

Abstract
The epidermal growth factor receptor family (ErbB2/Her2 and EGFR/ErbB1/Her1) often modulates the transcriptional program involved in promoting mammary tumorigenesis. In humans, more than 70% of ErbB2-positive sporadic breast cancers harbor p53 mutations, which correlate with poor prognosis. Also, the extremely high incidence of ErbB2-positive breast cancer in women with p53 germ-line mutations (Li-Fraumeni Syndrome) suggests the key role of mutant p53 specifically in ErbB2-mediated mammary tumorigenesis. To examine the role of mutant p53 during ErbB2-mediated mammary tumorigenesis we introduced a mutant p53 R172H allele into a (MMTV)-ErbB2/Neu mouse model. We show in heterozygous p53 mice that mutp53 R172H is a more potent activator of ErbB2-mediated mammary tumorigenesis than simple loss of p53. The more aggressive disease in mutant p53 animals was reflected by earlier tumor onset, increased mammary tumor multiplicity, and shorter survival. We provide molecular evidence that mutant p53 amplifies ErbB2 and EGFR signaling to promote the expansion of mammary stem cells and induce cancer cell proliferation. This study therefore identifies mutant p53 as an essential player in ErbB2 and EGFR-mediated breast cancer and indicates the potential translational importance of targeting mutant p53 in this subset of breast cancer patients.
Keywords: mutant p53, mammary stem cells, ErbB2, EGFR, Hsp90
Introduction
Breast cancer (BC) in women is the most frequent life-threatening disease, which have a complex, heterogeneous genetic and biochemical background. No single genomic alteration can be regarded as decisive for its formation and progression. However, a few key players have been identified and among them are the p53 tumor suppressor and members of the Epidermal Growth Factor Receptor family, such as Her2 (human EGF receptor 2, alias ErbB2) and EGFR (Epidermal Growth Factor Receptor, alias ErbB1/Her1).
According to a recent comprehensive analysis, mutations in p53 gene are the most frequent genetic alteration found in BC1, 2, with high occurrence in the Her2 positive (72%) and basal-like (80%) BC subtypes. Whereas mutations in p53 gene are rather rare in Luminal A (12%) and Luminal B (29%) BCs, suggesting that mutp53 may cooperate with certain oncogenenic pathways to promote mammary tumorigenesis. The significance of mutant p53 (mutp53) in Her2-positive BC initiation is also supported by its frequent occurrence in Li-Fraumeni Syndrome (LFS), a hereditary cancer disorder associated with p53 germ-line mutations. BC is the most common event in LFS, accounting for 49% of LFS women3. Importantly, LFS patients with germ line p53 mutations have more than 70% incidence of Her2 BC compared to the 20-30% of sporadic BC with deregulated Her23, 4. Therefore, mutp53 germ-line mutations predispose LFS women for Her2-positive BC development, suggesting a critical role of mutp53 in pathogenesis specifically of this subtype of BC. Additionally, mutp53 Her2 BCs have a worse prognosis and susceptibility to metastatic recurrence than wildtype p53 (wtp53) Her2 BC5, 6. Therefore, mutp53 and Her2 oncogenes appear to strongly cooperate. While indicating oncogenic synergy between mutp53 and Her2 in the clinic, the cellular and molecular basis of this cooperation is completely unknown.
A rising interest in cancer stem cells (SCs), combined with the idea that SCs and/or early progenitors might be targets of neoplastic transformation7, 8, 9, drew attention to the role of p53 and its tumor suppression activities in SC homeostasis. Recent findings suggest the intriguing possibility that wtp53 carries out its tumor suppressor function by inhibition of SCs symmetric division and blocking of reprogramming of somatic/progenitor cells into SCs10. The observation that p53 controls stem cell maintenance is coupled with the notion that p53 mutations may contribute to stem cell evolution. Compared to ‘simple’ p53 deficiency, the presence of mutp53 markedly enhances the efficiency of reprogramming somatic fibroblasts into induced pluripotent stem cells (iPS) 11. This suggests that reprogramming of somatic mutp53 cells may result in the generation of stem-like cells with malignant potential. Also, we9 and others12 have shown that mutp53 promotes expansion of normal progenitors of different tissue origins, compared to the p53 null allele in vivo. Yet, the underlying mechanisms of these observations remain to be elucidated.
ErbB2/Her2 is a tyrosine kinase transmembrane receptor that forms heterodimers with other EGFR family members, including EGFR/Her1, to stimulate oncogenic signaling 13. Overexpression of ErbB2 in BC activates pathways that promote cell proliferation, reduce apoptosis and increase metastasis14. Intriguingly, recent reports demonstrated the novel oncogenic function of ErbB2 signaling that, at least in part, drives mammary carcinogenesis through its effects on normal and malignant mammary stem cells15, 16, 17. Similarly, the sustained activation of EGFR signaling may play critical functions for high self-renewal potential, survival, invasion and metastases of cancer stem/progenitor cells and their progenies 18.
Here we demonstrate that mutp53 cooperates with oncogenic ErbB2 signaling in mammary tumor development using a newly generated (MMTV)-ErbB2/Neu mouse model containing a knock-in mutp53 R172H allele. In heterozygous animals, containing one wt p53 allele, the mutant p53 allele accelerated ErbB2-mediated mammary tumorigenesis compared to their null p53 allele counterparts. In the ErbB2 mouse model, the mutp53 allele induces multicentric mammary tumor formation, earlier tumor onset and shorter survival, which may be mediated by the expansion of normal mammary progenitors and/or cancer stem cells. Moreover, we elucidated a mechanistic underpinning of mutp53-Epidermal Growth Factor Receptors (EGFR/ErbB2) cooperation, which we propose is based on novel tumorigenic activity of mutp53. This study for the first time provides evidence that mutp53, via augmented ErbB2/EGFR signaling, promotes the proliferation of mammary cells and expands mammary stem cell populations. This establishes a new role of mutp53 in BC stem cell biology and opens the opportunities for targeting mutp53 as a therapeutic strategy in Her2/EGFR positive BC.
Results
Mutant R172H p53 promotes mammary tumorigenesis in ErbB2 mouse model
While ample clinical evidence suggests strong oncogenic cooperation between mutp53 and ErbB2/Her2 in human BC, its underlying mechanism remains unknown. To date, the best available LFS mouse models are ‘straight’ mutp53 knockin mice 1, 19. However, in contrast to human LFS patients, which mainly develop Her2 positive BC in women, these mice mainly develop hematopoietic malignancies and are not suitable for BC research1, 19. The extremely high incidence of Her2 positive BC in LFS women3, 4 suggests the essential role of mutp53 in specifically Her2-mediated mammary tumorigenesis. To faithfully recapitulate LFS disease and explore oncogenic activity of mutp53 in Her2-positive breast cancer we generated a novel mutp53 mouse model by crossing the hotspot mutp53 knockin allele p53 R172H (referred to as p53H hereafter) into the classic MMTV-ErbB2 Tg mouse20. Four experimental cohorts, all expressing a single copy of activated ErbB2, were generated: homozygous p53H/H; ErbB2 and p53-/-; ErbB2 littermates, and heterozygous p53H/+; ErbB2, and p53-/+; ErbB2 littermates.
Compared to heterozygous animals, homozygous mice (H/H;ErbB2 and p53-/-;ErbB2) show accelerated tumorigenesis, due to rapid development of aggressive mammary tumors and lymphomas. 50% of p53 -/-;ErbB2 and 37% of H/H;ErbB2 mice developed mammary tumors only, whereas 43% of p53-/-;ErbB2 and 52% of H/H;ErbB2 mice developed lymphomas concurrently with mammary tumors or only lymphomas (Table 1). The survival curves of p53-/-;ErbB2 and of p53H/H;ErbB2 mice were identical (median age of death 135.5 and 137 days, respectively), seemingly due to the aggressive nature of ErbB2-mediated disease and high incidence of lymphomas (Fig 1A). In contrast, there is a marked difference in both tumor free survival and overall survival (Fig 1A) between the heterozygous cohorts of p53H/+;ErbB2 and p53-/+;ErbB2 mice (median age of death 278 and 329 days, respectively). Heterozygous cohorts developed mainly mammary tumors (86% of p53-/+;ErbB2 and 95% of p53H/+;ErbB2 mice) (Table 2). A more aggressive phenotype caused the shorter survival of p53H/+;ErbB2 mice, reflected by development of twice the number of simultaneously initiated, multicentric tumors that arise in single or multiple mammary glands in p53H/+;ErbB2 females (average 6.1 tumors per mouse) compared to p53-/+;ErbB2 mice (average 2.7 tumors per mouse) (Fig 1B, Table 2). Strikingly, some p53H/+;ErbB2 animals developed up to 15 visible individual mammary tumors. Homozygous p53H/H;ErbB2 mice, which contained fewer mammary tumors compared to H/+;ErbB2 mice, also developed twice more mammary tumors (2.5 tumors/per mouse) compared to p53-/-;ErbB2 mouse (1.5 tumors/mouse). Apparently, the short lifespan of homozygous animals did not provide sufficient time to observe the drastic differences in both survival and tumor multiplicity between p53 homozygous genotypes.
Table 1.
Tumor Spectrum and tumor multiplicity in p53 -/-;ErbB2 and p53 H/H;ErbB2 mice.
| Tumor types | p53 -/-;ErbB2 (n=16) | p53 H/H;ErbB2 (n=19) |
|---|---|---|
| Mammary Adenocarcinoma | 8 (50%) | 7 (37%) |
| Lymphoma | 1 (6.25%) | 5 (26%) |
| Mammary + Lymphoma | 6 (37.5%) | 5 (26%) |
| Sarcoma | 1 (6.25%) | 2 (10%) |
| Average mammary tumor number per mouse | 1.55 | 2.5 |
Fig. 1. Mutant R172H p53 promotes mammary tumorigenesis in the ErbB2 mouse model.
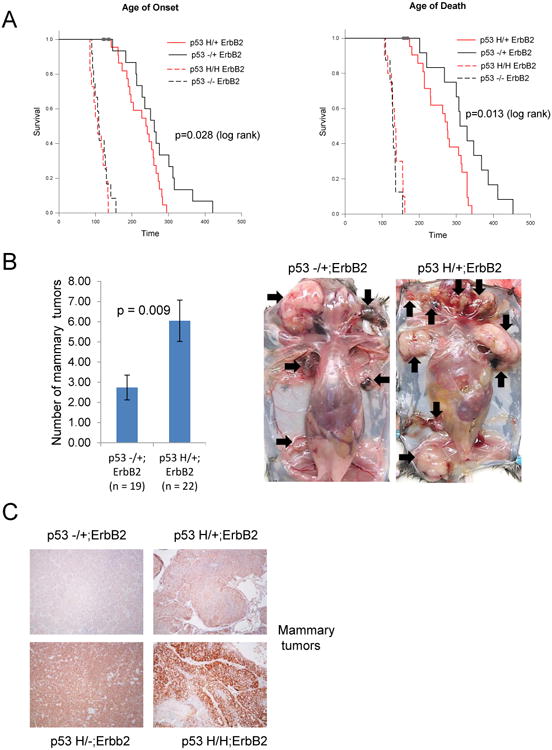
(A) Mutant p53 R172H allele (p53H) promotes earlier mammary tumors onset and death in ErbB2 mouse model. Kaplan-Meier tumor free survival curves of p53H/+;ErbB2 (red curve, n=22) vs p53-/+;ErbB2 (black curve, n=22) (solid lines) mice and p53H/H;ErbB2 (red curve, n=11) vs p53-/-;ErbB2 (black curve, n=12) mice (dashed lines). p-values are indicated. For survival curves only mice developed mammary tumors were selected.
(B) Left, Mutant p53 R172H allele promotes tumor multiplicity. Average number (±SD) of mammary tumors per mouse per genotype. Right, representative images of p53H/+ErbB2 vs p53-/+ ErbB2 mice. Arrows indicate individual mammary tumors.
(C) p53 levels in mammary tumors from animals with indicated genotypes were examined by immunohistochemistry. Paraffin-embedded mammary tumors were stained with a p53 (CM5)antibody. Representative images (10×).
Table 2.
Tumor Spectrum and tumor multiplicity in p53 -/+;ErbB2 and p53 H/+;ErbB2 mice.
| Virgins | Parous | |||
|---|---|---|---|---|
|
|
|
|||
| Tumor types | p53 -/+ (n=19) | p53 H/+ (n=22) | p53 -/+ (n=18) | p53 H/+ (n=18) |
| Mammary Adenocarcinoma | 12 (86%) | 13 (95%) | 17 (94%) | 17 (94%) |
| Lymphoma | 1 (7%) | 1 (7%) | 0 | 1 (6%) |
| Sarcoma | 2 (14%) | 0 | 0 | 0 |
| Non Mammary Carcinoma | 0 | 1 (7%) | 0 | 0 |
| Average mammary tumor number per mouse | 2.7 | 6.1 | 6.5 | 10.3 |
An important feature of mutp53 cancers is the tumor-specific stabilization of mutp53 proteins, which is thought to be critical for oncogenic activity of mutp5321, 22. Since the mechanisms and physiological consequences of mutp53 stabilization in cancer may have significant translational impact, we evaluated the levels of p53 in vivo in the mutp53/ErbB2 mouse model. Mutp53, but not wtp53, is stabilized in mammary tumors of both homozygous and heterozygous mice (Fig 1C). Mutp53 staining in mammary tumors of p53H/+;ErbB2 mice was heterogeneous with less than 50% of the tumor cells showing prominent nuclear mutp53 staining (Fig 1C). However, the majority of primary mammary tumors from p53H/H;ErbB2 animals have strong nuclear mutp53 expression (Fig 1C). To determine if wtp53 activity in p53H/+; ErbB2 animals prevents mutp53 stabilization, tumors from an additional cohort of p53H/-; ErbB2 mice were analyzed. p53 nuclear accumulation in p53H/-; ErbB2 tumors, on average, was higher than in p53H/+;ErbB2 tumors but lower than in p53H/H; ErbB2 tumors (Fig 1C). These results may imply that even in the presence of mutp53 allele remaining wtp53 allele preserves partial transcriptional activity and via MDM2 induction may to some extent promote mutp53 degradation. Nevertheless, elevated mutp53 protein levels in mammary tumors of p53H/+;ErbB2 mice appear to contribute to more aggressive disease compared to p53-/+;ErbB2 mice in the context of oncogenic ErbB2 stimulation (Fig 1A).
Mutant p53 expands the population of cells with stem-like properties
Stabilization of mutp53 in mammary tumors and increased mammary tumor multiplicity in the p53H/+; ErbB2 animals suggests a novel tumor promoting function of mutp53 in ErbB2 – driven mammary cancer initiation. Tumor initiation is thought to be attributed to the presence of specific population of cells, called Tumor Initiating Cells (TICs) with stem cells properties 8. Therefore, the observed phenotype in the mutp53-ErbB2 model may be a physiological consequence of an expansion of mammary SCs and/or progenitors. Thus, we hypothesize, that mutp53 promotes the acquisition of stem-like properties within mammary cell populations and confers a physiological expansion of SCs and/or progenitors in breast tissue.
First, we tested whether, similar to hematopoietic and mesenchymal progenitors9, mutp53 presence alone is sufficient to expand mammary luminal progenitors, identified as a main targets of ErbB2/Neu oncogenic transformation in vivo23. Thus, FACS cell sorting was used to analyze the number of luminal progenitors (CD61+CD24HighCD29Low) 23 derived from normal mammary of 8 wk old mutant p53H/H mice in comparison to p53-/- littermates. The luminal progenitor pool was significantly expanded in p53H/H mice, independent of the presence of the activated ErbB2 oncogene, compared to p53-/- and p53wt animals (Fig 2A). This suggests mutp53 alone can drive the expansion of mammary progenitors and possibly stem cell populations. Mutp53 `also induces stem-like properties of immortalized mouse mammary epithelial cells (MECs) compared to p53-/- MECs as shown by increased expression of aldehyde dehydrogenase (ALDH) (Fig 2B), which identifies mammary epithelial cells with stem cell characteristics24. Interestingly, the highest level of ALDH was detected in MECs expressing both mutp53 and ErbB2 (Fig 2B), which confirms the synergistic oncogenic cooperation of mutp53 and ErbB2 in expansion cell population with stem-like properties.
Fig. 2. Mutant p53 expands the population of cells with stem-like properties.
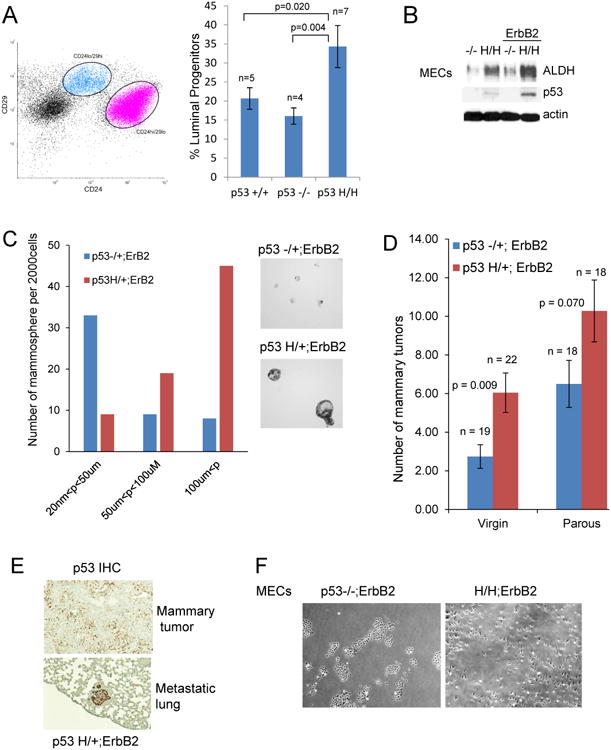
(A) Mutant p53 R172H promotes the expansion of luminal progenitors. Primary MECs isolated from mammary of wtp53, p53-/- and p53H/H mice are stained with CD61, CD24 and CD29. The luminal progenitor pool, defined by Lin- ;CD61+CD24high;CD29low. The presented data originates from MECs independently isolated from 7 p53 H/H mice, 4 p53-/- mice and 5 wtp53 mice.
(B) The presence of the mutant p53 allele increases ALDH+ levels in primary MECs in both straight p53H/H mice (lane 1, 2) and p53H/H;ErbB2 mice (lane 3,4) compared to p53 null littermates.
(C) The mutant p53 allele induces expansion of primary mammary epithelial stem cells in ErbB2 mice. Size comparison of mammospheres from MECs of p53H/+;ErbB2 vs p53-/+;ErbB2 mice (10×). Representative experiment shown.
(D) Parity induces higher tumor multiplicity in mice of both genotypes p53H/+;ErbB2 and p53-/+;ErbB2 compared to their virgin counterparts. Parous p53H/+;ErbB2 mice show the higher number of mammary tumors (10.3 tumors per mouse) compared to p53-/+; ErbB2 parous littermates (6.5 tumors per mouse).
(E) Mutant p53 is highly stabilized in lung metastasis compared to heterogeneous p53 staining in primary tumors from the same mouse. Representative image. Matching metastatic lungs/tumor specimens derived from 5 independent mice were analyzed. p53 IHC, 10×magnification.
(F) Mutant p53 allele changes cell morphology from epithelial in p53-/-;ErbB2 MECs to mesenchymal in H/H;ErbB2 MECs. 4× magnification.
Previous studies demonstrated that the number of mammospheres generated upon serial passaging provides an indirect measure of mammary stem cell self-renewal capacity, while mammosphere size reflects progenitor cell proliferation25. Therefore, mammospheres were generated from primary MECs isolated from normal mammaries of p53H/+;ErbB2 and p53-/+;ErbB2 littermates. p53H/+;ErbB2 MECs consistently produced a greater number and larger mammospheres (a 2-fold increase in average size) compared to p53-/+;ErbB2 MECs (Fig 2C). This provides evidence that mutp53 induces mammary stem cell presence and proliferation in ErbB2 animals.
The mammary gland is a highly dynamic tissue, undergoing significant morphological changes during puberty, pregnancy, lactation and involution. It is composed of two major epithelial populations, myoepithelial and luminal cells, which derive from myoepithelial and luminal progenitors, respectively26. A third and intriguing population of mammary progenitors, parity-induced mammary epithelial cells (PI-MECs), are pregnancy-responsive, apoptosis-resistant (in involution), self-renewing progenitors that undergo immense proliferation during pregnancy to give rise to milk-producing alveoli7, 27. A percentage of PI-MECs survive post-lactational involution after each round of pregnancy, which results in an accumulation of these progenitors in non-parous mammary glands compared to virgins27. The surviving PI-MECs self-renew and give rise to future ductal and alveolar structures7, 28. It was previously reported that PI-MEC progenitors are targets of ErbB2-mediated tumor transformation and animals garner an increased susceptibility to ErbB2-mediated tumorigenesis as the number of PI-MEC progenitors increase with each pregnancy29. In agreement with this observation, parous p53H/+;ErbB2 and p53-/+;ErbB2 have accelerated tumorigenesis compared to their virgin counterparts (Table 2 and Fig 2D). Parous mice of both genotypes developed primarily mammary tumors and had earlier tumor onset and shorter survival compared to virgins (data not shown). Interestingly, parity appeared to accelerate mammary tumorigenesis more prominently in the presence of the mutp53 allele. There was a trend of pregnancy having a greater impact on increasing mammary tumor multiplicity in the presence of the mutp53 allele of p53H/+; ErbB2 animals (10.3 tumors per mouse) compared to p53-/+; ErbB2 littermates (6.5 tumors per mouse) (Fig 2D). Pregnancy dependent acceleration of mammary tumorigenesis suggests mutp53 may target and promote the expansion of the PI-MEC progenitor pool, similar to its effect on other tissue progenitors9, 12. However, more rigorous studies are required to definitively address this question.
It is well established that primary and secondary tumor initiation sites, and the acquisition of epithelial-mesenchymal transition (EMT) features, are attributed to the presence of stem cells8, 30. Although p53 staining of primary mammary tumors from p53H/+;ErbB2 mice was heterogeneous (Fig 1C), secondary metastatic lesions in the lungs of the same mice had homogeneous and highly stabilized mutp53 (Fig 2E). In contrast, there was no stabilization of wtp53 in metastases in p53-/+;ErbB2 littermates (data not shown). This suggests that primary tumor cells with highly stabilized mutp53 stimulate invasion, have higher survival ability and/or tumor initiating potential at the metastatic site, which might be a physiological consequence of mammary SC expansion in the presence of the mutp53 allele.
In sum, our data suggests a shift to a more aggressive mammary tumor phenotype when the mutp53 allele is present in ErbB2 transformed mammary cells of both virgin and parous animals, which, at least in part, is mediated by the expansion of mammary progenitors and/or stem cells.
Mutp53 allele amplifies the ErbB2 signaling in vivo
To understand how mutp53 may enhance SC behavior in the ErbB2 context, we examined the pathways that are known to be involved in acquisition of stem-like properties in primary mammary tumors originating from p53H/+;ErbB2 and p53-/+;ErbB2 mice.
To this end we examined 68 primary tumors originating in virgin p53H/+;ErbB2 and p53-/+;ErbB2 mice. Although we did not observe statistically significant differences in Notch, Sox9, Slug, Oct4 8, 30, 31 by immunoblot analyses, we found that both total ErbB2 (p=0.0066) and activated p-ErbB2 (p=0.0005) levels were elevated in the presence of the mutp53 allele (Fig 3A, 3B). Concurrently, there was more activated forms of p-ERK, downstream effectors of ErbB2 activation13, in p53H/+;ErbB2 tumors compared to p53-/+;ErbB2 tumors (Fig 3A). This suggests mutp53 amplifies ErbB2 signaling in vivo.
Fig. 3. Mutant p53 promotes mammary cell proliferation.
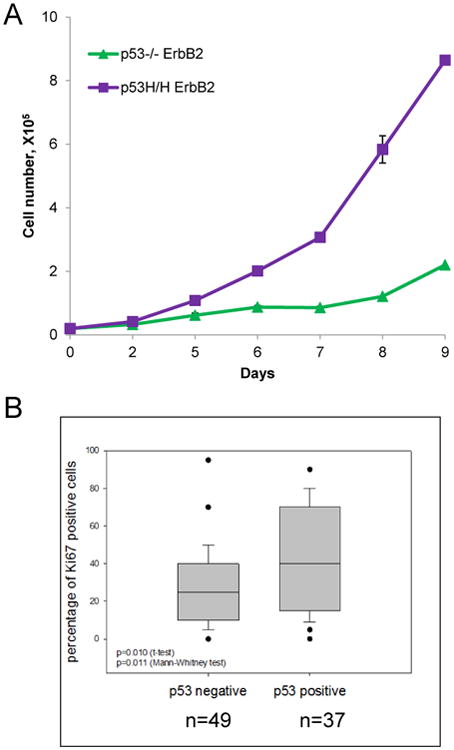
(A) To measure cell growth rates, MECs of different genotypes were plated at day 0 and counted at indicated time points. Representative graph from two independent experiments.
(B) Mutant p53 enhances cells proliferation in Her2 positive human cancers. Tissue microarrays of 150 breast cancers biopsies with known molecular status (ER+,PR+,Her2+ or triple negative) and percent of Ki67 positive cells in every individual core. p53 staining was used as surrogate marker for p53 mutation. p53 staining correlates with Ki67 positivity in Her2 positive breast cancer biopsies. (p= 0.01- t-test, p=0.011- Mann-Whithey test).
Previous studies provide strong evidence that ErbB2-mediated tumorigenesis and invasion might be determined by its effects on normal and malignant mammary stem/progenitor cells15, 16, 17. Consistent with our current in vivo data, we recently showed in vitro that RNAi mediated downregulation of mutp53 in ErbB2 positive SKBR3 human breast cancer cells promotes ErbB2 degradation32. Conversely, ectopic overexpression of the R175H p53 mutant increases ErbB2 levels in SKBR3 cells 32. Hence, mutp53, via amplification of ErbB2 signaling, may promote the expansion of the SC pool, leading to increased metastatic recurrence, as observed in mutp53/HER2 -positive breast cancer patients5.
Mutant p53 promotes mammary cells proliferation
The other tumor promoting function of mutp53 in ErbB2 context, which may not relate to SC biology, is a stimulation of mammary cells proliferation. Early stages of breast cancer (hyperplasia and ductal carcinoma in situ (DCIS), are characterized by an increased proliferation of epithelial cells, a loss of acinar organization and filling of the luminal space, followed by acquisition of invasive behavior at the later stages of mammary tumorigenesis. Activation of ErbB2 in acini induces the proliferation and generation of multi-acinar structures with filled lumen, indicating that ErbB2 activation overcomes the growth inhibitory effect of polarized epithelial cells33. Therefore, mutp53 mediated amplification of ErbB2 signaling may lead to enhanced proliferation of mammary luminal cells and thereby promote tumor initiation. Growth curve analysis of primary MECs isolated from p53H/H; ErbB2 and p53-/-;ErbB2 mammary glands confirmed that the presence of the mutp53 allele promotes more rapid proliferation compared to the p53 null (Fig 4A). Our data imply that mutp53 provides MECs a growth advantage allowing cells to bypass contact inhibition to greater extent than p53 null MECs. These hyperprolifereative properties of H/H;ErbB2 MECs may closely relate to disruption of mammary tissue architecture, characteristic for early stages mammary tumorigenesis in vivo.
Fig. 4. Mutant p53 allele amplifies the ErbB2 signaling in vivo.
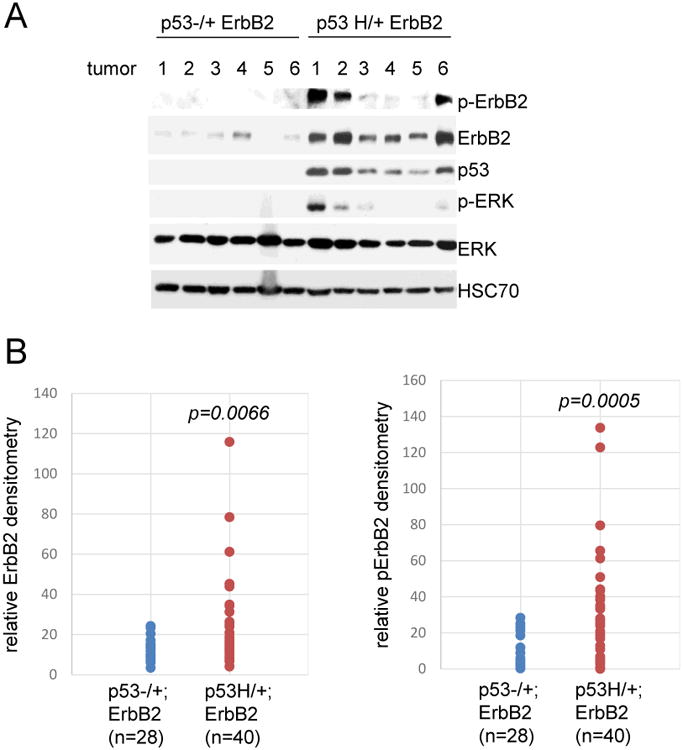
(A) Representative Western blot from primary mammary tumors derived from p53-/+;ErbB2 and p53H/+;ErbB2 mice. Cells were lysed and phosphorylatied p-ErbB2 (residue Y1248) and p-ERK (residue 202/204) were determined by immunoblot analysis using phospho-specific antibodies. HSC as a loading control.
(B) Histogram of relative densitometry of ErbB2 and activated p-ErbB2 Western blot analysis derived from p53-/+;ErbB2 (n=28) vs p53H/+;ErbB2 (n=40) primary mammary tumors. p values are indicated.
To test if mutp53-enhanced proliferation holds true in human mammary tumors, we examined tissue arrays of 150 breast cancers biopsies with known molecular status (ER+, PR+, Her2+ or triple negative) and the percent of Ki67 positive cells in every individual core. Stabilized p53 staining was used as a surrogate marker for mutant p53. p53 staining intensity was blindly scored from - to +++, and p53 staining ++ to +++ were selected as p53 positive cores. The intensity of p53 staining was compared to Ki67 staining of Her2 positive-, ER/PR-negative breast cancer biopsies. A clear correlation between p53 and Ki67 staining (p= 0.01- t-test, p=0.011-Mann-Whithey test) was determined by comparing 49 p53 negative and 37 p53 positive HER2-positive (3+) tumors. This convincingly showed that mutp53-Her2 positive breast cancer has a significantly higher proliferative index compared to p53 negative tumors in humans (Fig 4B). Thus, this indicates the presence of mutp53 enhances proliferation of both ErbB2 mouse mammary cells and human Her2 positive breast cancer cells, in addition to the expansion of mammary SCs, and this may underlie the mechanism for mutp53-ErbB2 oncogenic cooperation.
Mutant p53 modulates breast cancer stem cells via amplification of EGFR signaling
Independent of mutp53 synergizing with ErbB2 signaling, our data also indicates mutp53 can increase the luminal progenitor pool in the absence of ErbB2 oncogenic stimulation (Fig 2A). This suggests that other pathways, besides ErbB2, may be involved in the induction of stem-like properties of mammary cells in a mutp53 dependent manner. Epidermal growth factor receptor EGFR (ErbB1/Her1) is another member of the Epidermal Growth Factor Receptor family that is closely relate to ErbB213. Sustained activation of EGFR can play important role for high self-renewal potential, survival, invasion and metastases of cancer stem/progenitor cells and their progenies18. Previous our and others data revealed mutp53 can enhance EGFR signaling by increasing its recycling efficiency34 and/or stability32.
To test if mutp53 expands a population of cells with stem-like properties via modulation of EGFR signaling we used the EGFR positive human breast cancer cell line MDA231, which expresses the p53 R280K mutant protein. We first generated a stable MDA231 cell line that ectopically overexpressed native mutp53 (R280K), referred to as MDA231 R280K (Fig 5A). The elevated level of mutp53 increased the size and number of mammospheres, which are key characteristics of self-renewal ability of SCs. Moreover, the replicative potential of mammospheres derived from MDA231-R280K cells increased progressively during passaging (Fig 5A, right) indicating an enrichment of the cancer SC pool. Mutp53 overexpression in MDA231 cells also increased the Aldefluor-positive/ALDH population compared to vector transfected cells, indicating mutp53 can enhance the presence of stem cells in MDA231 cells (Fig 5B).
Fig. 5. Mutant p53 modulates breast cancer stem cells via amplification of Epidermal Growth Factor Receptor signaling.
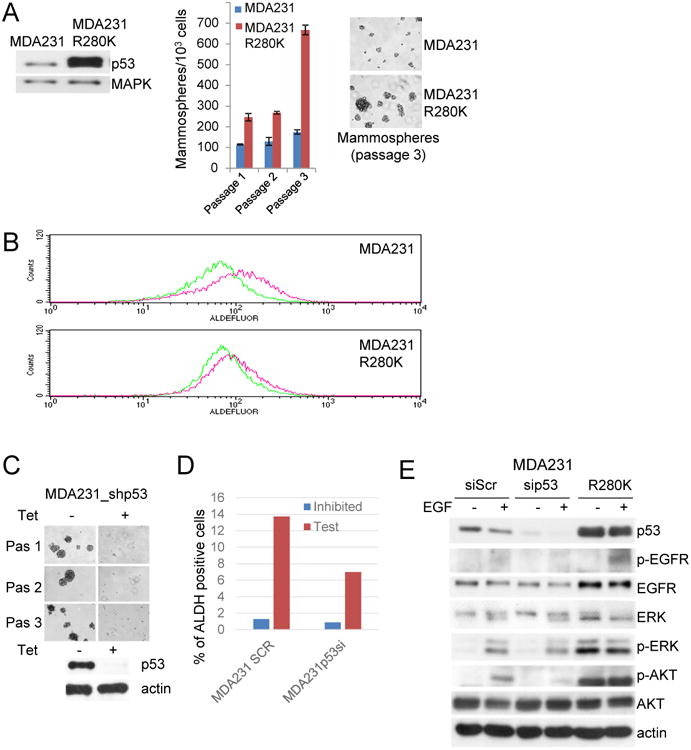
(A) Ectopic over-expression of native mutant p53 (R280K) (left) increases the mammospheres-initiating population and the size of MDA231 mammospheres (10×) (right). MAPK as a loading control.
(B) Mutant p53 (R280K) overexpression expands the Aldefluor-positive cell population in MDA231 cells, measured by FACS analysis. Representative experiment.
(C) Tet-inducible mutant p53 knockdown reduces the mammosphere forming ability of MDA231 cells. Top, mammosphere assay (10×). Bottom, Western blotting. Actin as a loading control.
(D) Mutant p53 downregullation decreases the Aldefluor-positive cell population in MDA231 cells. Bars represent percent of Aldefluor-positive cells (test) compared to substrate inhibited control (inhibited). Representative experiment.
(E) Mutant p53 allele amplifies EGFR signaling. MDA231 vector control, RNAi p53 depleted or cells with overexpressed mutant p53 were serum starved and then stimulated with EGF for 15 min. Cells were lysed and phosphorylation of EGFR (residue 845), AKT (residue 437), and ERK 1/2 (residue 202/204) were determined by immunoblot analysis using phospho-specific antibodies. Actin expression was used as loading control.
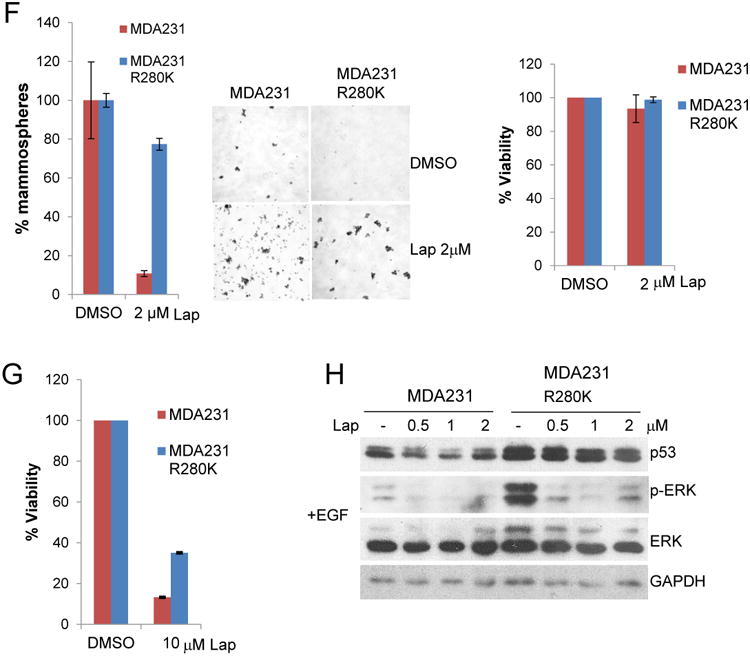
(F) Overexpression of mutant p53 impacts self-renewal ability of MDA231 cells upon EGFR inhibition. Lapatinib at low concentration (2μM), preserving cell viability (right), dramatically decreases mammosphere formation in MDA231 cells but not in MDA231cells overexpressing R280K mutant p53 (left). Middle, representative images of low adherent culture (10×).
(G) Inhibition of EGFR by high doses of Lapatinib (10 μM) has more profound effect on MDA231 cells while mutant p53 overexpression protects MDA231 cells from EGFR inhibition. CTB, cell viability assay.
(H) Inhibition of EGFR signaling by Lapatinib (24h with indicated concentrations) is partially rescued by mutp53 overexpression in MDA231R280K cells. Cells were serum starved overnight and then stimulated with EGF for 15 min before harvesting. GAPDH as a loading control.
An important question is whether mutp53 could be a relevant target for suppression of BC stem cells. Previous data from our lab and others have shown that acute RNAi or pharmacological downregulation of mutp53 inhibits the invasive properties of human MDA231 BC cells and tumorigenicity in xenograft assays35, 36. However, whether this effect is related to self-renewal properties of BC cells has not been evaluated. We generated MDA231 cells stably expressing tetracycline inducible shp53 in order to determine the effect of mutp53 expression on the cancer SC population. MDA231 cells containing tet-inducible shp53 were cultured in conditions that allow mammosphere formation and western blot analysis demonstrated the efficiency of mutp53 downregulation upon introduction of tetracycline (Fig 5C, bottom). Serial mammosphere re-plating of mock treated cells had a significant pool of cells with self-renewal properties, while mutp53 knockdown dramatically decreased mammosphere formation (Fig 5C, top). Consistent with the mammosphere assay, control shRNA transfected cells contained a 12% Aldefluor-positive population, which is indicative of stem cells, whereas mutp53 knockdown reduced this population by two-fold (Fig 5D). These results provide direct evidence that mutp53 regulates the self-renewal of BC stem cells.
Importantly, using these mutp53 overexpressing and knockdown lines we demonstrated mutp53 levels are a critical regulator of EGFR signaling. In MDA231 cells, total and phospho-activated EGFR levels were increased in the presence of over-expressed mutp53 and decreased after RNAi mediated ablation of mutp53 independent of exposure to ectopic epidermal growth factor (EGF) (Fig 5E). Mutp53 levels also proportionally modulate the activated downstream EGFR signaling pathways PI3K and MAPK, which phosphorylate AKT and ERK, respectively (Fig 5E).
In sum, our data implies that mutp53 expands the pool of mammary progenitors and BC stem cells, which, at least in part, is mediated by Epidermal Growth Factor Receptor family (EGFR and ErbB2) signaling.
Discussion
The p53 protein plays a key role in tumor suppression, supported by the observation that p53 is mutated in over half of all human cancers of different origins. In contrast to other tumor suppressors, which are typically inactivated by gene silencing, deletion or nonsense mutations, more than 75% of all p53 mutations are missense mutations suggesting a selective advantage for retaining the mutant allele over simple loss of wtp53 functions. It is generally accepted that mutant p53 may fuel carcinogenesis by exerting a dominant-negative effect over wild-type p53 protein, as well as by manifesting novel, oncogenic gain-of-function (GOF) activities 21. The best in vivo proof of mutp53 GOF are mutp53 knock-in (KI) mouse models. All 3 originally reported mutp53 KI (knockin) mice (R175H, R273H and R248W) manifest GOF with a broader tumor spectrum, including adenocarcinomas, higher tumor bulk with increased grade and invasion, and novel metastatic ability compared to the p53-null allele 1, 19, 37, 38. A recent report on a new mutp53 R248Q knock-in mouse model showed the strongest GOF phenotype, reflected by earlier tumor onset and shorter survival compared to p53 null littermates, a phenotype not seen in the 3 previously reported KI models. Importantly, accelerated tumorigenesis in mutp53 R248Q mice was associated with expansion of normal hematopoietic and mesenchymal progenitors9. Consistent with this data, a similar expansion of mouse mammary epithelial SC in the Wnt-1 mammary mouse model was observed when a human mutp53 R175H allele was present compared to the p53 null allele.12 The notion that mutp53 may have a positive role in regulating the mammary SC population is further supported by strong correlative clinical data. Compared to wtp53 BC, the presence of mutp53 in primary human BC tissues correlates with SC transcriptional signatures39. Furthermore, poorly differentiated Grade 3 BCs display the highest SC content and frequency of p53 mutations40. In addition, statistical analysis of human wtp53 and mutp53 BC tissues show a correlation between p53 mutations and concomitant acquisition of stem-like properties40. While these observations suggest a novel and intriguing oncogenic role of mutp53 in stem cell biology, the underlying mechanisms of this phenomenon remain to be defined.
To study the novel tumorigenic properties of mutp53 in BC in vivo we generated a mutp53 mouse model that harbors the mammary tumor virus (MMTV) driven ErbB/Neu/Her2 transgene (Tg), which drives mammary tumor formation. Strong clinical evidences of oncogenic cooperation between mutp53 and Her2 in human BC determined our choice to use the MMTV-ErbB2/Neu mouse model. There is extremely high incidence (>70%) of Her2 positive BC in LFS women compared to 25% of sporadic BC14, a high frequency of mutations in p53 specifically in Her2-positive BC compared to other subtypes (72%) 2, and mutations in the p53 gene indicate poor prognosis in Her2 positive BC due to more aggressive disease5, 6. A double transgenic mouse was made in a previous attempt to model mutp53; Her2- positive BC by combining a Whey Acidic Protein (WAP)-promoter driven mutp53 Tg with the MMTV-ErbB2 Tg 41. However, this model had significant limitations, such as expression of mutp53 from an unknown genomic locus and expression restricted to when the WAP promoter is active, which is only in late pregnancy41. Our improved mutp53 R172H;ErbB2 model has overcome these limitations. The mouse R172H p53 mutation corresponds to the R175H p53 mutation in humans, which frequently occurs in Her2-positive BC2 and therefore closely mimics the mutp53 subtype of human Her2+ BC and LFS.
Our newly generated R172H;ErbB2 mouse model closely recapitulates the clinical data observed in HER2 positive BC patients. Similar to humans, we found strong evidences for mutp53 and ErbB2 oncogenic cooperation. In contrast to ‘straight’ R172H KI mice1, 19, p53H/+;ErbB2 animals show accelerated mammary tumorigenesis as reflected by earlier tumor onset and shorter survival compared to p53-/+;ErbB2 counterparts (Fig 1A). The formation of multiple, simultaneously initiated, multicentric mammary tumors was significantly higher in the presence of the mutp53 allele, which most likely underscored this phenotype (Fig 1B). Since the presence of SC can attribute to tumor initiation8, we hypothesized that the observed phenotype is the physiological consequence of an expansion of mammary SC and/or mammary progenitors by the mutp53 allele. Functional analyses provided further support for this idea. First, mutp53 containing MECs displayed increased mammosphere formation ability (Fig 2C) and higher expression of the SC marker ALDH1 (Fig 2B). Second, pregnancy further increased mammary tumor multiplicity in the presence of mutp53 (Fig 2E), suggesting that mutp53 may expand a pregnancy responsive population of mammary progenitors (PI-MECs), which are thought to be the most important targets of ErbB2-induced mammary tumorigenesis7, 27. Finally, mutp53 is highly stabilized in metastatic lesions of H/+;ErbB2 mice in contrast to the heterogeneous staining found in primary mammary tumors.
Recently, cancer SCs have been invoked as the seed for distant metastases, which are ultimately responsible for end-stage disease and death 8. Numerous reports suggest a functional link between EMT and cancer SC8, 30, 40, 42. The concept of metastases as “migrating cancer stem cells” has been recently proposed42. Selection of cells with highly stabilized mutp53 in metastatic lesions implies that high levels of mutp53 induces the acquisition of SC-like properties and promotes metastatic seeding to distant organs. In an attempt to identify the signaling pathways leading to expansion of TICs in the presence of mutp53 we found that mutp53 tumors express the highest levels of ErbB2 and exhibit increased ErbB2 signaling compared with p53 null counterparts (Fig 3). According to recent reports, ErbB2 signaling drives carcinogenesis, at least in part, through regulation of the mammary stem/progenitor cell population. First, the TIC population of Her2- overexpressing BC cell lines express the highest Her2 levels16 and are directly responsive to Her2 inhibitors15, 16. Lapatinib, a dual ErbB2/EGFR inhibitor, depletes the cancer SC pool from tumors of Her2-positive BC patients17. Second, expression of ErbB2 in murine MECs increases the cancer SC pool and induces mammary tumorigenesis, indicating a functional link between ErbB2-mediated expansion of cancer SCs and BC development15. Therefore, mutp53-mediatated amplification of ErbB2 signaling may mechanistically underlie the expansion of mammary stem cells that can lead to a more aggressive disease.
The question arises as to how mutp53 amplifies ErbB2 signaling mechanistically. To overcome proteotoxic stress inherent to malignant transformation, cancer cells induce a range of adaptive mechanisms, where the master transcription factor Heat Shock Factor 1 (HSF1)-orchestrated heat shock response plays a essential role43. Our previous in vitro studies identified a novel mutp53–HSF1 feed-forward loop, which provides cancer cells superior survival32. We found that mutant p53, via constitutive stimulation of EGFR and ErbB2 signaling, hyperactivates the MAPK and PI3K cascades to induce stabilization and transcriptional phosphoactivation of HSF1 on Ser326. Moreover, mutp53 protein has direct interaction with activated p-Ser326 HSF1 to facilitate HSF1 recruitment to its specific DNA binding elements and stimulate transcription of heat shock proteins, including Hsp90. In turn, induced Hsp90 stabilizes its oncogenic clients including EGFR, ErbB2 and mutp53, thereby further reinforcing oncogenic signaling32. Thus, mutp53 initiates a feed forward loop leading to HSF1-mediated protein stabilization of Epidermal Growth Factor Receptor family members, including mutp53 itself, EGFR and ErbB2. Importantly, here we confirmed this axis in vivo and defined its physiological consequences in ErbB2-driven breast cancer mouse model.
The other physiological outcome of mutp53-mediated stimulation of ErbB2 signaling in vivo, which may not relate to SC biology, is enhanced proliferation of mammary cells. BC is thought to arise from mammary epithelial cells found in structures referred to as acini, which collectively form terminal ductal lobular units. Each acinus consists of a single layer of polarized luminal epithelial cells surrounding a hollow lumen 44.
Activation of ErbB2 induces proliferation of acinar cells causing alterations in its epithelial architecture, characteristic for early stages of mammary tumorigenesis33. Indeed, we found that mutp53 potentiates the proliferation of ErbB2 expressing cells in both murine mammary and human BC (Fig 4). Therefore, enhanced proliferation of luminal cells in the presence of mutp53 may fuel the initiation of premalignant lesions in the ErbB2 context, leading to increased tumor multiplicity observed in p53H/+;ErbB2 mice. In support, mutp53 depletion from EGFR expressing MDA468 human BC cells was shown to induce phenotypic reversion from highly disorganized structures to acinus-like structures with a hollow lumen in 3D culture45.
In summary, our findings provide evidence that mutp53 cooperates with Epidermal Growth Factor Receptor family EGFR and ErbB2 and amplifies their signaling, thereby inducing proliferation of mammary cells and expansion of mammary stem cells. Thus, our studies identify mutp53 as a key player and potential target in Her2 and EGFR positive BC.
Materials and Methods
Cells
Human mutp53 breast cancer cells MDA231 (p53R280K). Generation of stable Tet-On shp53 MDA231 was previously described46. MDA231 stably overexpressing native ectopic p53 (R280K) or vector only were generated by Lipofectamine (Invitrogen) transfection followed by selection in G418. All cells were cultured in 10% FCS/DMEM.
RNA interference
Pools of 4 different siRNA duplexes specific for p53 or scrambled controls were transfected with Lipofectamine (RNAiTM/RNAiMAX, Invitrogen). Cells were harvested 48 h later and analyzed.
Immunoblots and immunoprecipitations
For immunoblots, equal total protein of cell lysates (2.5–20μg) were detected with antibodies to mouse p53 (FL393), human p53 (PAb1801) and ALDH 1/2 (all Santa Cruz Biotechn), AKT, pAKT, Erk, pErk, EGFR, EGRF-Tyr845P (all Cell Signaling), ErbB2, actin (all Neomarkers).
Mammary epithelial cells cultures
Mammary glands were dissected from 8 wk-old virgin female mice and sequentially digested at 37°C for 2h in collagenase/hyaluronidase, 0.05% Trypsin, DNAse I and Dispase (Stem Cell Technology). The ensuing cell suspension was treated with red blood cell lysis buffer, rinsed with PBS, and passed through a 40μm mesh after resuspension in Opti-Mem medium (Gibco). Cells were plated on gelatin-coated plates and grown in CnT-BM1 medium (Cell-N-Tec).
Dissociation of normal mammary tissue and mammosphere assay
For mammosphere experiments, single cell suspensions of MECs or MDA231cells were plated on ultralow attachment 6-well plates at a density of 20×103 cell/well for 10 days. Subsequent cultures after dissociation of primary spheres were re-plated. Mammosphere cultures were grown in a serum-free mammary epithelium basal medium as previously described 25.
Mice
MMTV-ErbB2 mice harboring activated ErbB2 (strain FVBN-Tg(MMTV-ErbB2)NK1Mul/J) were from Jackson Labs. p53 R172H (called p53H/H) and control p53 null (p53-/-) mice (C57Bl6J background) were a gift from G. Lozano1. p53 mice were interbred to generate H/-mice. Compound p53H/-;ErbB2 mice were generated by crossing ErbB2 into the p53-/-background and then breeding the p53+/-;ErbB2 progeny with p53H/H animals. H/-;ErbB2 mice were then crossed to generate p53H/H;ErbB2 and p53-/-;ErbB2 females for analysis. These F2 mice were of mixed background. Littermates were used for all analyses. Mice were regularly monitored and killed when they became moribund. Careful necropsies were performed and tumors and all major organs collected, fixed in 10% formalin, embedded in paraffin and sectioned forhistopathologic analysis. For survival analysis, P-values were determined by log-rank analysis. Mice were treated according to guidelines approved by the Institutional Animal Care and Use Committee.
Aldefluor assay and flow cytometry
To measure ALDH activity the Aldefluor assay was performed according to manufacturer's (Stemcell Technologies, Durham, NC, USA) protocol. Dissociated single cells were suspended in Aldefluor assay buffer containing the ALDH substrate, Bodipy-aminoacetaldehyde (BAAA) at 1.5 mM and incubated for 45 min at 37 1C. To distinguish between ALDH-positive and ALDH-negative cells, a fraction of cells was incubated under identical condition in the presence of a 10-fold molar excess of the ALDH inhibitor, diethylaminobenzaldehyde (DEAB). The ALDH positive population of cells was measured by FACS according to manufacturer's guidelines.
Immunohistochemical analysis
Immunohistochemical (IHC) analysis was conducted as previously described 47. Protein expression was analyzed using rabbit p53 (CM5) antibodies (Vector Laboratories) at a 1:1000 overnight, and visualized using ABC and DAB kits from (Vector Laboratories). Slides were counterstained with hematoxylin.
Tissue Microarrays
Tissue arrays of breast invasive ductal carcinomas (75 cases/150 cores; BR1504, Biomax) with known TNM, pathologic grade and markers (ER, PR, HER2 and Ki67) were stained with antibodies to p53 (DO1, Santa Cruz). Correlation between p53 positivity and percent of Ki67 positive cells was determined by t-test and Mann-Whithey test.
Acknowledgments
This work was funded by grants from the American Cancer Society (RSG-11-188-01 to NM) and the Carol Baldwin Breast Cancer Research Fund (to NM). We thank Sulan Xu for excellent technical assistance.
Abbreviations
- BC
breast cancer
- SC
stem cells
- mutp53
mutant p53
- wtp53
wild type p53
- LFS
Li-Fraumeni Syndrome
- HSF1
Heat Shock Factor 1
- GOF
gain-of-function
- EGFR
Epidermal Growth Factor Receptor
- MEC
mammary epithelial cells
- MAPK
Mitogen Activated Protein Kinase
- PI3K
Phosphatidylinositol 3-Kinase
- TICs
tumor initiating cells
References
- 1.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119(6):847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masciari S, Dillon DA, Rath M, Robson M, Weitzel JN, Balmana J, et al. Breast cancer phenotype in women with TP53 germline mutations: a Li-Fraumeni syndrome consortium effort. Breast cancer research and treatment. 2012;133(3):1125–1130. doi: 10.1007/s10549-012-1993-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melhem-Bertrandt A, Bojadzieva J, Ready KJ, Obeid E, Liu DD, Gutierrez-Barrera AM, et al. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer. 2012;118(4):908–913. doi: 10.1002/cncr.26377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamashita H, Nishio M, Toyama T, Sugiura H, Zhang Z, Kobayashi S, et al. Coexistence of HER2 over-expression and p53 protein accumulation is a strong prognostic molecular marker in breast cancer. Breast cancer research : BCR. 2004;6(1):R24–30. doi: 10.1186/bcr738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A. A mutant TP53 gene status is associated with a poor prognosis and anthracycline-resistance in breast cancer patients. European journal of cancer. 2003;39(4):447–453. doi: 10.1016/s0959-8049(02)00499-9. [DOI] [PubMed] [Google Scholar]
- 7.Boulanger CA, Wagner KU, Smith GH. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-beta1 expression. Oncogene. 2005;24(4):552–560. doi: 10.1038/sj.onc.1208185. [DOI] [PubMed] [Google Scholar]
- 8.Liu S, Wicha MS. Targeting breast cancer stem cells. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(25):4006–4012. doi: 10.1200/JCO.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanel W, Marchenko N, Xu S, Xiaofeng Yu S, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell death and differentiation. 2013 doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138(6):1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 11.Sarig R, Rivlin N, Brosh R, Bornstein C, Kamer I, Ezra O, et al. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. The Journal of experimental medicine. 2010;207(10):2127–2140. doi: 10.1084/jem.20100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Liu DP, Xu Y. The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene. 2012 doi: 10.1038/onc.2012.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nature reviews Cancer. 2012;12(8):553–563. doi: 10.1038/nrc3309. [DOI] [PubMed] [Google Scholar]
- 14.Sachdev JC, Jahanzeb M. Blockade of the HER family of receptors in the treatment of HER2-positive metastatic breast cancer. Clinical breast cancer. 2012;12(1):19–29. doi: 10.1016/j.clbc.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, et al. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(6):2010–2021. doi: 10.1158/1078-0432.CCR-08-1327. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008;100(9):672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 18.Mimeault M, Batra SK. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Molecular aspects of medicine. 2013 doi: 10.1016/j.mam.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119(6):861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54(1):105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 21.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nature reviews Cancer. 2009;9(10):701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 22.Suh YA, Post SM, Elizondo-Fraire AC, Maccio DR, Jackson JG, El-Naggar AK, et al. Multiple stress signals activate mutant p53 in vivo. Cancer research. 2011;71(23):7168–7175. doi: 10.1158/0008-5472.CAN-11-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo PK, Kanojia D, Liu X, Singh UP, Berger FG, Wang Q, et al. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFbeta signaling. Oncogene. 2012;31(21):2614–2626. doi: 10.1038/onc.2011.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes & development. 2003;17(10):1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 2011;479(7372):189–193. doi: 10.1038/nature10573. [DOI] [PubMed] [Google Scholar]
- 27.Matulka LA, Triplett AA, Wagner KU. Parity-induced mammary epithelial cells are multipotent and express cell surface markers associated with stem cells. Developmental biology. 2007;303(1):29–44. doi: 10.1016/j.ydbio.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 28.Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development. 2002;129(6):1377–1386. doi: 10.1242/dev.129.6.1377. [DOI] [PubMed] [Google Scholar]
- 29.Henry MD, Triplett AA, Oh KB, Smith GH, Wagner KU. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23(41):6980–6985. doi: 10.1038/sj.onc.1207827. [DOI] [PubMed] [Google Scholar]
- 30.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer research. 2009;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Yallowitz A, Ozog L, Marchenko N. A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell death & disease. 2014;5:e1194. doi: 10.1038/cddis.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nature cell biology. 2001;3(9):785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139(7):1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell death and differentiation. 2011;18(12):1904–1913. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137(1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 37.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9(5):573–580. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- 38.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes & development. 2008;22(10):1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(52):22745–22750. doi: 10.1073/pnas.1017001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coradini D, Fornili M, Ambrogi F, Boracchi P, Biganzoli E. TP53 mutation, epithelial-mesenchymal transition, and stemlike features in breast cancer subtypes. Journal of biomedicine & biotechnology. 2012;2012:254085. doi: 10.1155/2012/254085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, Rosen JM, McMenamin-Balano J, Muller WJ, Perkins AS. neu/ERBB2 cooperates with p53-172H during mammary tumorigenesis in transgenic mice. Molecular and cellular biology. 1997;17(6):3155–3163. doi: 10.1128/mcb.17.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert opinion on therapeutic targets. 2009;13(4):469–478. doi: 10.1517/14728220902832697. [DOI] [PubMed] [Google Scholar]
- 44.Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation; research in biological diversity. 2002;70(9-10):537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148(1-2):244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Molecular cancer research : MCR. 2011;9(5):577–588. doi: 10.1158/1541-7786.MCR-10-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans SC, Viswanathan M, Grier JD, Narayana M, El-Naggar AK, Lozano G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene. 2001;20(30):4041–4049. doi: 10.1038/sj.onc.1204533. [DOI] [PubMed] [Google Scholar]