


Abstract
PARP1 and PARP2 are implicated in the synthesis of poly(ADP-ribose) (PAR) after detection of DNA damage. The specificity of PARP1 and PARP2 interaction with long DNA fragments containing single- and/or double-strand breaks (SSBs and DSBs) have been studied using atomic force microscopy (AFM) imaging in combination with biochemical approaches. Our data show that PARP1 localizes mainly on DNA breaks and exhibits a slight preference for nicks over DSBs, although the protein has a moderately high affinity for undamaged DNA. In contrast to PARP1, PARP2 is mainly detected at a single DNA nick site, exhibiting a low level of binding to undamaged DNA and DSBs. The enhancement of binding affinity of PARP2 for DNA containing a single nick was also observed using fluorescence titration. AFM studies reveal that activation of both PARPs leads to the synthesis of highly branched PAR whose size depends strongly on the presence of SSBs and DSBs for PARP1 and of SSBs for PARP2. The initial affinity between the PARP1, PARP2 and the DNA damaged site appears to influence both the size of the PAR synthesized and the time of residence of PARylated PARP1 and PARP2 on DNA damages.
INTRODUCTION
Poly(ADP-ribose) polymerase-1 (PARP1) and poly(ADP-ribose) polymerase-2 (PARP2) are nuclear proteins responsible for the synthesis of polymers of ADP-ribose using β-NAD+ as substrate (1,2). These enzymes are implicated in response to cell DNA damage, recognizing damaged DNA generated under genotoxic stress or DNA breaks as a result of the activity of DNA repair enzymes (3–7). Upon binding to damaged DNA, PARP1(2) catalyse PARylation of itself and/or of a number of nuclear proteins including DNA repair/replication factors (1,3,8). Although the catalytic domains of PARP1 and PARP2 have a high degree of homology, the PARP2 DNA binding domain is distinct from that of PARP1 (4,9,10). This could reflect differences between these proteins in their preferences for binding to damaged DNA, because their activity is regulated by their DNA-binding domains (5,10,11). PARP1(2) activity and DNA substrate specificity have been studied by an ensemble of biochemical approaches to estimate their preference for the damaged DNA (11–15). Previously, analysis of PARP1(2) binding to damaged DNA was carried out mainly using short DNA duplexes; however in this case it is difficult to analyse the interaction with a single DNA damage. The interaction of PARP1(2) with nicks and blunt ends on short DNA duplexes can overlap. Therefore long DNA duplexes with several DNA damages located at a distance from one another are better models to study the influence of blunt DNA ends, undamaged DNA sequences and a single damaged site on the specificity of protein–DNA interaction.
Here, we studied the interaction of PARP1(2) with long DNA fragments containing DNA breaks using atomic force microscopy (AFM) imaging and fluorescence assays to estimate the DNA binding characteristics of these proteins. We established new optimal conditions for adsorption of DNA–PARP1(2) complexes on mica surface mediated by putrescine (Pu2+). The conditions were exploited to detect the interaction of PARP1 and PARP2 with long DNA fragments (1200-bp) containing only DSB ends or DSBs together with a unique single strand break (SSB). Statistical analysis of the AFM data in view of localizing proteins bound to DNAs shows that PARP1 specifically binds to both SSB and DSB with a preference for nicks. At the same time, this protein binds to undamaged DNA, but to a lower extent than to breaks. In contrast to PARP1, PARP2 binds weakly to undamaged DNA and to DSB ends, and localizes mainly to SSBs. These results correlate with fluorescence data revealing that the binding affinity of PARP2 to 1200-bp DNA is 5-fold weaker in comparison to the same DNA fragment containing a nick. At the same time, the appearance of a SSB in DNA leads only to a 2-fold increase in the binding affinity of PARP1 to the DNA fragment. AFM data show that PARylated PARP1(2) are still able to interact with DNA. Thus, PARylated PARP2 was detected near nicks, while modified PARP1 was near blunt ends. The data are in the agreement with previous biochemical findings indicating that the PARylated PARP1 can interact with damaged DNA (16,17). The length of the PAR polymer formed by PARP1(2) appears to be influenced by the initial binding affinity of the proteins for the damaged site. Our observations by a single-molecule study implicate PARP2 in the recognition of nicks and suggest its role in SSB repair.
MATERIALS AND METHODS
Chemicals, reagents and proteins
Chemical compounds (MgCl2, putrescine and spermidine) were purchased from Sigma-Aldrich. ‘Vivaspin’ ultrafiltration spin columns were from ‘Sartorius Stedim. Biotech. GmbH’. Nb.BsmI, nicking endonuclease and plasmid pBR322 were purchased from ‘New England BioLabs’. Taq DNA polymerase was purchased from ‘Thermo Scientific’. Murine PARP2 was expressed in insect cells and purified according to (18). Human PARP1 was expressed in Escherichia coli and purified according to (19). The 1200-bp DNA fragment containing the Nb.Bsm1 target sequence in the middle of the chain was prepared by the polymerase chain reaction (PCR). Plasmid pBR322 was used as a template with the following primers: 5′-CGCCGCACTTATGACTGTCTTC-3′- forward primer and 5′-GCGTTAATGTCTGGCTTCTGA-3′- reverse primer. Although Taq DNA polymerase has a tendency to add an adenine nucleotide to the 3′-ends of PCR products, 1200-bp DNA was considered as DNA fragment with blunt ends. The PCR products were run on a 1% agarose gel and purified using a gel extraction kit (‘Fermentas’). Nicked DNAs were obtained by incubating the 1200-bp DNA or pBR322 with Nb.Bsm1 nicking endonuclease according to the recommended protocol. Formation of the nicked circular form of pBR322 was analysed by 0.8% agarose gel electrophoresis with EtBr staining. The 1400-bp DNA carrying ends with short overhangs (4 nt) was prepared using pBR322. The plasmid was cleaved with Pst1 (ACGTC)/Sal1(CAGCT) and the fragment was purified on a 1% agarose gel. DNA concentrations were determined by measuring the absorbance at 260 nm.
Sample preparation for AFM imaging
PARP1 or PARP2 (1.75–14 nM) and DNAs (1.5 nM for 1200-bp DNAs or 0.35 nM for nicked pBR322) were incubated in AFM deposition buffer (12.5 mM Hepes, pH 8.0, 12.5 mM KCl, 1 mM DTT) on ice for 1–5 min. Just before sample deposition, to adsorb the PARP1(2), DNA, DNA–PARP1 or DNA–PARP2 complexes, multivalent cations were added to the solution to final concentrations of 10 mM Mg2+, as well as 5 mM putrescine (Pu2+) or 50 μM spermidine (Spd3+), after which a 10-μl droplet was deposited on the surface of freshly cleaved mica at room temperature for 30 s. The mica surface was then rinsed with 0.02% uranyl acetate solution to stabilize the DNA–protein complexes in their conformations for AFM imaging in air (20). The sample was rapidly rinsed with pure water (Millipore) and air-dried before imaging. For experiments with auto-PARylation of PARP2, 3.5 nM PARP2 was incubated with 1.5 nM nicked 1200-bp DNA in the AFM deposition buffer in the presence of 100 μM NAD+ on ice for 5–120 min. For experiments with auto-PARylation of PARP1, 35 nM PARP1 was incubated with 3.5 nM nicked pBR, 3.5 nM supercoiled pBR or 13.7 nM 1200-pb DNA in the AFM deposition buffer in the presence of 100 μM NAD+ and 10 mM MgCl2 at 37°C for 15–120 min. After incubation the samples were diluted 10× in AFM deposition buffer and immediately deposited on mica. For AFM imaging, the samples were processed as described above.
AFM and image analysis
Sample imaging was performed in air at room temperature in the tapping Mode™ with a Multimode™ AFM (Veeco, Santa Barbara, CA, USA) operating with a Nanoscope IIIa™ controller. Olympus (Hamburg, Germany) silicon cantilevers AC160TS with resonance frequencies of about 300 kHz and nominal spring constants of 10–100 N/m were used. The scan frequency was typically 1.5 Hz per line and the modulation amplitude was a few nanometres. Data were acquired at a set point chosen to minimize tip–sample interaction force and a first or second order polynomial function served to remove the background. The ‘section’ tool in the Nanoscope Analysis software (version 1.50) was used to determine the molecular dimensions of the protein particles. Cross-sections of the individual particles imaged by AFM were made and the height and diameter at half-maximal height of each single particle were measured. The DNA length and the position of PARP1(2) on DNA were analysed using ‘ImageJ’ software. The length of the DNA contour was manually traced on AFM images. In the case of DNA–protein complexes, the contour length was manually traced as the shortest possible DNA path through the bound protein. The size of PARylated proteins were calculated using the following equation: S = πR2, where the R is the minimum radius of the circle in which PARylated proteins could be enclosed. The radius was measured from AFM images using the ‘section’ tool in the Nanoscope Analysis software. The percent of DNA bound to PARP1(2) was estimated as the ratio between the number of DNA fragments interacting with one or more protein molecules over the total DNA fragments adsorbed on mica. The binding specificity of PARP1(2) with DSB and SSB was quantified by estimating the percent of PARP1(2) complexes bound to the DNA lesion site (21). The protein–DNA complexes were classified according to whether they were located at the ends, at the nick site or at the non-damaged sites of the model DNA fragments (Supplementary Figure S1A–C), taking into account that the protein interacts with the DSB ends when it is located within the last 40 nm of each DNA end, and with SSB when the protein is located between 194 and 234 nm from one DNA extremity (Supplementary Figure S1D). The interval of 40 nm was selected on the basis of the mean ± SD contour length measured for PARP1(2)–DNA complexes (Supplementary Figure S1 E and F).
DNA binding constants and specificities
Binding specificities (S) and constants (Kd) were calculated as described previously (21). Briefly, the method is based on the determination of the average DNA fractional occupancy of PARP1(2) at non-specific (undamaged DNA) sites, specific (nick) sites on the DNA and the DNA ends. The counting of protein–DNA complexes and DNA fragments was used to calculate the fractional occupancy, that is the total number of DNA fragments (nFragment), the total number of PARP1(2)–DNA complexes at undamaged DNA contours (nComplex,Und), the total number of DNA termini (nComplex,Ends) and nicks (nComplex,Nick) bound by these proteins (Supplementary Table S1). Taking into account the size of PARP1(2) in AFM images (Supplementary Figure S1 D and F), the 1200-bp DNA fragment contains two DNA ends and 976 non-specific sites; nicked 1200-bp DNA contains one specific site, two DNA ends and 864 non-specific sites. The specificities and AFM-site specific binding affinities (Kd) of PARP1(2) to nicked, to non-specific sites and to DNA ends were calculated directly estimating the number of specific and non-specific complexes and fractional occupancies of PARP1(2) bound to different DNA sites (Supplementary Table S1). Composite macroscopic binding constants (Kd, AFM macro) of PARP1 and PARP2 to 1200-bp DNA fragments were calculated using values of Kd for specific and non-specific sites and DNA ends (21).
Fluorescent labelling of PARP1 and PARP2 and fluorescence titration assays
For protein labelling, 5(6)-carboxyfluorescein N-hydroxysuccinimide ester (FSE) was used, in which the carboxyl groups were activated with N-hydroxysuccinimide (NHS). The NHS ester is easily displaced by nucleophilic attack from primary amino groups of the N-terminal group of the protein (primary target at pH 8.0) and lysine side chains at the water-accessible surface of the protein, thus forming an amide bond with the original carboxyl group of fluorescein at physiological pH. Purified PARP1 or PARP-2 (1.8 pmol) were incubated with FSE (8 pmol) at 4°C overnight in buffer containing 50 mM Hepes, pH 7.5 and 100 mM NaCl. Unreacted FSE was removed by dialysis, followed by concentration of the labelled proteins on ultrafiltration spin columns. The concentration of PARP1(2)-fluorescein conjugates and labelling efficiency were determined using the extinction coefficients: ϵ280 = 120 000 M−1cm−1 for PARP1, ϵ280 = 70 415 M−1cm−1 for PARP2 and ϵ280 = 23 400 cm−1 M−1 and ϵ495 = 60 000 M−1 cm−1 for fluorescein. The degree of labelling was estimated as 25% for PARP1 and 28% for PARP2. Fluorescence titration experiments were performed by adding increasing amount of DNAs to a fixed concentration of protein (20 nM for PARP1-fluorescein and 90 nM for PARP2-fluorescein) in buffer containing 12.5 mM Hepes-KOH (pH 8.0), 12.5 mM KCl and 1 mM DTT. The excitation wavelength was set at 485 nm, and emission wavelength at 520 nm. All experiments were performed at 28°C on a POLARstar Optima multidetection microplate reader (BMG Labtech, Offenburg, Germany) in a 96-well assay ‘V’ bottom black plate (Axigen), and thus, one well contained one titration point. These experiments were carried out in the same buffer as used for AFM sample preparations. The volume of the reaction mixture was 50–100 μl. All reactions were mixed at room temperature. The plate was incubated at 28°C in the reader. Each experiment was repeated at least three times.
The degree of binding (Db) was estimated using the following equation:
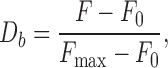 |
where F indicates the fluorescence intensity of the PARP1-fluorescein (or PARP2-fluorescein) conjugates at different DNA concentrations, and F0 and Fmax are the fluorescence intensities in the absence and at saturating levels of the DNA, respectively.
The dissociation constants (Kd) were calculated using the following equation:
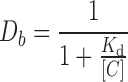 |
where [C] is the concentration of the DNA.
RESULTS
Imaging of PARP1(2)–DNA complexes in the presence of polyamines
AFM experiments make it possible to observe the interaction between single molecules whereas traditional biochemical techniques indicate only average values from a large number of interacting molecules (21–24). However, AFM imaging of DNA–protein complexes is not straightforward since the complexes can dissociate during their adsorption and immobilization on the surface (23,25). The absorption of DNA and of protein–DNA complexes on mica requires the presence of multivalent cations, such as magnesium (Mg2+), putrescine (Pu2+) or spermidine (Spd3+), in the deposition buffer or using fixing agents such as glutaraldehyde (25–28). However, it has been shown that the cations can affect protein stability during the absorption process (25,29–31). Therefore, at first, we determined the appropriate conditions for the deposition of free PARPs on mica in the presence of different cations, namely Mg2+, Pu2+ or Spd3+. Representative AFM images of free PARP1 and PARP2 are shown in Figure 1A. The average molecular heights and distribution of volumes for PARP1 and PARP2 molecules in the presence of different cations were estimated from these images (Figure 1B and Supplementary Figure S2). In contrast to Mg2+ or Pu2+, slight oligomerization of both proteins was observed in the presence of Spd3+ (Figure 1B and Supplementary Figure S2). Thus, using Spd3+ in the deposition process can induce protein aggregation that can complicate the analysis of DNA–PARP1(2) complexes by AFM.
Figure 1.

AFM images of PARP1 or PARP2 molecules deposited on mica surface in the presence of different counterions. (A) Images of PARP1 (2 nM) or PARP2 (2 nM) in the presence of 10 mM Mg2+, 5 mM Pu2+ or 50 μM Spd3+. Scale bar 500 nm; Z scale: 7 nm. (B) The average height of PARP1 or PARP2 measured in images shown in (A) (n, number of molecules analysed). Results are mean ± SD of three independent samples. P-values were obtained by comparing the results by t-test, *, P < 0.05; **, P < 0.01; ***, P < 0.005; ns, not significant.
PARP1 acts as a molecular sensor of DNA breaks and is involved in both SSB and DSB repair (1,7,32,33). PARP2 participates in SSB repair but there is no strong evidence that this protein is involved in DSB repair (8,34). In vitro, the interaction of PARP1(2) with DNA containing SSBs and DSBs has been studied by different techniques (10–15,35,36). However, AFM has never been employed in investigating binding of PARPs with DNA containing these types of lesions. Here we used a 1200-bp double-stranded DNA fragment (1200-bp DNA), and the same fragment with a nick in the middle of the chain (nicked 1200-bp DNA) to characterize PARPs and DNA complexes with AFM (Supplementary Figure S1D). The use of long DNA substrates can lead to a better discrimination of PARP1(2) binding to DSBs and nicks in the context of linear DNA duplexes.
Figure 2A and B show AFM images of 1200-bp DNA alone or after incubation with varying concentrations of PARP1(2) proteins. Sample depositions on mica were performed in the presence of Pu2+ or Mg2+. Under these conditions, PARP1(2)–DNA complexes were not detected by AFM using a large range of Mg2+ concentrations from 2 to 20 mM (data not shown). In contrast to Mg2+, the complexes were visualized in the presence of Pu2+ (Figure 2A). Consequently, Pu2+ was selected as counterion for PARP1(2)–DNA complex adsorption. Under high PARP1 concentrations, the formation of large aggregates and a decrease of the amount of isolated molecules adsorbed on the surface were observed (Figure 2A). PARP1 aggregation on mica has already been demonstrated by AFM using the chromatin system (37). In the same range of protein concentrations, PARP2 showed a completely different behaviour since the density of DNA molecules remained constant while the number of proteins adsorbed on the surface significantly increased (Figure 2A). Thus, a concentration of around 3.5 nM PARPs is more suitable for the comparison of the efficiencies of DNA–protein complex formation using AFM experiments for the following reasons; (i) aggregate formation for DNA–PARP1 complexes on mica surfaces is limited under these conditions, (ii) the number of PARP2 molecules adsorbed on the surface remains sufficiently low to limit the formation of non-specific DNA–PARP2 complexes and (iii) the ratio of DNA–protein complexes over the total adsorbed DNA is sufficiently high to perform statistical analyses of PARP interaction with DNA. Under the conditions developed here, a low level of complex formation between PARP2 and DNA was observed. Only 18% of DNA was involved in interaction with this protein. In contrast to PARP2, PARP1 is more effective in binding to dsDNA and the yield of complex was about 65% at 3.5 nM protein concentration (Figure 2C). The AFM results are in agreement with the results of fluorescence titration experiments showing that the affinity of PARP1 for 1200-bp DNA is about 5-fold higher than that of PARP2 (Figure 2D).
Figure 2.

Analysis of PARP1 and PARP2 complex formation with DNA containing DSB ends. (A) Binding of PARP1 or PARP2 to 1200-bp DNA analysed by AFM. A total of 1200-bp DNA was incubated with 1.75–14 nM PARP1 or PARP2 and imaged by AFM in air. All images were obtained using 5 mM Pu2+ for complex adsorption. White arrows indicate PARP1(2)–DNA complexes. Scale bar 1 μm; Z scale: 7 nm. (B) Image of 1200-bp DNA deposited on mica surface in the absence of PARP1 or PARP2. Scale bar 500 nm; Z scale: 7 nm. (C) Quantitative analysis of PARP1(2)–DNA complex formation depending on protein concentration, the number of DNA molecules analysed: 164 for 1.75 nM PARP1, 400 for 1.75 nM PARP2, 138 for 3.5 nM PARP1, 326 for 3.5 nM PARP2, 117 for 7 nM PARP1. At 7nM of PARP2, the detection of a specific interaction between the DNA fragment and the protein was complicated due to the high density of free PARP2 adsorbed on the surface. The results represent mean ± SD of three independent samples. (D) Fluorescence measurements of PARP1 and PARP2 binding to DNA. The reaction mixtures containing the fluorescein-labelled protein PARP1 or PARP2 were titrated with increasing amounts of 1200-bp DNA. Bars indicate the standard error of three independent experiments.
To analyse the interaction of PARP1(2) with the DSB ends, we estimated the position distributions of the proteins bound to 1200-bp DNA fragments using 3.5 nM PARP1 or PARP2 (Figure 3). Representative AFM images of 1200-bp DNA in the presence of PARP1 or PARP2 are shown in Figure 3A and B. Analysis of the position distributions of PARP1(2) molecules on DNA fragments shows that about 40% of PARP1(2) molecules are located at DNA ends while 60% interact with the internal region of the DNA molecules (Figure 3C). For detailed description of this DNA substrate and details of how the position of proteins along the DNA were defined and measured, see Supplementary Figure S1. Although high levels of PARP1–DNA complex formation and low levels in the case of PARP2 were observed (Figure 2C), both proteins bind to DNA ends with similar specificity (Table 1). At the same time, comparative analysis of the site specific Kd values of the proteins indicates that PARP1 has about five time more affinity to DNA ends and non-specific DNA sites than that of PARP2 (Table 1). Interestingly, the composite Kd values of PARP1(2)–DNA complexes calculated from AFM-site specific constants are consistent with the ones determined from fluorescence titration experiments (Table 1).
Figure 3.

AFM analysis of the interaction of PARP1 and PARP2 with undamaged DNA and DNA ends. Large scale AFM images (left) of PARP1 (A) and PARP2 (B) binding to 1200-bp DNA and zoomed images (right) of PARP1(2)–DNA complexes. A total of 1200-bp DNA was incubated with 3.5 nM PARP1 or PARP2 and imaged by AFM in air. Arrows indicate binding of PARP1(2) to DNA ends or undamaged DNA. Scale bar 1 μm; Z scale: 7 nm. (C) Position distributions of PARP1 and PARP2 on 1200-bp DNA; number of complexes analysed: 90 for PARP1 and 62 for PARP2. The results represent mean ± SD of three independent samples.
Table 1. DNA binding constants and specificities of PARP1 and PARP2 for the 1200-bp DNA fragment.
| PARP1 | PARP2 | |||
|---|---|---|---|---|
| Kd (nM) | Specificity | Kd (nM) | Specificity | |
| AFM-site specific constants: | ||||
| DNA ends | 17.5 ± 1.0 | 360 ± 36 | 90.9 ± 6.2 | 323 ± 29.3 |
| Non-specific | 6329 ± 518 | 29411 ± 1764 | ||
| Binding constants (AFM macro)a | 3.30 ± 0.23 | 16.10 ± 1.01 | ||
| Binding constants (Flu)b | 1.66 ± 0.02 | 7.65 ± 0.66 | ||
aComposite macroscopic binding constants of PARP1 and PARP2 to 1200-bp DNA fragments.
bMacroscopic binding constant of PARP1 and PARP2 to a 1200-bp DNA measured by fluorescence titration in this study (Figure 2D).
Collectively, these data indicate that PARP1 interacts more efficiently with both termini and undamaged parts of long linear DNA than PARP2.
Specificity of PARP1 and PARP2 interaction with SSBs and DSBs
Both PARP1 and PARP2 have been shown to be involved in BER/SSB repair interacting with repair proteins and DNA intermediates, including DNA with an apurinic/apyrimidinic site, nick, short gap or flap lesions (5,7,8,13,16,38–41). Although PARP2 recognizes and binds to gap- or flap-containing DNA structures (3,8,10,13), recently DNA with a 5′-phosphorylated SSB was identified as a preferential substrate for PARP2 activation (14). To test whether a single DNA nick influences PARP1 and PARP2 binding to long DNA molecules by AFM, nicked circular pBR or nicked 1200-bp DNA fragments were used. The circular shape of the pBR plasmid formed by endonuclease treatment was demonstrated by agarose gel and AFM imaging (Supplementary Figure S3). Both for PARP1 and PARP2, the presence of one protein molecule interacting with the nicked plasmid was detected (Supplementary Figure S4A). However, the compaction or crossover formation of the DNA on the AFM surface limits the detection of specific DNA–protein interactions and complicates the statistical analysis of PARP complex formation (Supplementary Figure S4A). Although nicked pBR does not appear to be a good model for AFM studies of interactions of PARP1 and PARP2 with the DNA nick site, we compared the affinity of these proteins for the substrate using fluorescence titration (Supplementary Figure S4B). Both proteins demonstrate similar Kd values with nicked pBR, although PARP1 and PARP2 show clear differences in binding affinity to 1200-bp DNA (Figure 2D and Supplementary Figure S4B). In contrast to nicked pBR, using linear 1200-bp DNA fragments carrying a single nick allowed to quantify the complexes of PARP1(2) with DNA and to determine the specificity of interactions of these proteins with individual lesions (Supplementary Figure S1D). Using nicked 1200-bp DNA results in a slight increase in the amount of PARP1–DNA complexes formed in comparison to the same DNA fragment without SSB (Figures 2C and 4C). About 65% of PARP1 molecules are detected on SSB/DSB sites and only 35% are bound to undamaged DNA. Interestingly, the presence of a single nick in the DNA significantly increases the amount of PARP2–DNA complexes detected by AFM, and the protein is clearly localized at the nick site (Figure 4 C and B). The resulting distributions of PARP2 on nicked DNA reveals its high preference for SSBs, since more than 70% of all complexes are formed by PARP2 molecules interacting with SSBs (Figure 4D). Thanks to high resolution imaging, it was also possible to determine the fractional occupancies of the proteins bound to different DNA sites and to calculate their binding affinity and the specificity for nicked sites (Supplementary Tables S1 and S2). The data show that PARP2 and PARP1 have a similar affinity for nicks; however PARP2 reveals a higher specificity for this type of DNA breaks (Table 2). Thus, PARP2 exhibits a significant preference for binding to nicked sites over DNA ends and undamaged DNA. Notably, binding of the protein to nicks influences the calculated composite macroscopic binding constants, and the affinity of PARP2 for nicked DNA fragments comes very close to that of PARP1 (Table 2). These results are consistent with the ones of the determination of the Kd values of the PARP1(2) complex with a nicked 1200-bp DNA using fluorescence titration (Figure 4E). Both proteins show a higher affinity for DNA carrying a single nick as compared to DNAs with only blunt ends (Figures 2D and 4E). When compared to PARP1, PARP2 has a weaker affinity for DNA containing only DSB ends, but a similar affinity for nicked DNA (Figures 2D and 4E, Tables 1 and 2). Thus, when 1200-bp DNA fragments were used, the affinity of PARP1 was higher than that of PARP2 due to the contribution of PARP1 binding to both blunt ends and undamaged DNA (Table 1). However, our data demonstrate that both proteins have a similar affinity for nick-containing DNA. Added to this, the high level of discrimination of nick by PARP2 detected by AFM indicates that binding of PARP2 to DNA is significantly facilitated by SSB formation.
Figure 4.

Comparative analysis of PARP1 and PARP2 binding to DNA containing a single nick. Large scale AFM images (left) of PARP1 (A) and PARP2 (B) binding to nicked 1200-bp DNA and zoomed images (right) of PARP1(2)–DNA complexes. Nicked 1200-bp DNA was incubated with 3.5 nM PARP1 or PARP2 and imaged by AFM in air. Arrows indicate binding of PARP1(2) to DNA ends, nick or undamaged DNA. Scale bar 200 or 500 nm; Z scale: 7 nm. (C) AFM analysis of PARP1(2)–DNA complex formation, number of DNA molecules analysed: 210 for PARP1 and 189 for PARP2. The results represent mean ± SD of three to six independent samples. (D) Analysis of the position distributions of PARP1 and PARP2 on nicked 1200-bp DNA, number of complexes analysed: 145 for PARP1 and 104 for PARP2. The results represent mean ± SD of three to six independent samples. (E) Fluorescence measurements of PARP1 or PARP2 binding to DNA. The reaction mixtures containing the fluorescein-labelled protein PARP1 or PARP2 were titrated with increasing amounts of nicked 1200-bp DNA. Bars indicate the standard error of three independent experiments.
Table 2. DNA binding constants and specificities of PARP1 and PARP2 for nicked 1200-bp DNA fragment.
| PARP1 | PARP2 2 | |||
|---|---|---|---|---|
| Kd (nM) | Specificity | Kd (nM) | Specificity | |
| AFM-site specific constants: | ||||
| DNA nick | 5.88 ± 0.51 | 1349 ± 134 | 3.7 ± 0.40 | 7297 ± 872 |
| Non-specific | 7936 ± 476 | 27027 ± 1270 | ||
| Binding constants (AFM macro)a | 2.3 ± 0.15 | 3.0 ± 0.29 | ||
| Binding constants (Flu)b | 0.84 ± 0.09 | 1.6 ± 0.12 | ||
aComposite macroscopic binding constants of PARP1 and PARP2 to nicked 1200-bp DNA fragments.
bMacroscopic binding constant of PARP1 and PARP2 to a 1200-bp DNA measured by fluorescence titration in this study (Figure 4E).
AFM imaging of PARP1 and PARP2 activation in the presence of DSBs and SSBs
PARP1(2) catalyse the synthesis of PAR using NAD+ as substrate (1,2,4–6). PARP1(2) activation results in the synthesis of PAR, the branched negatively charged polymer, that is covalently attached to the PARPs themselves or to other acceptor proteins (1,5). PARylation of PARP1 leads to suppression of its DNA binding activity and seems to be a factor regulating the interaction of the protein with DNA (1,5). As for PARP1, PAR synthesis catalysed by PARP2 is also stimulated in the presence of DNA damage (3,8,9). To address PARylation of PARP1(2) using AFM, imaging of these proteins after incubation with 1200-bp DNA, nicked 1200-bp DNA or nicked or supercoiled pBR in the presence of NAD+ was undertaken (Figures 5 and 6). When DNA substrates were incubated with PARP1 in the presence of NAD+, the formation of branched polymers was detected by AFM (Figure 5A and B). It should be noted that incubation of PARP1(2) with NAD+ without DNA did not lead to the appearance of PARylated proteins under the assay conditions used here (Supplementary Figure S5). The time course of PARP1 activation detected at the molecular level allowed us to measure the average size of the PARylated proteins in the presence of 1200-bp DNA, nicked 1200-bp DNA, and nicked or supercoiled pBR (Figure 5C). The size was estimated by determining the area of the circle with the radius, which encloses the auto-PARylated protein (Figure 5D). Three major points could be underlined. First, the size of the PAR increases mainly within the first 15 min, but the polymerization reaction proceeds for all the incubation period (Figure 5D). Second, PARylation of PARP1 was observed for all DNA substrates including supercoiled pBR, but the size of PAR and the amount of PARylated proteins increased significantly in the presence of DNA substrates with breaks (Figure 5D). Third, in the case of DNA substrates containing both DSB and SSB, PARylated PARP1 can still bind to DNA and has been mainly detected near DSB ends (Figures 5A and 7).
Figure 5.

AFM analysis of PARP1 activation in the presence of the different DNA substrates. AFM images show auto-PARylation of PARP1 in the presence of nicked 1200-bp DNA (A), and 1200-bp DNA, circular nicked pBR or supercoiled pBR (B). Arrows indicate PARylated proteins bound to DNA molecules. Scale bar: 500 nm; Z scale: 7 nm. (C) Comparative analysis of the size of PARylated PARP1 in the presence of different DNA substrates. Number of PARylated molecules analysed: 145 for nicked 1200-bp DNA, 202 for 1200-bp DNA, 114 for nicked pBR and 107 for supercoiled pBR. Each data point represents the mean area ± SD with R of PARylated molecules measured from images. All measurements of radius (R) were done from the three to ten images obtained from three independent samples for each DNA substrate. The size of PARylated PARP1 smaller than 1200 nm2 (R < 20 nm) were not taken into account. (D) Zoomed image of PARylated PARP1 bound to SSB. The minimum radius of the circle (R) enclosing the PARylated protein was used to estimate the area of PARylated proteins. Scale bar: 100 nm; Z scale: 7 nm.
Figure 6.

AFM analysis of PARP2 activation in the presence of SSBs. (A) AFM images show time course of auto-PARylation of PARP2 in the presence of nicked 1200-bp DNA. White arrows indicate PARylated proteins bound to DNA molecules. Scale bar: 500 nm; Z scale: 7 nm. (B) Large scale AFM images of PARylated PARP2 in the presence of nicked 1200-bp DNA. Insert: zoomed image of a large PARylated PARP2 formed under these conditions. Scale bar: 500 nm; Z scale: 7 nm. (C) Comparative analysis of the size of PARylated PARP2 in the presence of 1200-bp DNA with or without nick after 120 min of reaction. Number or PARylated molecules analysed: 354 for nicked 1200-bp DNA and 145 for 1200-bp DNA. Each data point represents the mean area ± SD with radius of PARylated molecules measured from images. All measurements of the radiuses were done from the five images obtained from three independent samples for each DNA substrate. The size of PARylated PARP2 smaller than 1200 nm2 (R < 20 nm) were not taken into account.
Figure 7.

AFM analysis of the position distributions of PARylated PARP1(2) on nicked 1200-bp DNA. PARP1 (3.5 nM) or PARP2 (3.5 nM) was incubated with 1200-bp nicked DNA (1.5 nM) at 37°C for PARP1 and on ice for PARP2 for 60 min in the presence of 100 μM NAD+. Number of complexes analysed: 89 for PARP1 and 55 for PARP2.
When PARP2 was incubated with DNA in the presence of NAD+ at 37°C, PARylation of this protein was not observed by AFM, although it was detected by SDS gel electrophoresis (Supplementary Figure S6). The visualization of PARP2 auto-PARylation by AFM was achieved by incubating the reaction mixtures on ice before sample loading on mica (Figure 6A and B). In the case of 1200-bp DNA, the PARylation catalysed by PARP2 was detected, but at a low level (Figure 6C) while PARP2 activation was clearly observed after 15 min of incubation in the presence of nicked 1200-bp DNA (Figure 6A and B). Thus, the presence of a nick in the DNA fragment led to a significant increase in both the amount of PARylated protein and the size of the PAR polymer observed under the assay conditions used here (Figure 6C). As for PARP1, the increase in size of the PAR polymer is mainly observed during the 15 min of the reaction; although the sizes of the PAR structures are increased at least 2-fold after incubation for 2 h on ice. It should be noted that interaction of PARylated PARP2, as also PARP1, with DNA was detected by AFM images, but mainly at the nick site (Figures 6A and 7). According to the literature, poly(ADP-ribosyl)ation of PARPs leads to a decrease of its DNA binding activity and facilitates its dissociation from DNA breaks (1–6). Although automodified PARPs can still bind to the DNA, the amount of complexes of PARylated proteins with DNA is strongly reduced, such that the percentage of nicked 1200-bp DNA bound to PARylated proteins is about 5.5 and 3.7% for PARP1 and PARP2 respectively. It is nearly one order of magnitude lower than the percentage of binding of unPARylated proteins to DNA (which correlates to a ≈50-fold effect on the binding affinity of the proteins to nicks) (Table 2 and Supplementary Table S2). The interaction of PARylated proteins with DNA raises the question of their preferential localization on the DNA. We examined the position distributions of PARylated PARP1(2) on nicked 1200-bp DNA by analysing a DNA molecules complexed with the modified proteins (Figure 7). Our results show that PARylated PAPR1 is mainly located at DNA ends and nick, while PARylated PARP2 is preferentially located on the nick site (Figure 7). This is in agreement with the mode of binding of unmodified PARP2 or PARP1 to individual sites observed by AFM for nicked 1200-bp DNA, when about 65% of PARP1 or 85% of PARP2 complexes were bound to DNA breaks (Figure 4D). The analysis of PARylation catalysed by the proteins as measured by AFM and gel electrophoresis shows the increase in size of the PAR polymer produced in the presence of DSBs and SSB for PARP1 and SSB for PARP2 (Figures 5C and 6C, Supplementary Figure S7). This suggests that the extent of PARylation can be influenced by initial binding affinity of PARPs for DNA sites (Tables 1 and 2). To determine by AFM the effect of different DNA binding sites on the extent of PARP1(2) PARylation, we measured the time course of changes in the size for the proteins activated by DSB, SSB or non-damaged DNA (Figure 8A). Figure 8B and D show the average size of PARylated PARP1 or PARP2 after incubation of the proteins with nicked 1200-bp DNA for 30, 60 and 120 min in the presence of NAD+. These data demonstrate that after 2 h of incubation the size of PARylated PARP1 detected at SSB and DSB was 25–40% greater than the size of modified PARP1 molecules located on undamaged DNA (Figure 8B). In the case of PARP2, its activation was detected mainly at DNA breaks (Figure 8D). Size of PARylated PARP2 detected at SSBs increases of 37% in comparison with the modified form of proteins detected near DSBs. Thus, types of DNA damage have an influence on the size of the PAR polymers produced by PARPs.
Figure 8.

AFM-based size determination of PARylated PARP1 and PARP2 according to position of the proteins on DNA. AFM images of PARylated PARP1 (A) and PARP2 (C) located at different sites of nicked 1200-bp DNA (from top to bottom: DNA end, nick and undamaged DNA for PARP1, DNA end and nick for PARP2). Scale bar: 100 nm; Z scale: 7 nm. (B) Time course of changes in the size of PARylated PARP1 during its activation at nick, DNA ends or undamaged DNA sites. (D) Time course of changes in the size of PARylated PARP2 during its activation at nick or DNA ends. Each data point represents the mean area ± SD. P-values were obtained by comparing the results by t-test, *, P < 0.05; **, P < 0.01; ***, P < 0.005; ns, not significant. All measurements of radiuses were done from more than 10 images obtained from three independent samples for each DNA substrate.
DISCUSSION
PARP1 and PARP2 have been proposed to play a critical role in the detection and repair of DNA breaks (1,3,5,6,41). Here, we analysed the interaction of PARP1(2) with long DNA molecules to estimate the specificity of PARP1 and PARP2 interaction with SSBs and DSBs using a combination of AFM imaging and biochemical approaches. Previously, the AFM technique was only applied to study the interaction of PARP1 with chromatin, plasmid and specific DNA structures such as loops, hairpins and cruciform (42–45). Due to its ability to image single molecules, AFM provides unique information on DNA–protein complexes such as specificity of the interaction and cooperative aspects of binding (21–25). However, the main problem of this approach remains the potential dissociation of DNA–protein complexes on mica surface during the adsorption step. As mica is a highly negatively charged surface as are DNA molecules, the adsorption and immobilization of nucleic acids alone or associated with proteins require the presence of multivalent cations in the deposition buffer (25,26). Magnesium and polyamines such as putrescine or spermidine can be used as cations in the buffer (26,28). Spermidine at sub-millimolar concentrations induces strong DNA or DNA–protein complex adsorption allowing the observation of protein–ssDNA complexes under various conditions of ionic strength (26,27). It was shown that the concentration of putrescine has to be in the millimolar range for adsorption of proteins with dsDNA on mica (26,28). We developed a procedure for absorption of PARP–DNA complexes on mica surface mediated by putrescine. In contrast to Mg2+ and Spd3+, using of Pu2+ as counterions allowed to obtain representative large-scale images of DNA–PARP1(2) complexes and compare them not only to specific interactions of these proteins with DNA breaks but also their activation in the presence of NAD+. Indeed, PAR synthesized by PARP1(2) is a negatively charged polymer and can be adsorbed on mica surface such as DNA molecules, allowing to estimate the size and structure of the poly(ADP-ribose) chains.
PARP1 is considered as a molecular sensor of both DNA SSBs and DSBs (1,5,32,33,41). Our AFM data show that about 60% of PARP1 complexes were detected on DNA breaks and 40% on undamaged DNA (Figure 4D). However, considering the fact that undamaged sites are ∼1000-fold in excess over breaks, PARP1 binds predominantly to SSB/DSB rather than to undamaged DNA (Supplementary Table S1). At the same time, distribution of PARP1 between DSBs and SSBs is relatively similar, although this protein has higher affinity and specificity to nick sites (Tables 1 and 2). Fluorescence titration assay confirmed that PARP1 binds to 1200-bp DNA containing both a SSB and DSB with Kd value two times lower that the one for 1200-bp DNA without nick (Figures 2D and 4E).
PARP2 interaction with damaged DNA is less well documented than PARP1 (10,13,14). The higher level of stimulation of PARP2 activity towards SSBs compared to DSBs was detected using short DNA duplexes representing DNA intermediates of the different DNA repair pathways (13,14). In our case, AFM detection at the single molecule level have demonstrated a strong influence of a single nick on PARP2 binding to DNA (Figures 2C and 4C), PARP2 molecules are observed at SSB on nicked 1200-bp DNA in 75% of the total quantity of complexes (Figure 4D). Simultaneously, AFM and fluorescence titration experiments prove that the presence of nick in the 1200-bp DNA fragment induces a ∼5-fold increase in the affinity of PARP2 for the DNA substrate (Tables 1 and 2). Thus, in contrast to PARP1, PARP2 has a low affinity for undamaged DNA, being more specialized in recognizing SSB (Table 2). This specificity of PARP2 for SSBs suggests the possibility that PARP2 contributes to the SSB repair mechanism. Thus, AFM imaging provides direct evidence that PARP1 and PARP2 indeed preferentially bind to DNA breaks in the context of an extended DNA structure.
Another interesting finding of this study is the visualization of the PARylated proteins by AFM. Detection of PARylated PARPs by AFM at high resolution allowed us to compare the size and structure of PAR synthesized by the proteins (Figures 5–8). AFM images of PAR structures formed under activation of PARP1 or PARP2 does not show differences since PARs adopt a highly branched ‘star’ shape for both PARPs used (Figures 5C and 6A). Likewise, PARylated PARP1 and PARP2 interacting with SSBs or DSBs do not show differences in the PAR structure detected by AFM. In addition, measuring the size of PARylated proteins shows that modified PARP1(2) with R-value larger than 40 nm still interacts with DNA (Figure 8B and D). It is possible that ADP-ribose chain lengths synthesized by PARP1(2) depend mainly on the initial affinity of the proteins for the site of DNA damage (Tables 1 and 2), i.e. high affinity binding leads to a longer residence time of the protein at the specific site and thus offers the possibility of synthesizing a larger polymer. The size and the location of PARylated PARP2 along DNA molecules in the presence of both SSBs and DSBs are in agreement with this hypothesis (Figures 7 and 8). The model of PARP1 ‘shuttling’ proposes the regulation of PARP1 interaction with DNA through auto-PARylation (1,5,46). This model implies that auto-poly(ADP-ribosyl)ation of PARP1 initiates dissociation of the enzyme–DNA complexes due to an electrostatic repulsion between anionic PAR and DNA (5,46). Association of PARylated PARP1(2) of varied size with different DNA sites observed by AFM may point to the weaker influence of the electrostatic force on the stability of PARP1(2)–DNA complexes during the PARylation reaction. These AFM results are in line with biochemical data wherein it was shown lowered affinity of PARylated PARP1 to DNA nick rather than an inability of the automodified protein to bind DNA lesions (16,17,39).
In conclusion, using analyses at the single molecule level and long DNA fragments, we detected efficient interaction of PARP2 with SSB (Table 2). These results were confirmed by biochemical data that validate our experimental conditions used in AFM experiments. We then took advantage of our approach to study PARP1(2) activation and it appears that the nature of strand interruption influences the efficiency of PARylation, while the polymer remains highly branched in the case of reactions catalysed by PARP1 or PARP2. Our data permit us to reach the important conclusion that PARylated PARP1 and PARP2 retain their interaction with DNA at their specific DNA sites (SSB or DSB) which initiated the reaction of PARP poly(ADP-ribosyl)ation. These data open perspectives on PAR structural studies depending on the nature of the DNA damage and may bring important information on the function of the two proteins in the nucleus.
Supplementary Material
Acknowledgments
We are thankful to Dr Valerie Schreiber (Université de Strasbourg, IREBS, Illkirch, France) for providing the recombinant plasmid coding PARP2 and Dr Masahiko S. Satoh (Laval University Medical Centre (CHUQ), Laval University, Québec, Canada) for providing the recombinant plasmid coding PARP1. We would like to thank Dr Anne-Lise Haenni (Jacques Monod Institute, Paris, France) for careful reading of the manuscript and useful comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Scientific Fund [14-24-00038 to O.I.L.]; Institut National de la Santé et de la Recherche Médicale; Genopole Evry. Funding for open access charge: Russian Scientific Fund [14-24-00038].
Conflict of interest statement. None declared.
REFERENCES
- 1.D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 2.Ame J.C., Spenlehauer C., de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber V., Dantzer F., Ame J.C., de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 4.Yelamos J., Schreiber V., Dantzer F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol. Med. 2008;14:169–178. doi: 10.1016/j.molmed.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T., Satoh M.S., Poirier G.G., Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 6.Satoh M.S., Poirier G.G., Lindahl T. NAD(+)-dependent repair of damaged DNA by human cell extracts. J. Biol. Chem. 1993;268:5480–5487. [PubMed] [Google Scholar]
- 7.Niedergang C., Trucco C., Flatter E., De La Rubia G., Oliver J., Rolli V., Ménissier de Murcia J., de Murcia G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999;81:69–75. doi: 10.1016/s0300-9084(99)80040-6. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber V., Amé J.C., Dollé P., Schultz I., Rinaldi B., Fraulob V., Ménissier de Murciam J., de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002;21:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 9.Ame J.C., Rolli V., Schreiber V., Niedergang C., Apiou F., Decker P., Muller S., Hoger T., de Murcia J.M., de Murcia G. PARP-2, a novel mammalian DNA damage dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber V., Ricoul M., Amé J.C., Dantzer F., Meder V., Spenlehauer C., Stiegler P., Niedergang C., Sabatier L., Favaudon V., et al. PARP-2, structure–function relationship. In: Burkle A, editor. Poly(ADP-ribosyl)ation. 2006. pp. 13–31. [Google Scholar]
- 11.Pion E., Ullmann G.M., Amé J.C., Gérard D., de Murcia G., Bombarda E. DNA-induced dimerization of poly(ADP-ribose)polymerase-1 triggers its activation. Biochemistry. 2005;44:14670–14681. doi: 10.1021/bi050755o. [DOI] [PubMed] [Google Scholar]
- 12.D'Silva I., Pelletier J.D., Lagueux J., D'Amours D., Chaudhry M.A., Weinfeld M., Lees-Miller S.P., Poirier G.G. Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta. 1999;1430:119–126. doi: 10.1016/s0167-4838(98)00278-7. [DOI] [PubMed] [Google Scholar]
- 13.Kutuzov M.M., Khodyreva S.N., Ame J.C., Ilina E.S., Sukhanova M.V., Schreiber V., Lavrik O.I. Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie. 2013;95:1208–1215. doi: 10.1016/j.biochi.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Langelier M.F., Riccio A.A., Pascal J.M. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–7775. doi: 10.1093/nar/gku474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lonskaya I., Potaman V.N., Shlyakhtenko L.S., Oussatcheva E.A., Lyubchenko Y.L., Soldatenkov V.A. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J. Biol. Chem. 2005;280:17076–17083. doi: 10.1074/jbc.M413483200. [DOI] [PubMed] [Google Scholar]
- 16.Lavrik O.I., Prasad R., Sobol R.W., Horton J.K., Ackerman E.J., Wilson S.H. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. J. Biol. Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 17.Sukhanova M., Khodyreva S., Lavrik O. Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase in long patch base excision repair. Mutat. Res. 2010;685:80–89. doi: 10.1016/j.mrfmmm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Ame J.C., Kalisch T., Dantzer F., Schreiber V. Purification of recombinant poly(ADP-ribose) polymerases. Methods Mol. Biol. 2011;780:135–152. doi: 10.1007/978-1-61779-270-0_9. [DOI] [PubMed] [Google Scholar]
- 19.Sukhanova M.V., Khodyreva S.N., Lavrik O.I. Poly(ADP-ribose) polymerase-1 inhibits strand-displacement synthesis of DNA catalyzed by DNA polymerase beta. Biochemistry (Mosc.) 2004;69:558–568. doi: 10.1023/b:biry.0000029855.68502.fa. [DOI] [PubMed] [Google Scholar]
- 20.Revet B., Fourcade A. Short unligated sticky ends enable the observation of circularised DNA by atomic force and electron microscopies. Nucleic Acids Res. 1998;26:2092–2097. doi: 10.1093/nar/26.9.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y., Sass L.E., Du C., Hsieh P., Erie D.A. Determination of protein-DNA binding constants and specificities from statistical analyses of single molecules: MutS-DNA interactions. Nucleic Acids Res. 2005;33:4322–4334. doi: 10.1093/nar/gki708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engel A., Müller D.J. Observing single biomolecules at work with the atomic force microscope. Nat. Struct. Biol. 2000;7:715–718. doi: 10.1038/78929. [DOI] [PubMed] [Google Scholar]
- 23.Lyubchenko Y.L., Gall A.A., Shlyakhtenko L.S. Visualization of DNA and protein-DNA complexes with atomic force microscopy. Methods Mol. Biol. 2014;1117:367–384. doi: 10.1007/978-1-62703-776-1_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustamante C., Rivetti C. Visualizing protein-nucleic acid interactions on a large scale with the scanning force microscope. Annu. Rev. Biophys. Biomol. Struct. 1996;25:395–429. doi: 10.1146/annurev.bb.25.060196.002143. [DOI] [PubMed] [Google Scholar]
- 25.Pastre D., Hamon L., Sorel I., Le Cam E., Curmi P.A., Pietrement O. Specific DNA-protein interactions on mica investigated by atomic force microscopy. Langmuir. 2010;26:2618–2623. doi: 10.1021/la902727b. [DOI] [PubMed] [Google Scholar]
- 26.Pastre D., Hamon L., Landousy F., Sorel I., David M.O., Zozime A., Le Cam E., Pietrement O. Anionic polyelectrolyte adsorption on mica mediated by multivalent cations: a solution to DNA imaging by atomic force microscopy under high ionic strengths. Langmuir. 2006;22:6651–6660. doi: 10.1021/la053387y. [DOI] [PubMed] [Google Scholar]
- 27.Hamon L., Pastre D., Dupaigne P., Le Breton C., Le Cam E., Pietrement O. High-resolution AFM imaging of single-stranded DNA-binding (SSB) protein–DNA complexes. Nucleic Acids Res. 2007;35:e58. doi: 10.1093/nar/gkm147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansma H.G., Laney D.E. DNA binding to mica correlates with cationic radius: assay by atomic force microscopy. Biophys. J. 1996;70:1933–1939. doi: 10.1016/S0006-3495(96)79757-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi W.X., Larson R.G. Atomic force microscopic study of aggregation of RecA-DNA nucleoprotein filaments into left-handed supercoiled bundles. Nano Lett. 2005;5:2476–2481. doi: 10.1021/nl051783v. [DOI] [PubMed] [Google Scholar]
- 30.Ristic D., Modesti M., van der Heijden T., van Noort J., Dekker C., Kanaar R., Wyman C. Human Rad51 filaments on double- and single-stranded DNA: correlating regular and irregular forms with recombination function. Nucleic Acids Res. 2005;33:3292–3302. doi: 10.1093/nar/gki640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoyer W., Cherny D., Subramaniam V., Jovin T.M. Rapid self-assembly of alpha-synuclein observed by in situ atomic force microscopy. J. Mol. Biol. 2004;340:127–139. doi: 10.1016/j.jmb.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 32.Wang M., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansour W.Y., Rhein T., Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010;38:6065–6077. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beck C., Robert I., Reina-San-Martin B., Schreiber V., Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014;329:18–25. doi: 10.1016/j.yexcr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Benjamin R.C., Gill D.M. Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA molecules containing different types of strand breaks. J. Biol. Chem. 1980;255:10502–10508. [PubMed] [Google Scholar]
- 36.Hengartner C., Lagueux J., Poirier G.G. Analysis of the activation of poly(ADP-ribose) polymerase by various types of DNA. Biochem. Cell Biol. 1991;69:577–580. doi: 10.1139/o91-086. [DOI] [PubMed] [Google Scholar]
- 37.Wacker D.A., Ruhl D.D., Balagamwala E.H., Hope K.M., Zhang T., Kraus W.L. The DNA binding and catalytic domains of poly(ADP-ribose) polymerase 1 cooperate in the regulation of chromatin structure and transcription. Mol. Cell Biol. 2007;27:7475–7485. doi: 10.1128/MCB.01314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Murcia J.M., Niedergang C., Trucco C., Ricoul M., Dutrillaux B., Mark M., Oliver F.J., Masson M., Dierich A., LeMeur M., et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khodyreva S.N., Prasad R., Ilina E.S., Sukhanova M.V., Kutuzov M.M., Liu Y., Hou E.W., Wilson S.H., Lavrik O.I. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1) Proc. Natl. Acad. Sci. U.S.A. 2010;107:22090–22095. doi: 10.1073/pnas.1009182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsons J.L., Dianov G.L. Monitoring base excision repair proteins on damaged DNA using human cell extracts. Biochem. Soc. Trans. 2004;32:962–963. doi: 10.1042/BST0320962. [DOI] [PubMed] [Google Scholar]
- 41.Parsons J.L., Dianova I.I., Allinson S.L., Dianov G.L. Poly(ADP-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. FEBS J. 2005;272:2012–2021. doi: 10.1111/j.1742-4658.2005.04628.x. [DOI] [PubMed] [Google Scholar]
- 42.Smulson M.E., Pang D., Jung M., Dimtchev A., Chasovskikh S., Spoonde A., Simbulan-Rosenthal C., Rosenthal D., Yakovlev A., Dritschilo A. Irreversible binding of poly(ADP)ribose polymerase cleavage product to DNA ends revealed by atomic force microscopy: possible role in apoptosis. Cancer Res. 1998;58:3495–3498. [PubMed] [Google Scholar]
- 43.Chasovskikh S., Dimtchev A., Smulson M., Dritschilo A. DNA transitions induced by binding of PARP-1 to cruciform structures in supercoiled plasmids. Cytometry. 2005;68:21–27. doi: 10.1002/cyto.a.20187. [DOI] [PubMed] [Google Scholar]
- 44.Soldatenkov V.A., Chasovskikh S., Potaman V.N., Trofimova I., Smulson M. E., Dritschilo A. Transcriptional repression by binding of poly(ADP-ribose) polymerase to promoter sequences. J. Biol. Chem. 2002;277:665–670. doi: 10.1074/jbc.M108551200. [DOI] [PubMed] [Google Scholar]
- 45.Potaman V.N., Shlyakhtenko L.S., Oussatcheva E.A., Lyubchenko Y.L., Soldatenkov V.A. Specific binding of poly(ADP-ribose) polymerase-1 to cruciform hairpins. J. Mol. Biol. 2005;348:609–615. doi: 10.1016/j.jmb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 46.Tulin A., Spradling A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science. 2003;299:560–562. doi: 10.1126/science.1078764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.