Ffh, DnaJ, DnaK, and HtpG was detected clearly and reproducibly are indicated by colored dots. (B,C) Analysis of cross-linked products by SDS-PAGE and immunoblotting with anti-σ32 (B) or anti-Ffh (C) antibodies. Cells of CAG48238/pEVOL-pBpF/pTTQ18-his6-rpoH(amb) were grown at 30 °C in L-medium supplemented with 0.02% arabinose and 1 mM pBPA, induced to express His6-σ32pBPA with 1 mM IPTG for 1 h, and UV-irradiated. A strain producing wild type (WT) σ32 (shown here as “pBPA none”) served as a control (left lane). Total cellular proteins were acid-precipitated and analyzed by 7.5% SDS-PAGE followed by immunoblotting. XL indicates cross-linked products. (D) Immunoprecipitation (IP) of cross-linked products with anti-σ32 or anti-Ffh antibodies. Extracts of sonically disrupted UV-irradiated cells were subjected to IP with anti-Ffh or control antibodies. Precipitates were analyzed by 7.5% SDS-PAGE followed by immunoblotting with anti-σ32 or anti-Ffh antibodies. Blue arrowheads in (C,D) indicate the σ32–Ffh cross-linked products obtained by anti-σ32 or anti-Ffh immunoblotting. Gray arrowheads indicate proteins non-specifically precipitated by the control (SecA6-peptide)46 or other antibodies. In (D) non-cross-linked Ffh was hardly detected due to the overlapping IgG heavy chains used for immunoprecipitation.
Following UV irradiation of cells expressing His6-σ32pBPA, a number of the σ32 variants migrated as multiple protein bands with higher apparent molecular masses when analyzed by SDS-PAGE and anti-σ32 immunoblotting (Fig. 2B, indicated by XL). Immunoblotting with anti-Ffh antibodies clearly detected putative σ32–Ffh cross-linked products at four positions (E48, K51, T52 and I54); weaker bands for putative cross-linked products were also detected at position of L41 (Fig. 2C). We should note that the bands that correspond to σ32–Ffh cross-linked products were not directly detected by anti-σ32 antibodies due to high backgrounds (Fig. 2B), but became detectable after prior purification by immunoprecipitation with anti-Ffh antibodies (Fig. 2D). These results extend our previous result that showed cross-linking of His6-σ32T52pBPA to Ffh, and clearly demonstrate that Ffh directly interacts with σ32 at several positions in the homeostatic control region in vivo.
Chaperones DnaK, DnaJ, and HtpG also interact with the homeostatic control region of σ32
Generation of multiple cross-linked products at several positions (including Leu-47 and Lys-51) (Fig. 2B) suggested that protein factors other than Ffh also interacted with the homeostatic control region and its vicinity of σ32. To identify these factors, we purified the putative cross-linked products and analyzed them individually by mass spectrometry. Initially, we analyzed the cross-linked products obtained with two His6-σ32pBPA proteins, His6-σ32L47pBPA and His6-σ32K51pBPA, both of which yielded multiple cross-linked products, whereas only His6-σ32K51pBPA was cross-linked to Ffh. To improve the efficiency of His-tag affinity purification, we used a longer tag (His10); this modification barely affected the cross-linking profiles (Supplementary Fig. S2A,B).
The cross-linked products were purified under SDS-denaturing conditions, separated by SDS-PAGE, and subjected to mass spectrometry analysis (Supplementary Fig. S2C,D). Consistent with the results of immunoblotting (Fig. 2B,C), Ffh fragments were detected from the His10-σ32K51pBPA sample, but not from the His10-σ32L47pBPA sample. In addition, amino acid sequences of chaperones, DnaK, DnaJ, and HtpG, were found in the products formed at both positions (Supplementary Fig. S2C,D). Cross-linking of His6-σ32pBPA with these chaperones at all the 17 positions was then examined by immunoblotting using appropriate anti-chaperone antibodies. Cross-linking with these chaperones was detected at seven positions, including L47 and K51 (Supplementary Fig. S3A–C). These results demonstrate that DnaK, DnaJ, and HtpG interact rather promiscuously with the homeostatic control region of σ32, as summarized in Fig. 2A.
σ32 mutations that compromise feedback control affect the interaction between σ32 and Ffh
To further investigate the significance of the observed σ32–Ffh interaction in the negative feedback control of σ32, we tested the effects of the previously characterized dysregulation mutations24, A50D, K51E, I54N, and R91P, on the σ32–Ffh cross-linking. These mutations, when introduced in cis, stabilized both His6-σ32K51pBPA and His6-σ32T52pBPA as expected, resulting in elevated and variable levels of protein accumulation (Supplementary Fig. S4A,B). This made it difficult to accurately evaluate the effects of these mutations on σ32–Ffh cross-linking efficiencies. Because the synthesis rates of His6-σ32pBPA variants with or without the dysregulation mutations were nearly equal, we performed cross-linking using cells pulse-labeled with radioactive methionine (Fig. 3A). Cells expressing the His6-σ32K51pBPA or His6-σ32T52pBPA protein (without dysregulation mutation) were first labeled with [35S]Met, UV-irradiated, and subjected to immunoprecipitation with anti-σ32 or anti-Ffh antibodies. The results showed that the profiles of the radioactive cross-linking products were very similar to those detected by immunoblotting (compare Figs 3A and 2B). We then examined His6-σ32K51pBPA and His6-σ32T52pBPA having each of the σ32 dysregulation mutations. Remarkably, these mutations altered the profiles and/or amounts of σ32–Ffh cross-linking products (Fig. 3A). Whereas the parental His6-σ32 T52pBPA generated a single cross-linked product of approximately 85 kDa, the same protein with the A50D, I54N, or R91P mutation generated a single cross-linked product of slightly higher mobility. The same protein with the K51E mutation generated two cross-linked products, one migrating faster and another migrating slower. Moreover, the two strong mutations, A50D and I54N, caused clear reduction in the relative amounts of cross-linked products, whereas only little effects were observed with weak mutations, K51E and R91P (Fig. 3B). In the case of His6-σ32K51pBPA, the parental protein generated two cross-linked products of approximately 80 and 90 kDa, whereas the A50D, I54N, and R91P mutations significantly altered the mobility, the number, and/or the relative amounts of cross-linked products (Fig. 3A,B).
Figure 3. σ32 dysregulation mutations affect the mode and extent of the σ32–Ffh interaction.
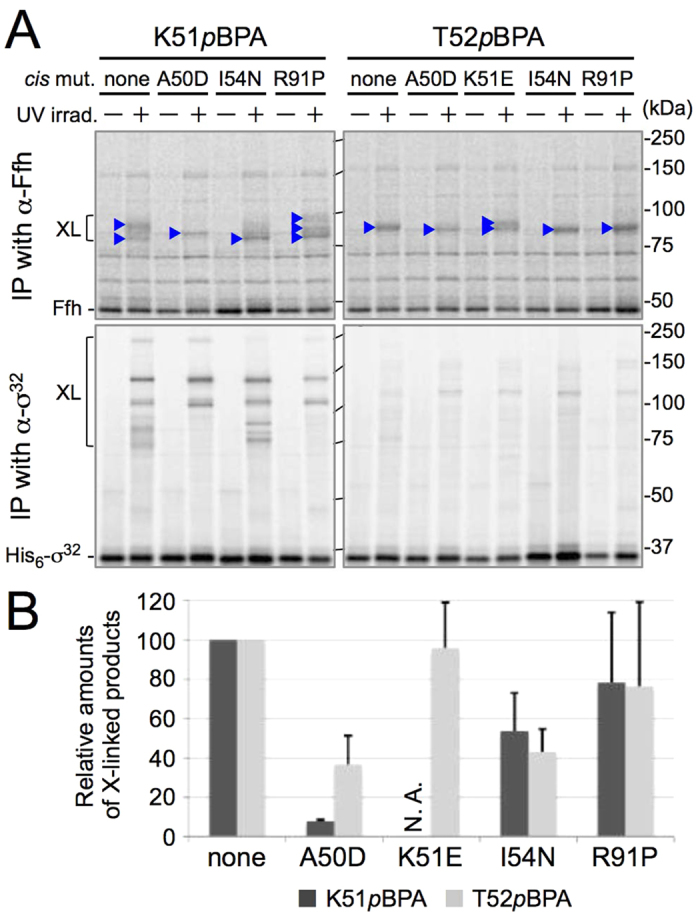
(A) In vivo photo-cross-linking using [35S]Met-pulse-labeled His6-σ32pBPA proteins. Plasmid pTTQ18-his6-rpoH(K51amb) or pTTQ18-his6-rpoH(T52amb), with or without additional dysregulation mutation, was transformed into CAG48238/pEVOL-pBpF/pRM83-ffh+ffs. Cells of the resulting strains were grown at 30 °C in M9-based medium with 1 mM pBPA, induced to express His6-σ32pBPA with 1 mM IPTG for 6 min, and labeled with [35S]Met for 1 min. Following UV irradiation, total cellular proteins were immunoprecipitated (IP) with anti-σ32 or anti-Ffh antibodies, and immunocomplexes were separated by 7.5% SDS-PAGE followed by phosphorimaging. (B) Quantification of His6-σ32–Ffh cross-linked products. The ratio of the sum of intensities of His6-σ32–Ffh cross-linked products to that of unirradiated His6-σ32 is shown (the values for His6-σ32pBPA carrying no dysregulation mutation were set to 100). Three independent experiments were performed, and mean values are shown along with standard deviations.
Cross-linking of two proteins reflects spatial proximity but not necessarily functional interactions between them. The observed changes in the cross-linking profiles by the σ32 dysregulation mutations suggest that each of the mutations caused a shift in the selection of cross-linking partner residue of Ffh. While the altered cross-linking profiles could have resulted from changes in the orientation of the photo-reactive side chain of pBPA, which was induced by the dysregulation mutations introduced into nearby positions in the primary structure, characteristic alterations in the number and mobility of the cross-linked products were also observed for the R91P mutation, which affects a position distant from that of pBPA in the primary structure but predicted to be nearby in the folded structure24. It should also be noted that, while the previous gel filtration experiments with purified proteins demonstrated that the I54N mutation apparently abolished the σ32–Ffh interaction, the current in vivo cross-linking results showed that the interaction was only moderately affected by the same mutation. This difference may be ascribed to different sensitivities of these methods; in vivo cross-linking could enable detection of weaker and/or transient interactions that would be missed by gel-filtration assays. In addition, the σ32 cis mutations exerted parallel effects on the negative regulation of σ32 and on the cross-linking of σ32 with Ffh; the stronger mutations exerted more severe effects. These results strongly implicate that the observed changes in the amounts and profiles of the cross-linking products are at least partly ascribable to some changes in the σ32–Ffh binding interfaces and that the σ32–Ffh cross-linking indeed represents functional client–machinery interactions involved in the homeostatic control of σ32.
DnaK/DnaJ chaperones are not required for in vivo interaction of Ffh with the homeostatic control region of σ32
Previous studies established that the DnaK/DnaJ chaperone system is involved in the negative feedback control of σ32 16,17,18,19,20,21. Because DnaK and Dn aJ directly interact with σ32 17,18, we considered the possibility that these chaperones induce conformational changes in σ32 to facilitate its recognition by Ffh. We addressed this possibility by using a dnaKJ deletion strain as a host for σ32–Ffh photo-cross-linking experiments involving the His6-σ32 K51pBPA and His6-σ32 T52pBPA proteins. Anti-Ffh immunoblotting revealed that the absence of DnaK/DnaJ chaperones caused no distinct alteration in the profiles and the levels of σ32–Ffh cross-linked products (Supplementary Fig. S5A,B). On the other hand, anti-σ32 immunoblotting analysis revealed that cross-linked products of about 150 kDa and 75 kDa disappeared from blots of His6-σ32 K51pBPA and His6-σ32T52pBPA, respectively (Supplementary Fig. S5A, lower panel). The corresponding bands were detected specifically with anti-DnaK and anti-DnaJ antibodies when the wild-type host was used, indicating that they represent the σ32–DnaK and σ32–DnaJ cross-linked products, respectively (Supplementary Fig. S3). These results show that the DnaK/DnaJ chaperones do bind to σ32, but they play no essential role in the interaction of Ffh with the homeostatic control region of σ32 observed in this study, consistent with our previous result that purified σ32 and SRP interact in the absence of DnaK/DnaJ25.
Ffh uses its signal peptide-binding site to bind the homeostatic control region of σ32
We next selected Ffh residues as a site of photo-cross-linker introduction in attempts to identify the residue(s) of Ffh that interact(s) with σ32 in vivo. The recently reported crystal structures of the Ffh homologs from Sulfolobus solfataricus (Fig. 4A)28 and M. jannaschii (Fig. 4B,C)29,30, each in complex with a signal peptide (SP), indicate that in each of these well conserved structures (Supplementary Fig. S6), the SP binds to the M domain of SRP54 in a distinct manner. Based on these structures, we selected 13 residues encompassing the possible SP-binding sites in the E. coli Ffh M domain as the sites of photo-cross-linker introduction. We used p-azido-L-phenylalanine (pAzPA)35 instead of pBPA to avoid cross-linking interference by nearby methionine residues in the M domain36. We confirmed that full-length, pAzPA-containing Ffh variants (FfhpAzPA) accumulated at similar levels across the different constructs (Supplementary Fig. S7A). All the FfhpAzPA variants tested supported growth of cells depleted for wild-type Ffh, indicating that they were functional (Supplementary Fig. S7B).
Figure 4. σ32 interacts with the SP–binding site of Ffh in vivo.

(A–C) Crystal structures of SsSRP54–SP (PDB:3kl4; A)28, MjSRP54–SP (PDB:3ndb; B)29, and MjSRP54 M–SRP19–SRP RNA (PDB:4xco; C)30 in complex with SP. Only the M domain of each Ffh homolog (SRP54) is shown with SP (in yellow). The positions corresponding to those in E. coli Ffh (see Supplementary Fig. S6) where pAzPA was incorporated are indicated by spheres: the positions where cross-linking with σ32 was detected are colored in blue (the corresponding residues of E. coli Ffh are indicated on the models), and the others are in light green. (D) Immunoblotting analysis of in vivo photo cross-linking using FfhpAzPA variant proteins. Cells of CAG48373 (ΔftsH sfhC21)/pEVOL-pAzF/pTTQ18-ffh(amb)+ffs were grown at 30 °C in L-medium supplemented with 0.02% arabinose and 1 mM pAzPA, induced to express FfhpAzPA with 1 mM IPTG for 1 h, and UV-irradiated as indicated. Total cellular proteins were analyzed by 7.5% SDS-PAGE and immunoblotted with anti-Ffh and anti-σ32 antibodies. (E) Immunoprecipitation (IP) of cross-linked products with anti-Ffh antibodies. Total cellular proteins of UV-irradiated cells were precipitated with anti-Ffh antibodies, and analyzed by 7.5% SDS-PAGE followed by immunoblotting with anti-σ32 and anti-Ffh antibodies.
To facilitate detection of Ffh–σ32 cross-linked products, we used as a host an FtsH-deletion strain that allowed increased levels of σ32 accumulation. Anti-σ32 immunoblotting analysis revealed that bands of ~80–100 kDa were generated in a UV-irradiation dependent manner when pAzPA was introduced in place of Met-341, Met-376, or Met-426 of Ffh, whereas no such bands were detected at other positions or with wild-type Ffh (no pAzPA incorporated) (Fig. 4D). The UV- and pAzPA-dependent generation of these bands, although less marked at positions 341 and 426, strongly suggests that they represent products of cross-linking between Ffh and σ32. These bands were not detectable by anti-Ffh immunoblotting, probably due to high background caused by overexpression of Ffh. However, these bands could be detected by anti-σ32 immunoblotting if preceded by immunoisolation with anti-Ffh antibodies, but not with control antibodies (Fig. 4E and Supplementary Fig. S8), indicating that they all contained both Ffh and the σ32 polypeptides. By contrast, no cross-linking of FfhM376pAzPA with σE, another alternative sigma factor involved in the extracytoplasmic stress response6, was observed (Supplementary Fig. S9). These results suggest that σ32 directly and specifically interacts with the SP-binding region of Ffh M domain in vivo.
The sites of σ32–Ffh interaction and the role of SP-binding site of Ffh were investigated further by means of disulfide-cross-linking experiments, featuring site-specifically introduced Cys residues in these proteins. As a σ32 derivative, we constructed His10-σ32T52C by replacing Thr-52 with Cys (note that WT-His10-σ32 has no intrinsic Cys residue, and that Thr-52 is the position where stable photo-cross-linking with Ffh was consistently observed) (Fig. 2). The mutant protein accumulated normally with unaltered activity (Supplementary Fig. S10A,B). As single Cys Ffh derivatives, we first constructed Cys-less Ffh (C406S), into which Cys was introduced at each of three positions, Met-341, Met-376 and Met-426, where we observed photo-cross-linking with σ32 (Fig. 4). None of the cysteine substitutions affected protein levels or activities (Supplementary Fig. S10C,D).
Cells expressing a combination of His10-σ32T52C and one of the single-Cys Ffh derivatives were treated with an oxidant, Cu2+(phenanthroline)3, to induce any possible disulfide bond formation. After quenching the oxidant, total cellular proteins were subjected to His-tag affinity isolation. Samples were then separated by SDS-PAGE in the presence or the absence of 2-mercaptoethanol (ME) and analyzed by anti-Ffh immunoblotting. The oxidant treatment generated a band of about 90 kDa for the combination of His10-σ32T52C and FfhM376C but not for the others (Fig. 5, no ME). The 90 kDa band disappeared when the sample had been treated with ME (Fig. 5, +ME). Its generation depended on the simultaneous presence of the Cys residues in His10-σ32 and Ffh. These results indicate that the residue 52 of σ32 and residue 376 of Ffh are within a distance that allows intermolecular formation of a disulfide bond when they are replaced with Cys. Taken together, our results demonstrate that Ffh directly binds the homeostatic control region of σ32 at the SP-binding site, such that the residue 376 of Ffh is in close proximity to the residue 52 of bound σ32 (see Fig. 4A–C and Discussion for possible modes of σ32–Ffh interaction).
Figure 5. Disulfide cross-linking between the homeostatic control region of σ32 and the SP-binding region of Ffh.
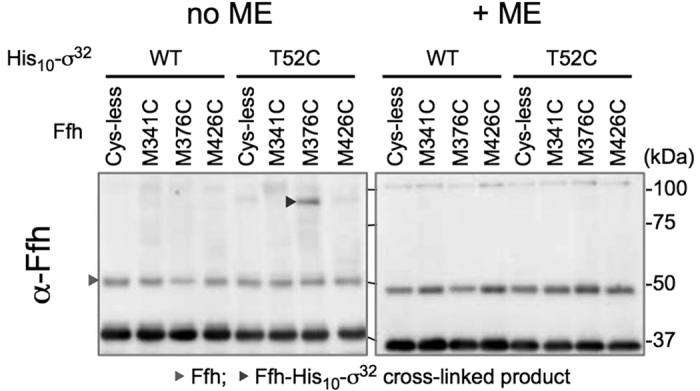
Cells of WAM121 (Δffh1::kan Para-ffh) carrying a combination of plasmids encoding Cys-less or single Cys-derivative of His10-σ32 and Ffh as indicated were grown in L-medium and induced to express His10-σ32 Cys and Ffh Cys with 1 mM IPTG for 1 h. Cells were treated with Cu2+(phenanthroline)3 at 37 °C for 5 min. After quenching the oxidant and blocking free thiol groups, extracts of sonically disrupted cells were subjected to TALON affinity purification. The purified proteins were treated with or without 2-mercaptoethanol (ME) and analyzed by 7.5% SDS-PAGE and immunoblotting with anti-Ffh antibodies.
Discussion
Our previous work revealed the unexpected involvement of the SRP-mediated membrane-targeting pathway in the HSR regulatory circuit governed by the transcription factor σ32, which connects cytoplasmic and IM proteostasis in E. coli25. Although we showed that SRP can directly bind σ32 in vivo and in vitro, it remained unclear how SRP and σ32 interact with each other. We investigated this issue using three variations of in vivo cross-linking techniques. The results reveal that the homeostatic control region of σ32 directly interacts with not only the DnaK, DnaJ, and HtpG chaperones but also Ffh at multiple positions, suggesting that this region of σ32 serves as a hub for molecular interactions involving SRP and chaperones for the homeostatic control of HSR (Fig. 2). The finding that several σ32 dysregulation mutations affect the extent and profile of cross-linking between σ32 and Ffh (Fig. 3) reinforces the notion that the observed σ32–Ffh interaction is important for the homeostatic control of σ32. In sharp contrast, this interaction was detected in the total absence of DnaK/DnaJ chaperones (Supplementary Fig. S5), indicating that these chaperones are not essential for the σ32–Ffh interaction observed.
The results of the photo- and disulfide-cross-linking experiments demonstrated that the homeostatic control region of σ32 directly and specifically interacts with the SP binding region in the M domain of Ffh in vivo. This observation is consistent with and complements the previous results of far western blotting analysis showing that purified σ32 binds to the M domain fragment of Ffh in vitro25. According to the published crystal structures of SRP54(Ffh)–SP complexes from archaeal species, the M domain of SRP54 invariably interacts with SP, but in slightly different manners (Fig. 4A–C)28,29,30. We detected photo cross-linking of σ32 at three (Met-341, Met-376, and Met-426) out of 13 positions tested in the E. coli Ffh M domain (Fig. 4D). In addition, Cys at position 376 of Ffh was disulfide-bonded with Cys at position 52 in the homeostatic control region of σ32. Among the three positions where we detected cross-linking, two (Met-341 and Met-426) are located near the bound SP in all the reported crystal structures (cf. Fig. 4A–C), whereas the other (Met-376) is located near SP only in the S. solfataricus SRP54–SP complex (Fig. 4A). These results and the structural information suggest that E. coli Ffh binds σ32 at the SP binding site, which could be shared by transmembrane segments of IM proteins, in a manner similar to the S. solfataricus SRP54–SP interaction. Such substrate-binding sites shared by IM proteins and σ32 would endow the cell with a robust and versatile capacity to respond to changes in protein folding status in the cytoplasm and IM. Thus, the dynamic change in the extent of interaction between SRP and σ32 may provide the basis for effective control of proteostasis during normal growth, as well as under stress.
Whereas SRP generally binds to hydrophobic polypeptide sequences, mostly transmembrane segments in bacteria37, the homeostatic control region of σ32 contains no consecutive array of hydrophobic residues. Sequence-based secondary structure prediction suggests that the homeostatic control region of σ32 forms an amphipathic α-helix (Supplementary Fig. S11A,B). This amphipathic helix might provide a hydrophobic surface for Ffh binding (Supplementary Fig. S11B). Consistent with this hypothesis, the strong dysregulation mutations A50D and I54N, which introduce a charged and a hydrophilic residue, respectively, into the hydrophobic surface of the predicted amphipathic helix, strongly affected Ffh binding (Fig. 3). In the S. solfataricus SRP54–SP complex, SP binds to the M domain in a partially unraveled configuration (Fig. 4A). The σ32 homeostatic control region might also be partially deformed to fit the SP binding site. Moreover, σ32K51pBPA generated two cross-linked products with Ffh (Fig. 3A), indicating that this position can contact two different sites in Ffh. This might reflect either flexibility in the interaction between the SP binding site and the σ32 homeostatic control region or the involvement of functionally distinct modes of interactions.
Our previous in vitro experiments using purified components showed that SRP can bind to folded σ32 25, demonstrating that SRP can recognize σ32 post-translationally. This study also supports the notion that σ32 interacts with SRP post-translationally in vivo, as full-length σ32 was able to cross-link with Ffh. Several earlier studies have suggested that SRP can target substrate proteins to the membrane post-translationally38,39. Post-translational SRP recognition of folded σ32 may be necessary to target accumulated σ32 to FtsH-mediated degradation during the recovery phase of HSR. On the other hand, σ32 may also interact with SRP co-translationally to allow targeting of its newly synthesized molecule to the membrane, where it is eventually inactivated and degraded. SRP-mediated co-translational membrane targeting of σ32 may well facilitate its rapid turnover under normal growth conditions. These possibilities should be addressed in future analyses.
In addition to Ffh, the DnaK, DnaJ, and HtpG chaperones interacted with the homeostatic control region of σ32. This finding is consistent with previous reports that DnaJ can bind to region 2.1 in vitro40,41. By contrast, DnaK has been suggested to interact in vitro with a region (around residues 198–200) C-terminal to the homeostatic control region40. Our results described here indicate that DnaK interacts with this control region with significant affinity in vivo. Indeed, a recent study suggested the existence of multiple DnaK binding sites in σ32 42. If SRP and DnaK/DnaJ chaperones act at distinct steps in the σ32 control circuit, these factors might bind to the homeostatic control region in a sequential manner, although we currently do not know whether SRP and DnaK/DnaJ can bind to the same σ32 molecule simultaneously or whether their binding is mutually exclusive. In vitro experiments with defined components will be needed to clarify these points. Although our results suggest that HtpG also interacts with σ32, this chaperone does not appear to play a critical role in the regulation of σ32, because a loss or an overexpression of HtpG has very little effect on the σ32 activity 16. Because HtpG can assist the DnaK/DnaJ chaperones 43, it could play a limited or auxiliary role in the σ32 regulation by modulating DnaK/DnaJ functions. Clearly, further work is needed to elucidate the significance of the observed binding between chaperones and the homeostatic control region in σ32 regulation.
It is intriguing that SRP can interact with a non-canonical substrate protein like σ32, which has no typical SP or transmembrane segment, to regulate its localization and function. Recent work showed that mammalian SRP also plays a critical role in the regulation of the unfolded protein response by promoting the Ire1α-mediated splicing of XBP1u mRNA44. In this case, SRP recognizes the ribosome-associated nascent XBP1u polypeptide to target it to the Ire1α/Sec61 complex on the ER membrane together with the XBP1u mRNA also in complex with the ribosome. Taken together with our findings in E. coli, these observations suggest that SRP plays an important novel role in the peripheral association of some proteins by assisting their targeting to the membrane. Like the homeostatic control region of σ32, the moderate hydrophobic region (HR2) required for SRP-mediated membrane targeting of XBP1u is predicted to form an amphipathic helix (Supplementary Fig. S11C). Thus, SRP, either prokaryotic or eukaryotic, could be able to recognize a certain class of amphipathic helices. It is important to study whether this new function of SRP requires any other properties such as the primary sequences, higher order structures, and separate cis-elements in the non-canonical substrates, as well as any other cellular components (trans factors). As shown here, the techniques of in vivo cross-linking are useful to capture transient substrates as well as more stable reaction partners of the system. Further systematic analysis targeted to Ffh may prove useful for identifying and analyzing some additional proteins that may be subject to and involved in the SRP-mediated localization/regulation. Such studies may reveal unexpected links between known cellular events and membrane functions.
In the case of HSR regulation in E. coli, it remains to be asked how this targeting event is productively coupled with subsequent events of feedback regulation, in which proteolysis and chaperone binding may be involved in a well-balanced fashion to poise the cell for forthcoming stresses. The new SRP pathway may contribute to the bidirectional regulation by allowing dual localization of σ32, a key regulator of bacterial proteostasis.
Methods
Bacterial Strains
Escherichia coli K12 strains used in this study are listed in Supplementary Table S1. CAG48238, a derivative of MG165524 carrying σ32-dependent reporter PhtpG-lacZ, was used as a wild-type strain. RM591 was a derivative of CAG48238 carrying a ΔdnaKJ::kan marker (a gift from C. A. Gross and B. Lim, University of California), but without the cat marker. WAM121 (Δffh1::kan Para-ffh)45 was a generous gift from G. J. Phillips (Iowa State University).
Plasmids and Primers
Plasmids and primers used in this study are listed in Supplementary Table S2 and S3, respectively, and details of the plasmid construction are described in Supplementary information.
Antibodies
Penta-His HRP conjugate was purchased from QIAGEN (Hilden), anti-σ32 antibody used for immunoprecipitation (RNA pol σH 3RH3) from Santa Cruz Biotechnology, and anti-σE antibody (Sigma E antibody) from MyBioSource. The control antibody used in Fig. 2D and Supplementary Fig. S8 was prepared from a rabbit immunized with a SecA peptide but recognized SecA very poorly46. Other antibodies used were kindly provided by various sources; anti-σ32 used for immunoblotting from M. Kanemori (Kanazawa University), anti-Ffh from C. A. Gross (University of California), anti-DnaK from R. McMacken (Johns Hopkins Bloomberg School of Public Health), anti-DnaJ from A. H. Becker (University of Heidelberg), and anti-HtpG from F. Motojima (Toyama Prefectural University).
In vivo Photo-Cross-Linking
In vivo photo-cross-linking experiments with pBPA-introduced His6-σ32 (His6-σ32pBPA) were carried out essentially as described previously25. For immunoblotting analysis, cells of CAG48238 carrying pEVOL-pBpF and one of the pTTQ18-his6-rpoH(amb) plasmids were grown at 30 °C in L-medium supplemented with 0.02% arabinose and 1 mM pBPA to an early log phase (0.2 TAITEC units), induced with 1 mM IPTG to express His6-σ32pBPA for 1 h, and UV-irradiated at 4 °C for 10 min on a petri dish using B-100 AP UV lamp (365 nm; UVP, LLC.) at a distance of 4 cm. Total cellular proteins were precipitated with 5% trichloroacetic acid (TCA), washed with acetone, and solubilized in SDS sample buffer. Proteins were analyzed by 7.5% Laemmli SDS-PAGE followed by immunoblotting and visualization using ECLTM Western Blotting Detection Reagents or ECLTM Prime Western Blotting Detection Reagents (GE Healthcare) and LAS4000 mini lumino-image analyzer (GE Healthcare). Band intensities were quantified with MultiGauge software (GE Healthcare). For pulse-labeling analysis, cells of strain CAG48238 carrying pEVOL-pBpF, pRM83-ffh+ffs and one of the pTTQ18-his6-rpoH(amb) derivatives additionally carrying the A50D, K51E, I54N or R91P mutation were grown at 30 °C in M9-medium supplemented with 2 μg/ml thiamine, 0.4% glycerol, 0.2% maltose, 18 amino acids (except Met and Cys; final concentration of 20 μg/mL each), 1 mM pBPA until early log phase (0.1 TAITEC units), and induced with 1 mM IPTG to express His6-σ32pBPA for 6 min. Cells were labeled with 370 kBq/ml [35S]Met (American Radiolabeled Chemicals) for 1 min. After addition of excess nonradioactive Met and Cys (final conc. 250 μg/ml each), cells were immediately chilled on ice, and UV-irradiated at 4 °C as described above. Total cellular proteins were precipitated with 5% TCA, washed with acetone, and solubilized in buffer containing 50 mM Tris-HCl (pH 8.1), 1% SDS, 1 mM EDTA. Samples with equal radioactivities were subjected to immunoprecipitation using an appropriate antibody essentially as described47. Proteins were separated by 7.5% SDS-PAGE, and visualized with BAS1800 phosphoimager (FUJIFILM). Band intensities were quantified with MultiGauge software.
In vivo photo-cross-linking experiments using pAzPA-introduced Ffh (FfhpAzPA) were carried out as follows. Cells of CAG48373 (ΔftsH3::kan sfhC21) carrying pEVOL-pAzF and one of the pTTQ18-ffh(amb)+ffs plasmids were grown at 30 °C in L-medium supplemented with L-0.02% arabinose and 1 mM pAzPA until early log phase (25 Klett units), and induced with 1 mM IPTG to express FfhpAzPA for 1 h. Cells were collected by centrifugation, suspended in L-medium supplemented with 0.02% arabinose at approximately 4 × 108 cells/ml, and UV-irradiated at 4 °C for 30 min in a 24-well plate using compact UV lamp 4 W (254 nm; UVP, LLC.) at a distance of 3 cm. Total cellular proteins were precipitated with 5% TCA, washed with acetone, solubilized in SDS sample buffer, and analyzed by 7.5% SDS-PAGE and immunoblotting.
Purification and Mass Spectrometry Analysis of Cross-linked Products
Cells of CAG48238 carrying pEVOL-pBpF and either pTTQ18-his10-rpoH(L47amb) or pTTQ18-his10-rpoH(K51amb) were grown at 30 °C in L-medium supplemented with 0.02% arabinose and 1 mM pBPA until early log phase (0.2 TAITEC units), and induced with 1 mM IPTG for 1 h. Cells were UV-irradiated as described above, disrupted by sonication in 10 mM Tris-HCl (pH 8.1) at 0 °C. After ultracentrifugation of cell lysates at 100,000 × g for 60 min, 3.8 ml of supernatants were mixed with 1.2 ml of Wash buffer A (50 mM Tris-HCl (pH 8.1), 150 mM NaCl, 0.1% SDS, 0.1 mM EDTA), and incubated with TALON resin (Takara Bio) at room temperature for 2.5 h with rotation. After washing the resin with Wash buffer A, proteins were eluted with wash buffer containing 81 mM EDTA. Eluted proteins were separated on SDS-PAGE and silver stained with Sil-Best Stain One (NACALAI TESQUE). The bands detected after UV radiation were excised and digested in gel with a TPCK-treated bovine trypsin (Worthington Biochemical). Then digest was analyzed by nano liquid chromatography–tandem mass spectrometry (LC-MS/MS) using Q Exactive mass spectrometer (Thermo Fisher Scientific). The peptides were separated using nano ESI spray column (75 μm [ID] × 100 mm [L], NTCC analytical column C18, 3 μm, Nikkyo Technos) with a linear gradient of 0%-35% buffer B (100% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL/min over 10 min (Easy nLC; Thermo Fisher Scientific). The mass spectrometer was operated in the positive-ion mode, and the MS and MS/MS spectra were acquired in a data-dependent TOP10 method. The MS/MS raw data set was searched against the NCBI-nr database using local MASCOT server (version 2.3; Matrix Science Ltd., London, UK). The taxonomy was selected as Escherichia coli and the variable modifications were selected as acetyl (protein N-term), deamidated (NQ), formyl (protein N-term), Gln->pyro-Glu (N-term Q) and oxidation (M).
Immunoprecipitation of σ32–Ffh Cross-linked Products
Anti-Ffh immunoprecipitation of cross-linked products of His6-σ32pBPA and FfhpAzPA were carried out as follows. For His6-σ32pBPA-cross-linked products, UV-irradiated cells were suspended in 10 mM Tris-HCl (pH 8.1) and disrupted by sonication at 0 °C. After removal of total membranes by ultracentrifugation, proteins were precipitated with 5% TCA. For FfhpAzPA-cross-linked products, UV-irradiated cells were suspended in 10 mM Tris-HCl (pH 8.1), and total cellular proteins were precipitated with 5% TCA. TCA-precipitated proteins were washed with acetone, solubilized in buffer containing 50 mM Tris-HCl (pH 8.1), 1% SDS and 1 mM EDTA and diluted 33-fold with NP40 buffer (50 mM Tris-HCl (pH 8.1), 150 mM NaCl, 1% NP40). After centrifugation, supernatants were incubated with True-Blot anti-Rabbit Ig IP Beads (eBioscience) and either anti-Ffh antibodies or control antibodies at 4 °C for 13 h with rotation. Immunocomplexes were isolated by centrifugation, washed 2 times with wash buffer and once with 10 mM Tris-HCl (pH 8.1), and dissolved in SDS sample buffer. Proteins were separated by 7.5% SDS-PAGE, and analyzed by immunoblotting using appropriate antibodies, TrueBlot anti-Rabbit IgG (eBioscience), and Can Get Signal immunoreaction enhancer solution (TOYOBO) as described previously25.
Disulfide-Cross-linking
Cells of WAM121 carrying pTTQ18-his10-rpoH or pTTQ18-his10-rpoH(T52C) in addition to one of the pSTD689-ffh(Cys)+ffs plasmids were grown overnight at 30 °C in L-medium supplemented with 0.2% arabinose. After washing the cells 3 times with L-medium, they were inoculated into L medium, grown at 30 °C for 3 h and induced with 1 mM IPTG to express His10-σ32 and Ffh derivatives for 1 h. Cells were washed with 10 mM Tris-HCl (pH 8.1), suspended in 10 mM Tris-HCl (pH 8.1) and treated with 50 μM Cu2+(phenanthroline)3 at 37 °C for 5 min. The oxidation reaction was terminated by incubation with 2.5 mM neocuproine for 5 min at 37 °C followed by additional incubation with 12.5 mM N-ethylmaleimide (NEM) for 10 min at 0 °C. Cells were washed with 10 mM Tris-HCl (pH 8.1), suspended in 10 mM Tris-HCl (pH 8.1) containing 10 mM NEM and disrupted by sonication at 0 °C. After removal of unbroken cells, 2 ml of cell extracts were mixed with 0.5 ml of Wash buffer B (50 mM Tris-HCl (pH 8.1), 150 mM NaCl, 0.1% SDS), and incubated with TALON resin at room temperature for 2.5 h with rotation. After washing the resin with Wash buffer B, proteins were eluted with Wash buffer B containing 81 mM EDTA. Eluted proteins were mixed with 2x SDS-sample buffer with or without 10% β-mercaptoethanol and incubated for 5 min at 98 °C. Purified proteins were analyzed by 7.5% SDS-PAGE followed by anti-Ffh immunoblotting.
Additional Information
How to cite this article: Miyazaki, R. et al. A Novel SRP Recognition Sequence in the Homeostatic Control Region of Heat Shock Transcription Factor σ32. Sci. Rep. 6, 24147; doi: 10.1038/srep24147 (2016).
Supplementary Material
Acknowledgments
We thank A. H. Becker, C. A. Gross, M. Kanemori, B. Lim, R. McMacken, F. Motojima, and G. J. Phillip for providing antibodies and/or strains, K. Ito for critical reading and editing of the manuscript and helpful comments, Y. Hizukuri for discussion, and M. Sano for technical support. This work was supported by JSPS KAKENHI (Grant Numbers 15J05262, 23657128, 15H04350) and MEXT KAKENHI (Grant Number 15H01532).
Footnotes
Author Contributions R.M., T.Y., H.M. and Y.A. designed the research. R.M., T.S. and N.D. performed the experiments. R.M., H.M., T.S., N.D. and Y.A. analyzed the data. R.M., T.Y., T.S., N.D. and Y.A. wrote the manuscript.
References
- Morimoto R. I. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 91–9 (2011). [DOI] [PubMed] [Google Scholar]
- Grossman A. D., Erickson J. W. & Gross C. A. The htpR gene product of E. coli is a sigma factor for heat-shock promoters. Cell 38, 383–90 (1984). [DOI] [PubMed] [Google Scholar]
- Landick R. et al. Nucleotide sequence of the heat shock regulatory gene of E. coli suggests its protein product may be a transcription factor. Cell 38, 175–82 (1984). [DOI] [PubMed] [Google Scholar]
- Taylor W. E. et al. Transcription from a heat-inducible promoter causes heat shock regulation of the sigma subunit of E. coli RNA polymerase. Cell 38, 371–81 (1984). [DOI] [PubMed] [Google Scholar]
- Yura T., Tobe T., Ito K. & Osawa T. Heat shock regulatory gene (htpR) of Escherichia coli is required for growth at high temperature but is dispensable at low temperature. Proc. Natl. Acad. Sci. USA 81, 6803–7 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M. S. & Gross C. A. Stress-induced remodeling of the bacterial proteome. Curr. Biol. 24, R424–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H., Yuzawa H. & Yura T. Interplay of two cis-acting mRNA regions in translational control of σ3.2. synthesis during the heat shock response of Escherichia coli. Proc. Natl. Acad. Sci. USA 88, 10515–9 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa H., Nagai H., Mori H. & Yura T. Heat induction of σ32 synthesis mediated by mRNA secondary structure: a primary step of the heat shock response in Escherichia coli. Nucleic Acids Res. 21, 5449–55 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M., Kanemori M., Yanagi H. & Yura T. Heat-induced synthesis of σ32 in Escherichia coli: structural and functional dissection of rpoH mRNA secondary structure. J. Bacteriol. 181, 401–10 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M. T. et al. Translational induction of heat shock transcription factor σ32: evidence for a built-in RNA thermosensor. Genes Dev. 13, 655–65 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus D. B., Walter W. A. & Gross C. A. The heat shock response of E. coli is regulated by changes in the concentration of σ32. Nature 329, 348–51 (1987). [DOI] [PubMed] [Google Scholar]
- Wild J., Walter W. A., Gross C. A. & Altman E. Accumulation of secretory protein precursors in Escherichia coli induces the heat shock response. J. Bacteriol. 175, 3992–7 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemori M., Mori H. & Yura T. Induction of heat shock proteins by abnormal proteins results from stabilization and not increased synthesis of σ32 in Escherichia coli. J. Bacteriol. 176, 5648–53 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman C., Thevenet D., D’Ari R. & Bouloc P. Degradation of σ32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc. Natl. Acad. Sci. USA 92, 3516–20 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoyasu T. et al. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor σ32. EMBO J. 14, 2551–60 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisbert E., Herman C., Lu C. Z. & Gross C. A. A chaperone network controls the heat shock response in E. coli. Genes Dev. 18, 2812–21 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamer J., Bujard H. & Bukau B. Physical interaction between heat shock proteins DnaK, DnaJ, and GrpE and the bacterial heat shock transcription factor σ32. Cell 69, 833–42 (1992). [DOI] [PubMed] [Google Scholar]
- Gamer J. et al. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor σ32. EMBO J. 15, 607–17 (1996). [PMC free article] [PubMed] [Google Scholar]
- Straus D., Walter W. & Gross C. A. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of σ32. Genes Dev. 4, 2202–9 (1990). [DOI] [PubMed] [Google Scholar]
- Tomoyasu T., Ogura T., Tatsuta T. & Bukau B. Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli. Mol. Microbiol. 30, 567–81 (1998). [DOI] [PubMed] [Google Scholar]
- Blaszczak A., Georgopoulos C. & Liberek K. On the mechanism of FtsH-dependent degradation of the σ32 transcriptional regulator of Escherichia coli and the role of the DnaK chaperone machine. Mol. Microbiol. 31, 157–66 (1999). [DOI] [PubMed] [Google Scholar]
- Horikoshi M., Yura T., Tsuchimoto S., Fukumori Y. & Kanemori M. Conserved region 2.1 of Escherichia coli heat shock transcription factor σ32 is required for modulating both metabolic stability and transcriptional activity. J. Bacteriol. 186, 7474–80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrist M. & Narberhaus F. Identification of a turnover element in region 2.1 of Escherichia coli σ32 by a bacterial one-hybrid approach. J. Bacteriol. 187, 3807–13 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yura T. et al. Analysis of σ32 mutants defective in chaperone-mediated feedback control reveals unexpected complexity of the heat shock response. Proc. Natl. Acad. Sci. USA 104, 17638–43 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B. et al. Heat shock transcription factor σ32 co-opts the signal recognition particle to regulate protein homeostasis in E. coli. PLoS Biol. 11, e1001735 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian D., Shen K., Zhang X. & Shan S. O. Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem. 82, 693–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luirink J., Yu Z., Wagner S. & de Gier J. W. Biogenesis of inner membrane proteins in Escherichia coli. Biochim Biophys Acta 1817, 965–76 (2012). [DOI] [PubMed] [Google Scholar]
- Janda C. Y. et al. Recognition of a signal peptide by the signal recognition particle. Nature 465, 507–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainzl T., Huang S., Merilainen G., Brannstrom K. & Sauer-Eriksson A. E. Structural basis of signal-sequence recognition by the signal recognition particle. Nat. Struct. Mol. Biol. 18, 389–91 (2011). [DOI] [PubMed] [Google Scholar]
- Hainzl T. & Sauer-Eriksson A. E. Signal-sequence induced conformational changes in the signal recognition particle. Nat. Commun. 6, 7163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. W., Martin A. B., King D. S., Wang L. & Schultz P. G. Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc. Natl. Acad. Sci. USA 99, 11020–4 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. W. et al. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 124, 9026–7 (2002). [DOI] [PubMed] [Google Scholar]
- Chin J. W. & Schultz P. G. In vivo photocrosslinking with unnatural amino Acid mutagenesis. Chembiochem 3, 1135–7 (2002). [DOI] [PubMed] [Google Scholar]
- Koide K., Ito K. & Akiyama Y. Substrate recognition and binding by RseP, an Escherichia coli intramembrane protease. J. Biol. Chem. 283, 9562–70 (2008). [DOI] [PubMed] [Google Scholar]
- Young T. S., Ahmad I., Yin J. A. & Schultz P. G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–74 (2010). [DOI] [PubMed] [Google Scholar]
- Lancia J. K. et al. Sequence context and crosslinking mechanism affect the efficiency of in vivo capture of a protein-protein interaction. Biopolymers 101, 391–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Rashid R., Wang K. & Shan S. O. Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science 328, 757–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstrom D. et al. Consequences of depletion of the signal recognition particle in Escherichia coli. J. Biol. Chem. 286, 4598–609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Henry R., Yuan J., Cline K. & Hoffman N. E. A chloroplast homologue of the signal recognition particle subunit SRP54 is involved in the posttranslational integration of a protein into thylakoid membranes. Proc. Natl. Acad. Sci. USA 92, 3789–93 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F. et al. Molecular basis for regulation of the heat shock transcription factor σ32 by the DnaK and DnaJ chaperones. Mol. Cell 32, 347–58 (2008). [DOI] [PubMed] [Google Scholar]
- Suzuki H. et al. Synergistic binding of DnaJ and DnaK chaperones to heat shock transcription factor σ32 ensures its characteristic high metabolic instability: implications for heat shock protein 70 (Hsp70)-Hsp40 mode of function. J. Biol. Chem. 287, 19275–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi A., Ikeda A., Mezaki M., Fukumori Y. & Kanemori M. DnaJ-promoted binding of DnaK to multiple sites on σ32 in the presence of ATP. J. Bacteriol. 196, 1694–703 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest O., Hoskins J. R., Camberg J. L., Doyle S. M. & Wickner S. Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc. Natl. Acad. Sci. USA 108, 8206–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb R., Zhang Z. R., Appathurai S. & Mariappan M. A functional link between the co-translational protein translocation pathway and the UPR. Elife 4, e07426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gier J. W. et al. Assembly of a cytoplasmic membrane protein in Escherichia coli is dependent on the signal recognition particle. FEBS Lett. 399, 307–9 (1996). [DOI] [PubMed] [Google Scholar]
- Nakatogawa H., Mori H., Matsumoto G. & Ito K. Characterization of a mutant form of SecA that alleviates a SecY defect at low temperature and shows a synthetic defect with SecY alteration at high temperature. J. Biochem. 127, 1071–9 (2000). [DOI] [PubMed] [Google Scholar]
- Narita S., Masui, C., Suzuki, T., Dohmae, N. & Akiyama, Y. Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. USA 110, E3612–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.