


Abstract
During class switch recombination (CSR), antigen-stimulated B-cells rearrange their immunoglobulin constant heavy chain (CH) loci to generate antibodies with different effector functions. CSR is initiated by activation-induced deaminase (AID), which converts cytosines in switch (S) regions, repetitive sequences flanking the CH loci, to uracils. Although U/G mispairs arising in this way are generally efficiently repaired to C/Gs by uracil DNA glycosylase (UNG)-initiated base excision repair (BER), uracil processing in S-regions of activated B-cells occasionally gives rise to double strand breaks (DSBs), which trigger CSR. Surprisingly, genetic experiments revealed that CSR is dependent not only on AID and UNG, but also on mismatch repair (MMR). To elucidate the role of MMR in CSR, we studied the processing of uracil-containing DNA substrates in extracts of MMR-proficient and –deficient human cells, as well as in a system reconstituted from recombinant BER and MMR proteins. Here, we show that the interplay of these repair systems gives rise to DSBs in vitro and to genomic deletions and mutations in vivo, particularly in an S-region sequence. Our findings further suggest that MMR affects pathway choice in DSB repair. Given its amenability to manipulation, our system represents a powerful tool for the molecular dissection of CSR.
INTRODUCTION
Antigen-dependent antibody diversification occurs in two stages: somatic hypermutation (SHM) and class switch recombination (CSR) (1,2). During SHM, antibody-expressing B cell clones that bind antigens are stimulated to proliferate and the variable regions of their immunoglobulin (Ig) genes acquire a large number of mutations. Cell clones expressing mutated immunoglobulins with higher affinities for the antigen undergo positive selection and further rounds of maturation. In contrast to SHM, which yields antibodies with higher affinity through altering the variable region, CSR gives rise to antibodies of a different isotype through an irreversible rearrangement of the heavy chain (CH) locus. The human CH locus consists of several genes (Cμ, Cδ, Cγ3, Cγ1, Cα1, Cγ2, Cγ4, Cϵ and Cα2), but mature B cells express on their surface initially only IgM or IgD (the latter arising through alternative splicing). During CSR, the antigen/cytokine combination in the environment of the B cell activates transcription of specific switch (S) regions, highly-repetitive sequences flanking the 5′ termini of the CH genes. Transcription then triggers a recombination process during which the Ig variable region is joined to one of the downstream constant genes with a concomitant loss of the intervening DNA. In this way, the B-cell switches from expressing the high-avidity IgM to producing high-affinity IgG, IgA or IgE with different biological effector functions (3).
SHM and CSR are initiated by activation-induced cytidine deaminase (AID), a protein expressed in antigen-activated B cells (4,5). AID converts numerous cytidines within the Ig locus to uracils, in a reaction that is dependent on transcription (6). In the absence of AID, both SHM and CSR are abrogated (7,8). In humans, CSR malfunction causes Hyper-IgM syndrome, characterized by elevated IgM levels and a concomitant decrease or complete absence of IgG, IgA and IgE (9).
That SHM and CSR depend not only on the generation of uracils but also on their metabolism was demonstrated by the finding that ageing mice lacking uracil-DNA N-glycosylase (UNG), an enzyme that excises uracil from DNA (10), develop B-cell lymphomas (11) and that SHM and CSR are severely attenuated in these animals (12). In humans, recessive mutations in the UNG gene cause Hyper-IgM syndrome (13).
Genetic evidence implicated also the mismatch repair (MMR) pathway in these processes: disruption of the mouse MMR genes Msh2, Msh6, Mlh1, Pms2 or Exo1 led to altered SHM and to a reduction in CSR that ranged from 2- to 7-fold, depending on the gene and the Ig serotype (14–21). Similarly, patients lacking PMS2 or MSH6 were diagnosed with a profound CSR defect (22,23). These findings were unexpected. Deamination of deoxycytidine, both AID-catalyzed and spontaneous (24), gives rise to U/G mispairs in DNA, however, even though these structures are recognized and bound by the human mismatch binding factor MutSα (heterodimer of MSH2 and MSH6) (25), they should not be addressed by MMR. Postreplicative MMR has evolved to remove mispaired nucleotides from the newly-synthesized strand during replication. To achieve this goal, MMR proteins need not only detect the mispair, but also direct its repair to the nascent strand. In eukaryotes, this strand is distinguished from the template by pre-existing termini, such as gaps between Okazaki fragments, where EXO1 initiates the degradation of the error-containing nascent strand up to and ∼150 nucleotides past the mispair (26). Because AID-induced U/G mispairs arise in G1 phase of the cell cycle, i.e. in DNA devoid of EXO1 loading sites, they should not trigger MMR. Instead, they should be repaired to C/G by base excision repair (BER) (27).
In all organisms, short-patch BER of uracil is initiated by the removal of this aberrant base, catalyzed primarily by UNG (10), although mammalian cells encode also the uracil-processing enzymes TDG (28), SMUG1 (29) and MBD4 (30). The resulting apyrimidinic (AP) site is then incised at its 5′ phosphate by an AP-endonuclease (APE1 in humans), which thus provides an entry site for polymerase-β (pol-β) that extends the 3′-OH terminus of the break by a single dCMP and concurrently removes the baseless sugar-phosphate residue by β-elimination. The remaining nick is then sealed by DNA Ligase III/XRCC1. Uracils can also be addressed by long-patch BER, which differs from the short-patch process in that the repair synthesis catalyzed by pol-β, pol-δ or pol-λ generates repair tracts of 2–6 nucleotides through stand displacement. This process requires, in addition to the BER enzymes, also the replication factors RFC, PCNA and FEN1 (31–33). BER-mediated repair of uracils is generally extremely efficient, possibly also due to the redundancy between UNG, TDG, SMUG1 and MBD4, however, only UNG has to date been implicated in SHM/CSR.
BER and MMR are highly-effective guardians of genomic integrity. So why are these processes linked to mutagenesis and chromosomal deletions at the Ig locus? One possible reason could be the high density of uracils generated by AID at its preferred target sequences WRCY (where the underlined C is the target of deamination and W = A or T; R = A or G; Y = C or T) in the regions undergoing SHM and CSR. In an earlier study, we were able to demonstrate that MMR can interfere with BER-mediated uracil repair on substrates containing a U/G and a G/T mispair in close proximity (34). We could also show that processing of substrates containing two U/G mispairs in close proximity activated PCNA monoubiquitylation and recruitment to chromatin of the error-prone DNA pol-η (35), events required for SHM (36–38). It was proposed that a similar interference between BER and MMR might give rise to the double-strand breaks (DSBs) that arise in the switch regions during the G1 phase of the cell cycle and that are necessary for CSR (39,40), but experimental evidence supporting this hypothesis is not available to date.
In an attempt to gain novel insights into the CSR process, we made use of a series of circular phagemid substrates containing uracil residues in one strand and a nick at a defined site in the other strand, the latter representing an intermediate of BER-catalyzed uracil repair and an entry point for MMR. We then studied the appearance and location of DSBs upon incubation of these molecules with MMR-proficient or -deficient extracts of human cells, as well as with purified recombinant BER and MMR proteins. We now show that DSBs can arise through processing of a combination of uracils and nicks situated on opposite strands. We also show that DSB induction is absolutely dependent on UNG, but that the dependence on MutSα is limited to substrates in which the uracils and the nick are separated by more than ∼50 nucleotides. MutLα was required only in a subset of events and this function required its endonucleolytic activity. We also provide evidence that processing of these substrates in vivo gives rise to deletions delineated by the uracil and/or strand break sites, and that this process is not limited to B-cells.
MATERIALS AND METHODS
Antibodies and reagents
The rabbit polyclonal anti-TDG antibody was a generous gift of Primo Schär, MSH2 antibody was from Calbiochem and the MSH6 and Lamin B antibodies were from Abcam. Restriction enzymes (NotI, XmnI, AclI and Nt.BstNBI), UDG, APE1 and the UDG inhibitor Ugi were obtained from New England Biolabs and the Protein A Dynabeads were obtained from Dynal Biotech. The purified recombinant MutLα D699N protein was a generous gift of Farid A. Kadyrov; additionally, MutLα and MutSα wt and MutSα KR (41), RPA and EXO1 were purified in our laboratory.
Cell culture
Burkitt lymphoma cells (BL2) were cultured in RPMI medium 1640 (GIBCO) with 10% fetal calf serum (GIBCO), 10 000 U/ml penicillin and 10 mg/ml streptomycin (Gibco-BRL). LoVo cells were grown in 50% DMEM (GIBCO), 50% F12 (GIBCO) supplemented with 10% fetal calf serum and penicillin-streptomycin, 293T Lα were cultured in DMEM supplemented with 10% Tet System-approved fetal calf serum (Clontech), 300 μg/ml hygromycin B (Roche) and 100 μg/ml zeocin (InvivoGen). To abrogate MutLα expression, 50 ng/ml doxycyclin (Clontech) was added to the media. siRNA transfections were carried out using the calcium phosphate transfection. Synthetic siRNA oligonucleotides sequences (all 5′ to 3′) were as follows: siLuc sequence CGTACGCGGAATACTTCGATT, siMSH2 UCCAGGCAUGCUUGUGUUGAATT and MSH6 CGCCATTGTTCGAGATTTA (Microsynth).
DNA substrates and DSB/mismatch repair assays
Isolation of the supercoiled homo- and heteroduplex substrates and the MMR assays were carried out as previously described (42). A heteroduplex DNA substrate (referred to as ‘304’, Figures 1A, and 3A) containing a U/G-U/G mismatch (one within an AclI restriction site) in the 89-bp polylinker of a pGEM13Zf(+) derivative pRichi was constructed by primer extension, using the primer.
Figure 1.

Processing of the U/G-U/G substrate in vitro gives rise to DSBs. (A) Schematic representation of the circular heteroduplex substrates carrying two U/G mispairs, one located in the recognition site of AclI endonuclease at nucleotide 47 and the other at position 3182. The positions of the Nt.BstNBI sites at nucleotide 351 (5′ substrate) or 2865 (3′ substrate), where a nick can be introduced selectively into the inner strand, and the position of the XmnI cleavage site are indicated. Digestion of the phagemid DNA with XmnI resulted in its linearization (3194 bp). Upon incubation of the substrates (either covalently-closed or nicked as shown in the respective figures) with the nuclear extracts, recovery and enzymatic digestion, appearance of DSBs in the vicinity of the uracils (mimicked here by additional restriction with NotI that cuts between the two uracils) gave rise to two smaller fragments of about 1900 bp and 1300 bp. The efficiency of DSB induction was estimated by ImageQuant from scans of GelRed-stained agarose gels. (B) Minimal requirements for DSB induction, analyzed by incubation of substrates containing two uracils in one strand and, where indicated, a nick either 3′ or 5′ from the mispaired Gs with BL2 nuclear extracts (NE). The upper panel shows UV shadowing of an agarose gel stained with GelRed. Black arrows indicate the fragments arising due to the presence of DSBs. (C) DSBs arising in the U/G-U/G substrate nicked 3′ from the mispaired Gs upon incubation with extracts of BL2 cells, LoVo extracts supplemented with recombinant MutSα or HEK 293TLα+ (MMR-proficient) extracts. Quantifications of three independent experiments are shown in the lower panels. Error bars show mean +/− S.D. (n = 3).
Figure 3.

Lesion density and sequence context affect DSB induction. Schematic representation of the circular heteroduplex substrates 304 (A) and 58 (B) carrying two U/G mispairs and a Nt.BstNBI site 58 or 304 nucleotides 5′ from the nearest mispaired G. (C) The Nt.BstNBI-nicked ‘58’ or ‘304’ substrates were incubated with BL2 extracts pretreated with Ugi where indicated. (D) The Nt.BstNBI-nicked ‘58’ substrate was incubated with LoVo extracts pretreated where indicated with Ugi and supplemented or not with recombinant MutSα. (E) The presence of uracils and a nick located within a sequence fragment from an Ig switch region renders the phagemid more prone to DSB induction in a time course experiment. (F) Same as in C, but the experiment was performed with ‘304’ substrates containing an abasic site, a U/G mispair and a nick in the G-strand 3′ from the U/G mispairs as indicated. Bar graphs show quantifications of three independent experiments. Error bars show mean +/− S.D. (n = 3).
CCAGTGAATTGTAATAUGAACACTATAGGGCGAATTGGCGGCCGCGATCTGATCAGA TCCAGACGTCTGTCAAUGTTGGGAAGCTTGAG. In this substrate, the distance between the nick and the closest uracil was 304 nucleotides. In order to create a substrate with a distance of 58 nucleotides between the nicking site and the closest uracil in the top strand (referred to as ‘58’, Figure 3B), the following primer was used:
U/G-U/G 235–306 GCCCGCTTTCUAGTCGGGAAACCTGTCGTGCCAGCTGCATTAATG
AATCTGCCAACGCGUGGGGAGAGGCGG
To assess the impact of sequence context, a 95-mer oligonucleotide containing nucleotides 267–344 sequence of the human Sμ region (GenBank/EMBL/DDBJ accession no. X54713) was cloned into the PshAI site of the polylinker of plasmid pRichi. The Sμ substrate was then generated by primer extension on the phagemid ssDNA, using the oligonucleotide CCAGACGTGGGCTAAGTTGUACCAGGTGAGCTGAGCTGAGCTAGGGCTTGGCTGCACTAACTGGGCTGAGUTGGGCAGGGCTGGGCCTGTCAACG. (The Sμ region sequence is shown in italics.) In this substrate, the Us are mispaired with Gs in the complementary strand.
To generate substrates with abasic sites, the following primers were used: U/G-ab: CCA GTG AAT TGT AAT AUG AAC ACT ATA GGG CGA ATT GGC GGC CGC GAT CTG ATC AGA TCC AGA CGT CTG TCA AXG TTG GGA AGC TTG AG and ab-U/G: CCA GTG AAT TGT AAT AXG AAC ACT ATA GGG CGA ATT GGC GGC CGC GAT CTG ATC AGA TCC AGA CGT CTG TCA AUG TTG GGA AGC TTG AG. In all sequences, uracils are shown in bold. X represents the abasic site analog inserted with using dSpacer CE phosphoramidite (Glen Research, 10–1914–90). The desired supercoiled substrates were purified on a CsCl gradient by standard methods. The nick was introduced by incubation with Nt.BstNBI.
The DSB assays were carried out using 100 ng (47.5 fmol) heteroduplex DNA substrate and 100 μg of nuclear extracts from BL2, 293TLα+ (MLH1+), 293TLα− (MLH1−) or LoVo cells in a total volume of 25 μl in a buffer containing 20 mM Tris HCl pH 7.6, 110 mM KCl, 5 mM MgCl2, 1 mM glutathione, 1.5 mM ATP, 50 μg/ml BSA. The reactions were incubated for 25 min at 30°C, followed by 60 min incubation with a stop solution (final concentrations: 0.5 mmol/l EDTA, 1.5% SDS, 2.5 mg/ml proteinase K). The DNA was purified using Qiagen MinElute Reaction Cleanup Kit followed by digestion with XmnI and treatment with 200 μg/ml RNase A (Sigma-Aldrich, Germany).
MMR efficiency was estimated as described previously (34), by digestion of the recovered DNA with AclI. In order to be able to visualize the repair tracts, the nuclear extracts were supplemented with [α-32P]dATP. The DNA fragments were then separated on 1% agarose gels containing GelRed and visualized on a UV trans-illuminator. The gels were then dried and exposed to PhosphoImager screens or X-ray films.
UDG inhibition and immunodepletions
Where indicated, UDG was inhibited by the addition of Ugi peptide (4.8 units) per 100 μg nuclear extract and pre-incubation for 10 min at 37°C. Immunodepletions were carried out as described previously (34).
Primer extension reactions
Phagemid DNA recovered from the DSB assay was subjected to primer extension reactions, using the 5′-labeled ‘Blue’ GCCTCCCTCGCGCCATCAGCTTAATGCGCCGCTACAGGG or ‘Red’ CTCATTAGGCACCCCAGGC primers (Figure 5). The reactions were performed by adding 0.2 μM 5′-labelled primer to 50 ng DNA, 1 mM dNTPs, 1x thermobuffer and 0.3 μl Taq polymerase. The following parameters were used for the annealing and extension reactions: 15 min at 95°C, 15 min at 50°C and 35 min at 72°C. The reaction products were admixed with an equal volume of formamide and separated on 5% denaturing polyacrylamide gels. The gels were fixed in 50% EtOH, 40% H2O and 10% acetic acid, dried and exposed to PhosphoImager screens.
Figure 5.

Breakpoints in the U/G-U/G substrate are at or near the uracil residues. U/G-U/G substrate nicked 5′ from the mispaired Gs was incubated with HEK 293T-Lα+/− extracts pretreated with Ugi where indicated. The DNA was recovered and subjected to primer extension using 5′ labeled primers ‘blue’ and ‘red’ (see Materials and Methods). The reaction products were separated on 5% denaturing polyacrylamide gels. Markers were products of primer extensions on plasmids digested with AclI (A) or NotI (N). The ‘blue’ primer was complementary to the G strand (A), the ‘red’ primer was complementary to the uracil-bearing strand (B) as shown schematically between the panels. The figure shows representative autoradiographs from four independent experiments.
Transfection/transformation experiments
HEK 293 cells were grown to 60% confluence and transfected with 1 μg U/G-U/G or C/G-C/G (ccc or nicked 5′ from the mispaired Us) plasmids (Amp resistant) and 500 ng of a control plasmid (Kan resistant) using calcium phosphate transfection. Twenty four hours post transfection, the plasmids were recovered using a DNA extraction kit (Qiagen). As DSB generation and religation was expected to be rare, we used positive selection. Half of the recovered plasmid DNA was digested with PshAI, the restriction site of which is situated in the vicinity of the uracils. Deletions were expected to eliminate this cleavage site and hence render the plasmid refractory to cleavage, while making it also unable to form colonies. DH10B bacteria were transformed with the digested or undigested plasmid DNA and plated on kanamycin or ampicillin plates. Single ampicillin-resistant colonies were picked and taken forward to colony hybridization.
Colony hybridization
The colony hybridizations were performed essentially as described (43). The colonies picked from the agar plate (see above) were streaked on a fresh plate as shown in Figure 6B. The bacteria were then transferred to a nitrocellulose membrane by placing the membrane on top of the agar plate for 30 s. The membrane was then denatured and fixed as described (43). The bacterial plate was kept at 4°C. The clones identified as mutated or deleted were then sequenced.
Figure 6.

In vivo repair of DSBs in the U/G-U/G substrate leads to mutations and deletions. (A) Western blot of extracts of HEK 293 cells showing efficiency of knock-down of MSH2 and MSH6 with siRNA. (B) Autoradiograph of a representative filter carrying plasmid DNA recovered from transfections of HEK 293 cells with the U/G-U/G substrate nicked 5′ from the mispaired Gs, hybridized with a radiolabeled oligonucleotide complementary to the wild type (unmodified) plasmid sequence flanking the uracil residues. (C) Mutated and deleted plasmids recovered from multiple transfections of HEK 293 cells with the listed substrates. (D) As in C, but the substrates were transfected into HEK 293 cells depleted of MutSα. (E) As in C, but the substrates were transfected into HEC 59 + chr2 cells (MMR-proficient). (F) As in C, but the substrates were transfected into HEC 59 cells. (G) Incidence of microhomologies in plasmids recovered from transfections of the indicated substrates into HEK 293 cells depleted or not of MutSα. (H) Incidence of microhomologies in plasmids recovered from transfections of the indicated substrates into HEC 59 + chr2 or HEC 59 cells.
Reconstituted DSB assay
For the in vitro DSB-assay, the reaction contained 100 ng of the substrate, 375 fmol human MutSα, 460 fmol human MutLα, 478 fmol human RPA, 26 fmol human EXOI, 1.2 fmol bacterial Uracil-DNA glycosylase and 0.378 fmol human AP Endonuclease. Briefly, the substrates were incubated with the indicated proteins in the presence of 20 mM Tris-HCl (pH 7.6), 3 mM ATP, 1 mM glutathione, 5 mM MgCl2, 0.05 mg/ml BSA and 100 mM KCl. After incubation at 37°C for 20 min, the samples were heat-inactivated for 10 min at 80°C, digested with XmnI and analyzed on a 1% agarose gel containing GelRed.
RESULTS
U/G processing in B-cell extracts promotes DSB formation
AID was reported to act in a processive manner (44) and to target both strands in switch regions, albeit with different efficiencies (45). In an attempt to mimic the substrates that might arise in vivo in our in vitro assays, we set out to generate circular plasmids containing uracils in both DNA strands. However, U/G mispairs arising through AID-catalyzed deamination of cytosines are substrates for both BER and MMR and processing of substrates carrying several such mispairs could therefore potentially initiate at any uracil residue, which would complicate the analysis of the products. In order to simplify the situation, we generated substrates containing two U/G lesions in the same strand, representing the processive action of AID, and a nicking site for Nt.BstNBI on the opposite strand, either 3′ or 5′ from the uracils (Figure 1A, left panel), representing a cleaved abasic site generated upon removal of a uracil by UNG and hydrolysis by APE1 that would provide an entry site for MMR as shown in our earlier work (34). Restriction digest of these substrates with XmnI gives rise to the linearized full-length plasmid molecule of 3193 bp (Figure 1A, top right panel), but a DSB arising in the vicinity of the uracils would give rise to fragments migrating close to the 1923 and 1270 bp fragments generated by XmnI/NotI cleavage (Figure 1A, bottom right panel). When we incubated the closed-circular U/G-U/G substrates (which should be addressed by BER but not by MMR due to the absence of a pre-existing nick) with nuclear extracts (NE) of BL2 (human Burkitt lymphoma) B cells and subjected the recovered DNA to XmnI digest, only the full-length fragment was detectable (Figure 1B, lane 1). Introduction of a single nick (MMR entry site) in the opposite strand either 5′ or 3′ from the U/G mispairs prior to incubation of the substrates with the extracts yielded ∼10% of molecules cleaved by XmnI into two fragments of ∼2000 and ∼1200 bp in size, indicative of the presence of DSBs in the vicinity of the uracil residues (Figure 1B lane 1 versus lanes 2, 3).
Induction of DSBs in the nicked U/G-U/G substrate is not limited to B-cell extracts
In order to learn whether the DSBs arising as a consequence of uracil processing require B-cell specific factors, we compared the processing of the above substrates in B-cell extracts to that in extracts of LoVo and HEK293T-Lα cells. The former is a MMR-deficient human colon cancer cell line mutated in MSH2, but the extracts can be made MMR-proficient by complementation with purified recombinant MutSα. The latter cells are of human embryonic kidney origin. The MLH1 MMR gene is silenced by methylation in these cells, but we have modified them to express exogenous MLH1 upon treatment with doxycycline (46). As shown in Figure 1C, DSBs were generated in nuclear extracts derived from BL2 cells as shown previously (lane 1), and even somewhat more efficiently in the MMR-proficient extracts of LoVo cells supplemented with recombinant MutSα (lane 2) or in HEK293T-Lα+ extracts expressing MLH1 (lane 3). This finding demonstrates that DSB generation in this system is not limited to B-cell extracts.
UNG and MutSα play key roles in DSB induction
As mentioned above, genetic evidence showed that both BER and MMR participate in CSR. We therefore wanted to test whether the induction of DSBs in our system was dependent on these repair pathways. MMR-deficient extracts of LoVo cells generated no DSBs in the U/G-U/G substrate containing a single nick in the G strand 5′ from the mispairs (Figure 2A, lane 1) and addition of the uracil DNA glycosylase inhibitor Ugi did not alter the situation (lane 2). When the MMR defect in these extracts was complemented with MutSα, DSBs were formed (lane 3), but their formation was effectively inhibited by Ugi (lane 4). This showed that the breaks were generated by a process requiring uracil excision. Similar observations were made when the substrate contained a nick in the G strand 3′ from the mispairs (Figure 2A, lanes 5–8). In order to test whether DSB induction required enzymatically-active MutSα, we supplemented the LoVo extract with the MutSα KR variant, which is mutated in the ATP binding motifs of both MSH2 and MSH6. This variant is unable to undergo the ATP-driven transition to a sliding clamp and hence remains bound at the mismatch (47). Unlike the wild type protein (Figure 2A, lane 7), MutSα KR failed to restore DSB induction in LoVo extracts (lane 9). The generation of DSBs thus requires not only the binding of MutSα to DNA, but also its ATPase activity.
Figure 2.

UNG and MutSα play key roles in DSB induction. (A) Lanes 1–4: DSB formation in a U/G-U/G substrate, nicked 5′ from the mispaired Gs, upon incubation with NE of LoVo cells supplemented with recombinant MutSα and pretreated with Ugi where indicated. Lanes 5–10: Incubation of the U/G-U/G substrate, nicked 3′ from the mispaired Gs, with LoVo extracts supplemented either with wild type MutSα or the ATPase-dead KR mutant, and pretreated with Ugi where indicated. (B) Lanes 1–4: DSB formation in a U/G-U/G substrate, nicked 5′ from the mispaired Gs, upon incubation with MMR-proficient (HEK 293T-Lα+) and -deficient (HEK 293T-Lα-) extracts. UNG was inhibited by the addition of Ugi where indicated. Lanes 5–10: DSB formation in a U/G-U/G substrate, nicked 3′ from the mispaired Gs, upon incubation with MMR-proficient (HEK 293T-Lα+) or -deficient (HEK 293T-Lα−) extracts. The MMR-deficient extracts were supplemented with either wild type MutLα or its endonuclease-deficient mutant (DN). Extracts were pretreated with Ugi where indicated. Quantifications of three independent experiments are shown in the lower panels. Error bars show mean +/− S.D. (n = 3).
We next studied the requirement for MutLα (a heterodimer of MLH1 and PMS2). When the nick was positioned 5′ from the mispaired guanines, DSBs were induced independently of MutLα (Figure 2B, lanes 1 and 3). As above, their formation was inhibited by Ugi (lanes 2 and 4). In contrast, MutLα was indispensable for the induction of DSBs in a substrate containing the nick 3′ from the mispaired guanines (Figure 2B, lane 9). No DSBs were formed when the extracts were supplemented with a MutLα variant lacking endonuclease activity (lane 7). These results conform to our understanding of MMR mechanism (see also Discussion), in which excision initiated at a 3′ nick displays an absolute requirement for MutLα, whereas excision starting at a 5′ nick does not (48). The requirement for UNG showed that DSB formation in these substrates required an intermediate of uracil processing, most likely a strand break arising through APE1-mediated cleavage of the abasic site generated by the UNG-catalyzed removal of one of the uracils. DSBs might then arise through the collision of a U/G-activated MMR-catalyzed excision tract in the nicked strand with a cleaved abasic site in the other strand.
The requirement for MMR in DSB formation is dependent on the distance between the nick and the U/G mispairs
In the Ig loci of activated B-cells, AID carries out multiple deaminations (44) in both strands (45). In an attempt to gain further insight into the criteria necessary to give rise to BER- and MMR-dependent DSBs in vivo, we varied the distance between the uracils and the nick in the opposite strand. By using uracil-containing oligonucleotides in the primer extension reaction that annealed closer to the Nt.BstNBI site, we were able to generate an additional substrate (Figure 3B), in which the nicking site was 58 nucleotides 5′ from the G of the nearest U/G mispair rather than 304 (Figure 3A). When the nick was 58 nucleotides away, the DSBs were formed with similar efficiency as in the substrate where it was 304 nucleotides away (Figure 3C). [NB: In the ‘58’ substrate, a DSB generated close to the uracils and XmnI cleavage generate two fragments of similar size, hence only a single band is seen in the agarose gel, as opposed to two different size bands generated from the ‘304’ substrate.]
Because CSR efficiency is only decreased and not abolished in MMR-deficient cells, we wanted to test whether the DSBs generated in the ‘58’ substrate were MMR-dependent, like those detected in the ‘304’ substrate (Figure 2). As shown in Figure 3D, incubation of the former substrate with MMR-deficient extracts of LoVo cells resulted in the generation of DSBs when the extract was supplemented with purified recombinant MutSα (lane 3), but were detectable, albeit only weakly, even in the unsupplemented extract (lane 1). Because DSB formation was still absolutely dependent on UNG (lanes 2,4), we postulated that they must have arisen by MMR-independent exonucleolytic degradation between the nick and the cleaved abasic site(s) in the opposite strand. This was substantiated by the finding that no DSBs were detected upon incubation of this substrate with extracts of EXO1-deficient cells (Supplementary Figure S1).
The DSB formation is dependent on sequence context
As mentioned in the Introduction, deletions occurring at the Ig locus during CSR initiate and terminate at the intergenic S regions. In order to learn whether these repetitive sequence elements affect the efficiency of DSB formation, we cloned a fragment of the Sμ-region into our phagemid and positioned the U/G mispairs 52 nucleotides apart within this sequence (see Materials and Methods). As shown in Figure 3E, DSB induction in the switch region sequence context was more efficient than in the original phagemid (compare lanes 1,3,5,7 with lanes 2,4,6,8). This result suggested that the S regions might be highly recombinogenic in vivo not only because they contain numerous AID consensus sequences, but possibly also because the highly-repetitive sequences might form secondary structures upon degradation of one strand. This would hinder repair synthesis and thus increase the time window for a collision of the MMR and or BER degradation tracts, which could result in DSB formation.
MMR involvement in DSB generation requires U/G mispairs
Exonucleolytic degradation of the error-containing strand during postreplicative MMR is initiated by loading of EXO1 at the nick by mismatch-activated MutSα. We wanted to confirm that the MMR-dependent DSB formation observed in our assays was indeed triggered by MutSα binding to the U/G mispairs. We also wondered whether the low efficiency of DSB formation was linked to the rapid removal of the uracils by UNG, which would convert our U/G-U/G substrate into a DNA molecule carrying two abasic sites, which are not substrates for MutSα. We therefore generated a substrate carrying only a single U/G mispair and a synthetic abasic site (a tetrahydrofuran ring) in place of the second uracil. This substrate mimics the U/G-U/G substrate from which one uracil had been removed by UNG. When a nick was introduced 3′ or 5′ from the mispair into the unmodified G strand, DSBs arose only in the MMR-proficient extract and upon inhibition of UNG with Ugi (Figure 3F, lanes 4,8). This suggested that UNG-catalysed removal of the uracil was more rapid than recognition of the U/G mispair by MutSα. Once the uracil was removed, the substrate contained either two abasic sites (intact or cleaved), or two C/G pairs generated by successful BER (see next section). As neither abasic sites nor C/G pairs are substrates for MMR, the strand degradation process was not activated.
U/G mispairs are processed by BER more rapidly than by MMR
We could test the above hypothesis directly. The 3′ uracil in the U/G-U/G substrate is situated in an AclI site such that it renders the substrate refractory to cleavage by this enzyme (Figure 4A). Correction to C/G, which can be mediated either by BER or by MMR directed to the uracil-containing strand, restores the AclI site (35). Because the repaired plasmid contains three AclI sites as opposed to only two in the U/G-U/G substrate, digestion of the repaired plasmid with AclI gives rise to three fragments of 1515, 1305 and 373 bp, whereas the unrepaired plasmid is cleaved into only two, of 2820 and 373 bp. As shown in Figure 4B (lane 1), incubation of the U/G-U/G plasmid nicked in the G strand 5′ from the mismatch in the AclI site with MMR-proficient extracts of HEK293TLα+ cells resulted in ∼60% repair to C/G. Importantly, much of this repair was mediated by BER, as its efficiency was reduced by maximally 50% (lanes 2,3) in the presence of aphidicolin, an inhibitor of DNA polymerase δ that carries out repair synthesis in the in vitro MMR reaction (26). Moreover, the MMR-mediated events were directed into the U-containing strand, most likely through repair tracts initiating at the cleaved abasic site generated by BER at the 5′ uracil (34), because only U/G to C/G repair restores the AclI site. This was further confirmed by the incorporation of [α-32P]dAMP into the 1515 and 1305 bp AclI fragments that only arise upon U/G to C/G repair (Figure 4C, lane 1).
Figure 4.
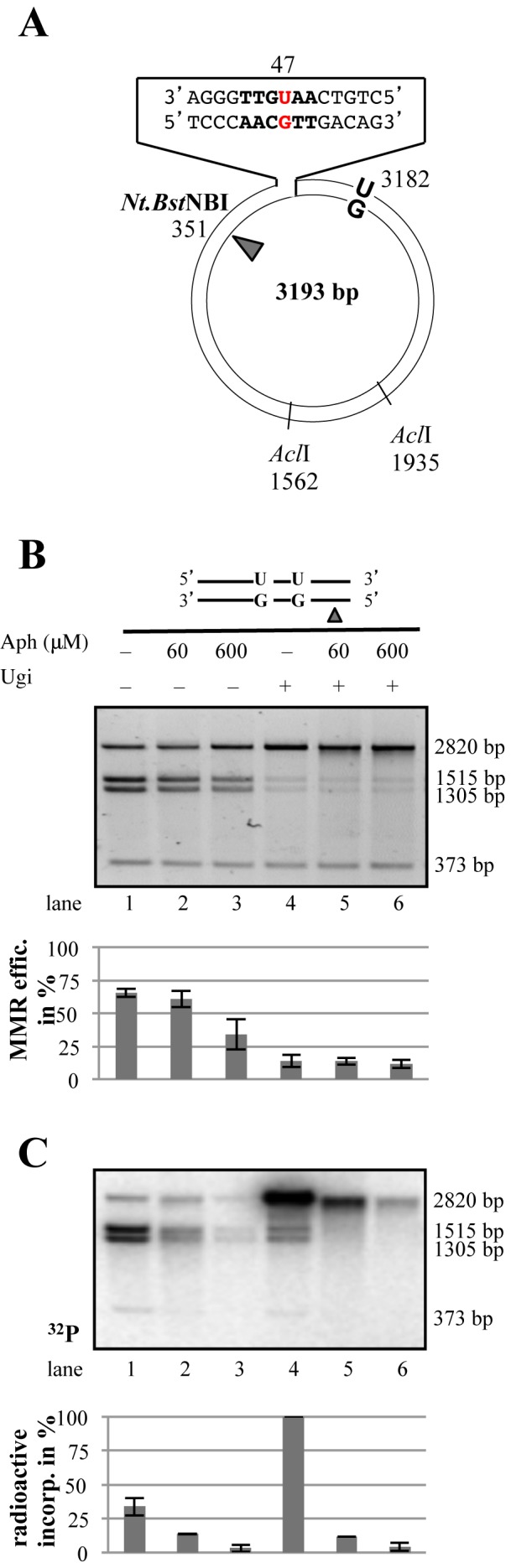
Both BER and MMR are activated on the nicked U/G-U/G substrate. (A) Schematic representation of the circular heteroduplex MMR substrate carrying two U/G mispairs, one located in the recognition site of AclI endonuclease at nucleotide 47 and the other at position 3182. The positions of the Nt.BstNBI site, where a nick can be introduced selectively into the inner strand 5′ from the mispaired Gs, and the position of the other two AclI cleavage sites are indicated. (B) U/G to C/G mismatch repair efficiency estimated by conversion of AclI-resistant to AclI-susceptible substrate (34). Aph, aphidicolin. The panel shows a representative image of a 0.8% agarose gel stained with GelRed. (C) Autoradiograph of the dried 0.8% agarose gel shown in panel B above. Bar graphs in panels B and C show quantifications of three independent experiments. Error bars show mean +/− S.D. (n = 3).
In contrast, inhibition of UNG with Ugi inhibited AclI cleavage almost completely (Figure 4B, lanes 4–6). Importantly, MMR was activated on this substrate more efficiently than in the absence of Ugi, as witnessed by the greater incorporation of [α-32P]dAMP into the phagemid molecules (Figure 4C, lane 4). Moreover, this process was mostly directed to the G-strand, given that the labeled molecules were refractory to AclI cleavage. This is indicative of U/G-activated MMR repair tracts initiating at the Nt.BstNBI nicking site, which would convert the U/G mispairs into U/A pairs.
Taken together, the latter evidence helps explain why DSB incidence in our system is relatively low. First, to generate a DSB, the MMR repair tract must collide with a BER intermediate in the opposite strand. Second, in order for MMR to be activated, some U/G mispairs must remain in the DNA, which is likely to occur only rarely due to the high efficiency of BER.
In vitro-generated DSBs cluster mostly in the proximity of the uracil sites
Using primer extension assays, we mapped the position of the DSBs arising in the U/G-U/G substrate nicked 3′ from the mispairs in the G-strand upon incubation with extracts of MMR-proficient HEK 293TLα+ cells. The most prominent breakpoint in the G-strand (mapped with the ‘blue’ primer) was located in close proximity of the AclI site containing the distal (3′) uracil residue (Figure 5A, lane 1). Upon inhibition of UNG with Ugi (lane 2), several weaker bands were detected, which were most likely generated by the MutLα heterodimer (49) upon MMR activation by the U/G mismatch.
The major breakpoint in the uracil-containing strand, mapped by extension of the ‘red’ primer, also resided at or close to the uracil within the AclI site (Figure 5B, lane 1). As addition of Ugi to the extract substantially reduced the incidence of this breakpoint (lane 2), we propose that it arose through APE1-catalyzed cleavage of the abasic site generated by UNG. Under the latter conditions, a second, slightly shorter, product was also detected. This fragment might represent an intermediate of uracil processing by long-patch BER initiated by other glycosylases present in the extracts (MBD4, SMUG1 or undepleted TDG).
DSBs in the U/G-U/G substrate are induced also in vivo
The sizes of the plasmid fragments generated from the U/G-U/G substrates in our in vitro assay indicated that the DSBs were occurring in close proximity to the uracils. We wanted to learn whether these findings reflected the processing of these substrates in vivo. DSBs arising upon transfection of the U/G-U/G plasmids into human cells should be repaired by non-homologous end-joining (NHEJ), a process that generally leads to a small loss of genetic information at the break (50,51). We therefore designed a protocol that enabled us to identify the mutated plasmids among the large excess of molecules that were corrected by BER to C/G-C/G without mutations. We carried out these experiments in human kidney 293 (HEK 293) cells, because they can be transfected with very high efficiency. Moreover, we showed above that DSBs form in uracil-containing substrates also in non-B cell types, and ectopic expression of AID in non-B cells was shown to trigger both SHM and CSR (52–54), suggesting that AID is the only B-cell-specific factor required for these processes.
We transfected the U/G-U/G or the control C/G-C/G substrate into the HEK293 cells, either wild-type or depleted of MutSα with siRNAs targeting MSH2 and MSH6 (Figure 6A), recovered the plasmid DNA 24 h later and used it to transform competent E. coli after digestion with PshAI. The cleavage site of this enzyme is situated in close proximity of the uracils and it was therefore anticipated that all plasmid molecules carrying the wild-type sequence at this site would be linearized and thus made transformation-incompetent. [NB: The enrichment for mutant molecules by digestion with PshAI was necessary, because the formation of DSBs and/or uracil misrepair in vivo are extremely rare.] In six independent experiments, we generally obtained ∼20 times more colonies from transformations with the recovered PshAI-digested U/G-U/G plasmid than with the C/G-C/G homoduplex control (data not shown). However, DNA sequencing revealed that the majority of these molecules in both populations were devoid of mutations, which implied that the PshAI digest was not quantitative. We therefore introduced a second screening step for the mutants: colony hybridization with a radiolabeled oligonucleotide complementary to the sequence flanking the PshAI site. We then sequenced only those colonies that indicated the presence of point mutations or deletions (Figure 6B).
As shown in Figure 6C and Supplementary Figure S2A, of 326 sequenced colonies from control transformations of HEK 293 cells with the covalently-closed circular (ccc) C/G-C/G plasmid, 7 (2.1%) contained point mutations and 7 (2.1%) contained deletions. Because the mutants were selected with a probe located between the two uracil residues, all examined deletion mutants lost this site. DNA sequencing revealed that these very rare deletions in the unnicked C/G-C/G substrate were several hundred base pairs long and did not share their end points. The presence of a nick in the G strand of this substrate trebled the occurrence of these large deletions to 6.3% and, as anticipated, one end point of these deletions was often (∼30%) close to the position of the nick. Transfection of the ccc U/G-U/G substrate yielded both mutations (8.5%) and deletions (7%), which were generally short, mostly <200 bp. Transfection of the nicked U/G-U/G substrate yielded plasmids containing a similar amount of mutations (8.7%), but substantially more deletions (16%), largely spanning the distance between the position of the uracils and the Nt.BstNBI nick. Most of the end-joining events occurred at sequences with no apparent homologies. This resembled analysis of hybrid S-S junctions in immunoglobulin switch regions, which showed that the sequences were fused at or very close to AID hotspots (40). We also detected several translocation events between the U/G-U/G plasmid substrate and genomic DNA (Supplementary Figure S2A, yellow lines). This finding shows that DSBs arising through the interplay of MMR and BER during the processing of proximal uracil residues in our substrate are recombinogenic and provides independent support for the hypothesis that chromosomal translocations arising in activated B cells (55) might have arisen through a similar mechanism.
We next wanted to study the effect of the MMR system on the above events. To this end, we treated the HEK 293 cells with siRNA targeting MSH2 and MSH6 to deplete MutSα (Figure 6A). Transfections of the above substrates into these cells yielded some unanticipated results (Figure 6D, Supplementary Figure S2B), inasmuch as the proportion of deletions was similar in all four substrates (8% for ccc C/G-C/G, 9.5% for nicked C/G-C/G, 5.3% for ccc U/G-U/G and 9.3% for nicked U/G-U/G), but—most remarkably—the number of point mutants was substantially reduced (0.8% for ccc C/G-C/G, 1% for nicked C/G-C/G, 1.9% for ccc U/G-U/G and 2.7% for nicked U/G-U/G). In all these mutants, the deletions were large and the influence of the nick was much less pronounced, which confirms that the nick is a specific signal for MMR-catalyzed strand degradation events.
We then transfected these substrates into the MMR-deficient human endometrial cancer cell line HEC 59, which is mutated in MSH2, or HEC 59 + chr2 cells, in which the MMR defect was corrected by transfer of chromosome 2 that carries the wild type MSH2 gene. Unexpectedly, the number of point mutants identified in both cell lines was very low, but the number of deletions in the MMR-proficient cells (Figure 6E, Supplementary FIgure S2C) was more than 2.5-fold higher than in the MMR-deficient line (Figure 6F, Supplementary FIgure S2D), particularly in the nicked U/G-U/G substrate. Their size and end-points reflected those observed in HEK 293 cells. Interestingly, sequence analysis of the clones revealed an increase in the number of microhomologies around the break points in the plasmids recovered from MMR-deficient MutSα-depleted HEK 293 (Figure 6G) and HEC 59 cells (Figure 6H), which suggests that the molecular mechanism of DSB repair is affected by the MMR status of the cells. [N.B.: The sequencing data are not shown due to space limitations, but are available upon request.]
DSBs arise in the nicked U/G-UG substrate in a system reconstituted from purified proteins
Based on the results of the above experiments and on published biochemical (34,35) and genetic (40,56,57) data, we postulated that the DSBs in the nicked U/G-U/G substrate might arise by a mechanism outlined in Figure 7. To substantiate that the DSBs in our system arose by this mechanism and that the process did not involve additional, as yet unidentified, factor(s) present in human cell extracts, we reconstituted the repair process from purified recombinant UNG, APE1, MutSα, MutLα, RPA (required to stabilize the single-stranded DNA gap) and EXO1. As shown in Figure 8, DSB generation in the substrate nicked in the G-strand 5′ from the mispairs was rather efficient.
Figure 7.

Putative scheme of BER- and MMR-dependent DSB induction in Ig loci. The first step of CSR is the active recruitment to the Ig locus of AID, which deaminates several cytosines in both DNA strands. The U/G mispairs generated at these sites are substrates for both BER and MMR. In a scenario explored in the present study, removal of a uracil in the bottom strand by UNG followed by APE1-mediated cleavage of the abasic site would generate the substrate shown in (A). Recognition of one of the remaining U/G mispairs by MutSα/MutLα causes activation of the complex and its translocation along the DNA contour. Interaction with PCNA loaded at the cleaved abasic site (B) activates the endonuclease activity of MutLα, which, upon complex formation with PCNA, introduces additional nicks into the discontinuous (bottom) strand (C). One of these is used as an entry site for EXO1, which commences to degrade the DNA in a 5′ to 3′ direction (D). If the remaining Us in the upper strand were in the meantime addressed by UNG (E), APE1 will cleave the abasic sites (F). Collision of the EXO1-catalyzed excision tract in the lower strand with the APE1-induced break in the upper strand will give rise to a DSB and EXO1 dissociation (G). [N.B. In this scenario, DSB formation is absolutely dependent on MutLα, which has to introduce additional nicks into the bottom strand, some of which must be situated 5′ from the mispaired Gs. Only then will EXO1 be able to degrade this strand toward the U/G mispairs. Had the process been initiated by the removal of a uracil residue 5′ from the two mispaired Gs, the process would have been largely independent of MutLα.]
Figure 8.

Reconstitution of the minimal BER/MMR interference system. The minimal mismatch repairosome was assembled from purified recombinant MutSα, MutLα, RPA, EXO1, RFC and PCNA. Uracil processing was mediated by purified recombinant UNG and APE1. Following incubation of the U/G-U/G and nicked U/G-U/G substrates with the indicated protein combinations, the recovered plasmids were subjected to electrophoresis on 0.8% agarose gels. The panel shows an image of a representative gel stained with GelRed. Bar graph shows quantification of three independent experiments. Error bars show mean +/− S.D. (n = 3).
DISCUSSION
Antigen-induced B cell activation leads to the expression of AID, which is targeted to the Ig loci, where it deaminates a subset of cytosines in its preferred recognition sequences to uracils. Metabolism of the AID-generated uracils then triggers SHM, during which the rearranged VDJ regions undergo extensive mutagenesis, and CSR that affects the switch regions between the constant genes. Here, uracil processing results in the generation of not only mutations, but also DSBs that trigger non-homologous end-joining and cause an irreversible loss of the intervening DNA sequences. Genetic evidence implicated BER and, to a lesser extent, also MMR in both these processes, even though the absence of UNG resulted in a substantially more severe phenotype than MMR deficiency. However, the absence of both MMR and BER abolishes CSR completely (58,59). What is the nature of the molecular transactions that allow SHM and CSR to subvert antimutator DNA repair processes such as BER and MMR to give rise to mutations and genomic rearrangements?
We argued that one answer to this question might lie in the processive action of AID. Because this enzyme is targeted to the variable and switch regions of the Ig locus (60), uracils arise at these sites frequently and are clustered together, rather than being dispersed and rare, as is the case when they arise through spontaneous hydrolytic deamination. This unique situation provides both BER and MMR with substrates in the same region of DNA and, because the two repair systems do not normally encounter each other, it is likely that there are no evolutionary safeguards in place that prevent them from interfering with one another. Our experiments suggest that the incidence of DSBs arising through BER and MMR interference is rather low, even in our system that was designed to increase the likelihood of their detection. In vivo, the mutagenic events triggering SHM/CSR also occur rather infrequently, but they are detectable due to a process of positive selection of the mutated and/or rearranged clones. Moreover, CSR can be triggered by only a few breaks, providing that they occur at sites conducive to recombination, such as the Ig switch regions. Experimental evidence in support of the hypothesis that strategically-placed DSBs are sufficient for CSR was provided by replacement of the switch regions with yeast I-SceI endonuclease cleavage sites (61).
The finding that CSR-associated DSBs arise outside of S-phase (40) rules out the possibility that these DSBs are generated when replication forks encounter single-strand breaks arising through APE1-mediated cleavage of abasic sites left behind after uracil removal. In this study, we provide evidence supporting this hypothesis, inasmuch as the DSBs arising in our in vitro assay arise in DNA molecules that do not replicate and that therefore lack strand discrimination signals (such as Okazaki fragment termini) that MMR uses to direct excision to the nascent DNA strand. Indeed, no DSBs were generated in the covalently-closed U/G-U/G substrate even when BER was active (Figure 1), nor were they formed in the same substrate when BER was blocked by the addition of Ugi, even though a nick was present in the G-strand (Figures 2–4). In contrast, when BER was active, DSBs were readily detected, providing that the substrate was nicked and that MMR was also active. This shows that the breaks in our system were generated by interplay of the two pathways, where MMR-dependent strand excision initiated at the nick in one strand collided with a cleaved abasic site arising after uracil removal and in the opposite strand. This hypothesis is experimentally substantiated by our break- and deletion-mapping experiments, which showed that the DSBs were predominantly positioned at or close to one of the uracils (Figure 5) and that the deletions most often spanned the distance between the nick and the closest uracil (Supplementary Figure S2). It should be remembered that the nick in our model substrates acts as a surrogate for an APE1-cleaved abasic site arising through UNG-catalyzed uracil removal, which we could show previously to act as a loading site for MMR-activated EXO1 (34).
During processive AID-catalyzed cytosine deamination of the Ig loci, uracil residues would be generated in both strands in close proximity. Although BER is normally extremely efficient, uracil repair in antigen-activated B cells appears to be sluggish. This leads on the one hand to mutations caused probably to Rev1/pol-ζ-catalyzed error-prone by-pass of a subset of abasic sites arising at the sites of deamination upon uracil removal by UNG and, on the other hand, to strand breaks generated by APE1-catalyzed cleavage of another subset of these abasic sites. Thus, AID-deaminated S region might contain intact U/G and G/U mispairs, as well as incised and intact abasic sites. The intact mispairs would activate MMR, which would load EXO1 at the strand discontinuities to generate a single-stranded excision tract that could be up to a kilobase long. If this tract were to collide with a cleaved abasic site or indeed with a MMR-activated EXO1-catalyzed excision tract in the opposite strand, a DSB would arise.
Our transfection experiments yielded also an unexpected finding, in that the frequency of microhomologies was higher in MMR-deficient cells compared to the MMR-proficient controls (Figure 6). The requirement for MMR in the control of HR fidelity has long been known (62), but its involvement in NHEJ or microhomology-mediated end-joining (MMEJ) has to our knowledge not been reported to date in recombination-proficient cells.
DSBs are a frequent cause of chromosomal rearrangements that play a key role in cancer, particularly in instances where recombination is disrupted, such as in case of BRCA1/2 mutations that cause breast cancer predisposition (63). In B-cells, deregulation of AID expression and its aberrant targeting induces translocations and results in lymphomagenesis (64), and it would appear highly likely that the latter chromosomal rearrangements are triggered by DSBs arising through MMR- and BER-mediated processing of AID-generated uracils. As shown above, the interference of MMR and BER is neither specific to B-cells, nor to U/G processing. We therefore posit that this phenomenon could come into play in instances where DNA modifications, such as U/G mispairs, addressed by both systems are generated at high density. One analogous example is methylation damage induced by SN1 type methylating agents, such as the cancer therapeutics temozolomide, dacarbazine or streptozotocin, which generate substrates for both BER and MMR and which are highly toxic to MMR-proficient, but not –deficient cells (46). It is also possible that the interference of MMR and BER may be the cause of the MMR-dependent DNA damage-induced chromosomal instability in colon cancer cells (65). Indeed, MMR-deficient colon cancer cell lines are generally diploid, in contrast to MMR-proficient cells, which are frequently aneuploid (8).
DNA repair processes are generally thought of as guardians of our genome. In this work we demonstrate that their canonical roles can—under certain circumstances—cause genomic instability. In this particular case, mutagenesis and genetic deletions that accompany SHM/CSR are beneficial to the organism, given that they protect it from infections. Moreover, these processes affect B cells that are programmed to die, which minimizes the possibility of deleterious side effects. Thus, even though aberrant recombination linked to antibody diversification may in rare instances cause cancer, the benefits offered by SHM/CSR in evolutionary terms far outweigh the drawbacks. It is our hope that an understanding of the molecular mechanisms of the latter processes will help us find the Achilles heel of cells in which antibody diversification went awry and, in the long run, identify ways of eliminating them.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Anne Durandy for helpful advice and numerous stimulating discussions, to Anja Saxer and Kalpana Surendranath for expressing and purifying the recombinant MutSα and MutLα, and to Stefano Ferrari and Pavel Janscak for the gift of the EXO1 and RPA expression vectors, respectively.
FUNDING
Swiss National Science Foundation [310030B-133123 and 31003A-149989]; European Research Council [‘MIRIAM’ 294537 to J.J.]. Funding for open access charge: University of Zurich.
Conflict of interest statement. None declared.
REFERENCES
- 1.Vaidyanathan B., Yen W.F., Pucella J.N., Chaudhuri J. AIDing chromatin and transcription-coupled orchestration of immunoglobulin class-switch recombination. Front. Immunol. 2014;5:120. doi: 10.3389/fimmu.2014.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saribasak H., Gearhart P.J. Does DNA repair occur during somatic hypermutation. Semin. Immunol. 2012;24:287–292. doi: 10.1016/j.smim.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manis J.P., Tian M., Alt F.W. Mechanism and control of class-switch recombination. Trends Immunol. 2002;23:31–39. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 5.Muramatsu M., Sankaranand V.S., Anant S., Sugai M., Kinoshita K., Davidson N.O., Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri J., Alt F.W. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 7.Revy P., Muto T., Levy Y., Geissmann F., Plebani A., Sanal O., Catalan N., Forveille M., Dufourcq-Labelouse R., Gennery A., et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 8.Lengauer C., Kinzler K.W., Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 9.Durandy A., Revy P., Imai K., Fischer A. Hyper-immunoglobulin M syndromes caused by intrinsic B-lymphocyte defects. Immunol. Rev. 2005;203:67–79. doi: 10.1111/j.0105-2896.2005.00222.x. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl. Acad. Sci. U.S.A. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nilsen H., Stamp G., Andersen S., Hrivnak G., Krokan H.E., Lindahl T., Barnes D.E. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene. 2003;22:5381–5386. doi: 10.1038/sj.onc.1206860. [DOI] [PubMed] [Google Scholar]
- 12.Rada C., Williams G.T., Nilsen H., Barnes D.E., Lindahl T., Neuberger M.S. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 13.Imai K., Slupphaug G., Lee W.I., Revy P., Nonoyama S., Catalan N., Yel L., Forveille M., Kavli B., Krokan H.E., et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 14.van Oers J.M., Roa S., Werling U., Liu Y., Genschel J., Hou H., Jr, Sellers R.S., Modrich P., Scharff M.D., Edelmann W. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc. Natl. Acad. Sci. U.S.A. 2010;107:13384–13389. doi: 10.1073/pnas.1008589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martomo S.A., Yang W.W., Gearhart P.J. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J. Exp. Med. 2004;200:61–68. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rada C., Ehrenstein M.R., Neuberger M.S., Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- 17.Neuberger M.S., Lanoue A., Ehrenstein M.R., Batista F.D., Sale J.E., Williams G.T. Antibody diversification and selection in the mature B-cell compartment. Cold Spring Harb. Symp. Quant. Biol. 1999;64:211–216. doi: 10.1101/sqb.1999.64.211. [DOI] [PubMed] [Google Scholar]
- 18.Ehrenstein M.R., Neuberger M.S. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J. 1999;18:3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrader C.E., Edelmann W., Kucherlapati R., Stavnezer J. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 1999;190:323–330. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z., Scherer S.J., Ronai D., Iglesias-Ussel M.D., Peled J.U., Bardwell P.D., Zhuang M., Lee K., Martin A., Edelmann W., et al. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. J. Exp. Med. 2004;200:47–59. doi: 10.1084/jem.20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardwell P.D., Woo C.J., Wei K., Li Z., Martin A., Sack S.Z., Parris T., Edelmann W., Scharff M.D. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 2004;5:224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- 22.Peron S., Metin A., Gardes P., Alyanakian M.A., Sheridan E., Kratz C.P., Fischer A., Durandy A. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J. Exp. Med. 2008;205:2465–2472. doi: 10.1084/jem.20080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardes P., Forveille M., Alyanakian M.A., Aucouturier P., Ilencikova D., Leroux D., Rahner N., Mazerolles F., Fischer A., Kracker S., et al. Human MSH6 deficiency is associated with impaired antibody maturation. J. Immunol. 2012;188:2023–2029. doi: 10.4049/jimmunol.1102984. [DOI] [PubMed] [Google Scholar]
- 24.Lindahl T. DNA repair enzymes. Annu. Rev. Biochem. 1982;51:61–87. doi: 10.1146/annurev.bi.51.070182.000425. [DOI] [PubMed] [Google Scholar]
- 25.Hughes M.J., Jiricny J. The purification of a human mismatch-binding protein and identification of its associated ATPase and helicase activities. J. Biol. Chem. 1992;267:23876–23882. [PubMed] [Google Scholar]
- 26.Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 27.Krokan H.E., Bjoras M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neddermann P., Jiricny J. Efficient removal of uracil from G.U mispairs by the mismatch-specific thymine DNA glycosylase from HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1642–1646. doi: 10.1073/pnas.91.5.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsen H., Haushalter K.A., Robins P., Barnes D.E., Verdine G.L., Lindahl T. Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J. 2001;20:4278–4286. doi: 10.1093/emboj/20.15.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendrich B., Hardeland U., Ng H.H., Jiricny J., Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 31.Visnes T., Doseth B., Pettersen H.S., Hagen L., Sousa M.M., Akbari M., Otterlei M., Kavli B., Slupphaug G., Krokan H.E. Uracil in DNA and its processing by different DNA glycosylases. Philos. Trans. R Soc. Lond. B Biol. Sci. 2009;364:563–568. doi: 10.1098/rstb.2008.0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robertson A.B., Klungland A., Rognes T., Leiros I. DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell. Mol. Life. Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frosina G., Fortini P., Rossi O., Carrozzino F., Raspaglio G., Cox L.S., Lane D.P., Abbondandolo A., Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 34.Schanz S., Castor D., Fischer F., Jiricny J. Interference of mismatch and base excision repair during the processing of adjacent U/G mispairs may play a key role in somatic hypermutation. Proc. Natl. Acad. Sci. U.S.A. 2009;106:5593–5598. doi: 10.1073/pnas.0901726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pena-Diaz J., Bregenhorn S., Ghodgaonkar M., Follonier C., Artola-Boran M., Castor D., Lopes M., Sartori A.A., Jiricny J. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. Cell. 2012;47:669–680. doi: 10.1016/j.molcel.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Langerak P., Nygren A.O., Krijger P.H., van den Berk P.C., Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delbos F., De Smet A., Faili A., Aoufouchi S., Weill J.C., Reynaud C.A. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2005;201:1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayorov V.I., Rogozin I.B., Adkison L.R., Gearhart P.J. DNA polymerase eta contributes to strand bias of mutations of A versus T in immunoglobulin genes. J. Immunol. 2005;174:7781–7786. doi: 10.4049/jimmunol.174.12.7781. [DOI] [PubMed] [Google Scholar]
- 39.Stavnezer J., Schrader C.E. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. 2006;22:23–28. doi: 10.1016/j.tig.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Schrader C.E., Guikema J.E., Linehan E.K., Selsing E., Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J. Immunol. 2007;179:6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- 41.Raschle M., Dufner P., Marra G., Jiricny J. Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J. Biol. Chem. 2002;277:21810–21820. doi: 10.1074/jbc.M108787200. [DOI] [PubMed] [Google Scholar]
- 42.Fischer F., Baerenfaller K., Jiricny J. 5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology. 2007;133:1858–1868. doi: 10.1053/j.gastro.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Grunstein M., Hogness D.S. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Proc. Natl. Acad. Sci. U.S.A. 1975;72:3961–3965. doi: 10.1073/pnas.72.10.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pham P., Bransteitter R., Petruska J., Goodman M.F. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 45.Shen H.M., Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12997–13002. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cejka P., Stojic L., Mojas N., Russell A.M., Heinimann K., Cannavo E., di Pietro M., Marra G., Jiricny J. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003;22:2245–2254. doi: 10.1093/emboj/cdg216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iaccarino I., Marra G., Dufner P., Jiricny J. Mutation in the magnesium binding site of hMSH6 disables the hMutSalpha sliding clamp from translocating along DNA. J. Biol. Chem. 2000;275:2080–2086. doi: 10.1074/jbc.275.3.2080. [DOI] [PubMed] [Google Scholar]
- 48.Kadyrov F.A., Dzantiev L., Constantin N., Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 49.Pluciennik A., Dzantiev L., Iyer R.R., Constantin N., Kadyrov F.A., Modrich P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casellas R., Nussenzweig A., Wuerffel R., Pelanda R., Reichlin A., Suh H., Qin X.F., Besmer E., Kenter A., Rajewsky K., et al. Ku80 is required for immunoglobulin isotype switching. EMBO J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manis J.P., Gu Y., Lansford R., Sonoda E., Ferrini R., Davidson L., Rajewsky K., Alt F.W. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshikawa K., Okazaki I.M., Eto T., Kinoshita K., Muramatsu M., Nagaoka H., Honjo T. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 2002;296:2033–2036. doi: 10.1126/science.1071556. [DOI] [PubMed] [Google Scholar]
- 53.Okazaki I.M., Kinoshita K., Muramatsu M., Yoshikawa K., Honjo T. The AID enzyme induces class switch recombination in fibroblasts. Nature. 2002;416:340–345. doi: 10.1038/nature727. [DOI] [PubMed] [Google Scholar]
- 54.Bowers P.M., Horlick R.A., Neben T.Y., Toobian R.M., Tomlinson G.L., Dalton J.L., Jones H.A., Chen A., Altobell L. 3rd, Zhang X., et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc. Natl. Acad. Sci. U.S.A. 2011;108:20455–20460. doi: 10.1073/pnas.1114010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greisman H.A., Lu Z., Tsai A.G., Greiner T.C., Yi H.S., Lieber M.R. IgH partner breakpoint sequences provide evidence that AID initiates t(11;14) and t(8;14) chromosomal breaks in mantle cell and Burkitt lymphomas. Blood. 2012;120:2864–2867. doi: 10.1182/blood-2012-02-412791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrader C.E., Linehan E.K., Mochegova S.N., Woodland R.T., Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J. Exp. Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrader C.E., Guikema J.E., Wu X., Stavnezer J. The roles of APE1, APE2, DNA polymerase beta and mismatch repair in creating S region DNA breaks during antibody class switch. Philos. Trans. R Soc. Lond. B Biol. Sci. 2009;364:645–652. doi: 10.1098/rstb.2008.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rada C., Di Noia J.M., Neuberger M.S. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 59.Shen H.M., Tanaka A., Bozek G., Nicolae D., Storb U. Somatic hypermutation and class switch recombination in Msh6(-/-)Ung(-/-) double-knockout mice. J. Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- 60.Basu U., Meng F.L., Keim C., Grinstein V., Pefanis E., Eccleston J., Zhang T., Myers D., Wasserman C.R., Wesemann D.R., et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zarrin A.A., Del Vecchio C., Tseng E., Gleason M., Zarin P., Tian M., Alt F.W. Antibody class switching mediated by yeast endonuclease-generated DNA breaks. Science. 2007;315:377–381. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]
- 62.Flores-Rozas H., Kolodner R.D. Links between replication, recombination and genome instability in eukaryotes. Trends Biochem. Sci. 2000;25:196–200. doi: 10.1016/s0968-0004(00)01568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paul A., Paul S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front. Biosci. 2014;19:605–618. doi: 10.2741/4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robbiani D.F., Bunting S., Feldhahn N., Bothmer A., Camps J., Deroubaix S., McBride K.M., Klein I.A., Stone G., Eisenreich T.R., et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol. Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bardelli A., Cahill D.P., Lederer G., Speicher M.R., Kinzler K.W., Vogelstein B., Lengauer C. Carcinogen-specific induction of genetic instability. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5770–5775. doi: 10.1073/pnas.081082898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.