


Abstract
The exosome plays an important role in RNA degradation and processing. In archaea, three Rrp41:Rrp42 heterodimers assemble into a barrel like structure that contains a narrow RNA entrance pore and a lumen that contains three active sites. Here, we demonstrate that this quaternary structure of the exosome is important for efficient RNA degradation. We find that the entrance pore of the barrel is required for nM substrate affinity. This strong interaction is crucial for processive substrate degradation and prevents premature release of the RNA from the enzyme. Using methyl TROSY NMR techniques, we establish that the 3′ end of the substrate remains highly flexible inside the lumen. As a result, the RNA jumps between the three active sites that all equally participate in substrate degradation. The RNA jumping rate is, however, much faster than the cleavage rate, indicating that not all active site:substrate encounters result in catalysis. Enzymatic turnover therefore benefits from the confinement of the active sites and substrate in the lumen, which ensures that the RNA is at all times bound to one of the active sites. The evolution of the exosome into a hexameric complex and the optimization of its catalytic efficiency were thus likely co-occurring events.
INTRODUCTION
The exosome is a large molecular machine that plays a role in the processing and degradation of the 3′ end of a large variety of RNA molecules (1). Complexes that belong to the exosome and exosome-like family share the same three-dimensional architecture and are found in all three domains of life. The simplest form of the complex is the bacterial RNase PH that has a 3′ to 5′ exoribonuclease activity (2) (Supplementary Figure S1A). The biological unit of this complex is a homo-hexamer that comprises three RNase PH dimers that assemble into a ring with six active sites (3). During the degradation reaction, the enzyme uses inorganic phosphate to release nucleotide di-phosphates from the 3′ end of the RNA. The second exosome-like complex is the polynucleotide phosphorylase (PNPase) (Supplementary Figure S1B) that is found in bacteria, chloroplasts and mitochondria. The building block of this enzyme contains two consecutive RNase PH domains, a KH and an S1 domain that are linked in one protein chain (4). Six RNase PH domains from three PNPase monomers assemble into a hexameric ring structure that contains three active sites. The exosome complex itself is found in archaea (5) and eukaryotes (6). In archaea, the core of the complex contains the two RNase PH domain proteins Rrp41 and Rrp42 (7). Three Rrp41:Rrp42 dimers assemble into a hexameric ring structure with three active sites (8,9)(Figure 1, Supplementary Figure S1C). The active sites are located in Rrp41, whereas the Rrp42 protein has lost its catalytic activity. The archaeal exosome core recruits three copies of the cap proteins Rrp4 or Csl4 that contain the RNA binding domains (9,10). The interaction of the archaeal exosome core with these cap proteins enhances the RNA degradation rates and provides substrate specificity (11,12). Besides the similar structures of the PNPase, RNase PH and archaeal exosome, these complexes share a similar phosphorolytic mechanism. The eukaryotic exosome (6) has evolved further into a fully asymmetric complex where all protein chains that form the core and all protein chains that form the cap are different (Supplementary Figure S1D and E). In plants (13), only a single subunit in the core (Exo-9) appears to be catalytically active. In other eukaryotes, all exosome subunits are inactive and form a scaffolding complex (14–16). Catalytic activity is added to the Exo-9 complex by the Rrp44 protein that harbors both exoribonucleolytic and endoribonucleolytic activity (16). Interestingly, the catalytic mechanism of the eukaryotic complex moved from phosphorolytic to hydrolytic. The removal of the phosphorolytic catalytic activities in the eukaryotic exosome barrel might have occurred to prevent polymerase activity that could result in non-specific 3′ elongation of RNA (17).
Figure 1.

Structure of the Sulfolobus solfataricus exosome in complex with a short RNA. (PDB: 2C38) (27). Rrp41 subunits are colored gray, Rrp42 subunits are colored in light brown and the 4 bases of the RNA substrate that are visible in the crystal structure are colored in red. Left: topview of the complex, where the substrate entrance pore is indicated with a circle. Right: sideview of the complex, where the substrate entrance path is indicated with an arrow. One Rrp41:Rrp42 dimer is shown transparent to allow visualization of the inside of the barrel.
RNase PH (3), PNPase (4), the archaeal and eukaryotic exosome complexes (8,9,15,18) all assemble into barrel-like structures with a (pseudo) 3-fold symmetry (Supplementary Figure S1). The active sites are located inside the lumen of these barrels and these are thus secluded from the cellular environment. As a result access to the active sites can be regulated and erroneous RNA degradation can be prevented. For the exosome and exosome-like complexes, RNA-binding domains that are not part of the catalytic RNase PH ring can be used to provide substrate selectivity (19). Based on previous biochemical data and on published crystal structures, it can be concluded that the RNA substrate is threaded to the catalytic chamber through a central pore (the neck region), which is only large enough to accommodate one single-stranded RNA (10) (Figure 1). The functional advantage of substrate selectivity that results from the formation of the quaternary structure is, however, counterbalanced by catalytic disadvantages, as oligomerization of an enzyme into a multimeric complex reduces the number of substrates that can be degraded at the same time. As an example, three isolated Rrp41:Rrp42 dimers will be able to degrade three substrate RNAs simultaneously, whereas a trimer of Rrp41:Rrp42 dimers, as found in the archaeal exosome, is only able to degrade a single substrate at a time. The oligomerization of enzyme complexes, as is seen in the exosome family of exonucleases, is thus a trade-off between a decrease in the number of available active sites per substrate and an increase in substrate selectivity.
Here, we address whether the quaternary structure of the archaeal exosome complex from Sulfolobus solfataricus provides catalytic advantages. We focus on two aspects in the exosome complex that arise due to the oligomerization of the enzyme: the creation of the neck region and the establishment of a high local concentration of active sites in the lumen of the barrel. In brief, we combine methyl TROSY NMR, RNA degradation and binding experiments and find that the neck region is essential for the processivity of the enzyme. In addition, we conclude that the sequestering of active sites inside a small lumen of the complex favors RNA degradation as it ensures that the substrate is always in contact with one of the active sites. The formation of a hexameric complex thus provides significant functional advantages for the exosome and exosome-like complexes.
MATERIALS AND METHODS
Protein expression and purification
The genes for the Rrp41 and Rrp42 proteins from Sulfolobus solfataricus were cloned into modified pET vectors that carried an N-terminal TEV cleavable His6-tag. In addition, a construct for the coexpression of both proteins was constructed in a modified pET vector, where only the Rrp41 protein carried an N-terminal TEV cleavable His6-tag. Point mutations were introduced using standard site-directed mutagenesis methods.
Escherichia coli BL21 codon plus cells were transformed with the appropriate plasmids (Supplementary Table S1). Cells were grown at 37°C and proteins were over-expressed at 25°C by addition of 1 mM IPTG (Isopropyl β-D-1-thiogalactopyranoside) when an OD 600 of 0.8 was reached. Twelve hours later, the cells were pelleted by centrifugation and lysed in buffer A (50 mM NaPO4 pH 7.5, 150 mM NaCl, 1 mM DTT) complemented with 10 mM imidazole, lyzozyme and 0.1% Triton X100. The cell lysate was cleared from insoluble debris by centrifugation and the supernatant was loaded on Ni-NTA resin. The resin was washed with buffer A that was complemented with 10 mM imidazole. The protein bound to the resin was eluted with buffer A complemented with 300 mM imidazole. TEV protease was added to the eluted protein and dialyzed overnight into buffer A. To remove the His6-tagged TEV protease and the cleaved His6-tag, the dialysate was applied to Ni-NTA resin. The final purification step was performed using size exclusion chromatography on a Superdex 200 column in buffer B (30 mM Hepes pH 6.9, 100 mM NaCl, 1 mM DTT).
The exosome complex was reconstituted from separately purified Rrp42 (after size exclusion chromatography) and Rrp41 (after dialysis and TEV cleavage). Equal amounts of both proteins were mixed and incubated for several hours at room temperature. Uncomplexed Rrp41 or Rrp42 was removed by incubation at 50°C for 2 h. After removal of the precipitated proteins, the sample was further purified using size exclusion chromatography as described above.
Exosome complexes with a different number of active sites (on average) were obtained by mixing catalytically active and catalytically inactive Rrp41 before the addition of Rrp42. The percentage of active Rrp41 was varied between 10 and 100%. After reconstitution, the complexes were purified as described above.
Exosome complexes with exactly one, two or three active sites were obtained with plasmids containing the gene coding for Rrp42 together with three copies of the Rrp41 gene (Supplementary Table S1). These coexpression plasmids were designed as previously described (20). The first copy of Rrp41 contains a His6-tag, the second one contains a MBP-tag and the third one contains a Strep-tag. For exosomes with a single active site, the His6- and Strep-tagged versions of Rrp41 were catalytically inactive; for exosomes with two active sites, the His6-tagged version of Rrp41 was catalytically active; for the exosome with three active sites, all Rrp41 versions were catalytically active (Supplementary Table S1). Cells that coexpressed Rrp42 and the three versions of Rrp41 were grown and induced as described above. After lysis, the proteins were purified using Ni-affinity chromatography as described above. The protein that eluted from the Ni column contained at least one His6-tagged version of Rrp41. These complexes were then applied to amylose resin to select for complexes that contained a MBP-tagged version of Rrp41 in addition to a His6-tagged version of Rrp41. The complexes that were eluted from the amylose resin were subsequently applied to Strep-Tactin resin to select for exosome complexes that contained all three tagged versions of Rrp41. The eluted complex was subsequently treated with TEV protease to remove all affinity tags and dialyzed, prior to performing a size exclusion chromatography as described above. It is worth mentioning that the yield of the exosome complexes purified this way is significantly reduced as only 22% of the exosome complexes that are formed during over-expression contain all three different tags.
Labeling with NMR active nuclei was achieved by over-expression in minimal medium that was based on 100% D2O. 12C2H glucose was used as the carbon source and methyl labeling was achieved by addition of 100 mg/l U-[1H,13C] methionine, 60 mg/l 4-methyl 13CH3 α-ketobutyric acid (labeled isoleucine precursors) or 100 mg/l methyl 13CH3 α-ketoisovaleric acids (labeled valine/leucine precursors) 1 h before induction with 1 mM IPTG.
Coexpression of Rrp41 and Rrp42 was used for the preparation of the complex that contained NMR active methyl groups in both Rrp41 and Rrp42. To that end, both proteins were coexpressed in NMR active growth medium (Supplementary Table S1). Purification was performed as for the single proteins.
RNA in vitro transcription and purification
RNA was prepared using in vitro transcription with T7 polymerase. The DNA template was obtained from a linearized vector. RNAs used in NMR experiments were transcribed with an HDV-ribozyme that cleaves at the end of the target RNA sequence, resulting in the presence of a 3′ cyclic phosphate. The 3′ cyclic phosphate prevents degradation of the RNA by the exosome and thus allows for long-term NMR measurements. RNAs used in binding and degradation experiments were produced using run-off transcription, where the final RNA contained a 3′ GCT that resulted from the linearization of the template vector with the HindIII restriction enzyme. All RNA constructs contained a hairpin structure (GGCCCCCCCCGAAAGGGGGGGG) followed by 32, 63, 92 or 118 adenines (Supplementary Table S1). The DNA vector containing 63, 92 or 118 adenines were obtained from gene-synthesis (GenScript USA Inc.). In vitro transcribed RNA was purified natively with weak ion exchange chromatography using a DEAE-sepharose column as described (21). The pooled fractions were concentrated and buffer exchanged into H2O with a PD10 column, followed by SpeedVac concentration.
Degradation assays
RNA degradation assays were performed in 180 μl reaction buffer (20 mM Hepes pH 6.5, 60 mM KCl, 0.1 mM EDTA, 2 mM DTT, 8 mM MgCl2, 10 mM Na2HPO4) that contained 60 nM exosome (hexameric complex) and 25 μM RNA. The 10 μl samples were taken at different time-points and the reaction was quenched by addition of 10 μl 8M Urea, 20 mM EDTA, 2mM Tris pH 8.
HPLC analysis
Ten microliters of the quenched reaction were automatically injected onto an analytical DNAPac PA100 column (Dionex) that was heated to 80°C. Substrate and product were separated using a linear gradient from buffer A (5 M Urea, 20 mM Tris pH 8, 100 mM NaCl) to buffer B (5 M Urea, 20 mM Tris pH 8, 2 M NaCl) and detected using the absorption at 260 nm. To convert peak intensities to absolute concentrations, the detector response was calibrated by injecting known amounts of RNA (Supplementary Figure S6).
Analysis of degradation data
For each time-point the product concentration was divided by the total concentration [product + substrate] to normalize the signal. The progression of the reaction was then fitted from data at several time-points (Supplementary Figures S4 and S5). Based on the known amounts of enzyme and substrate together with the length of the substrate, the progression curves were translated into number of nucleotides cleaved per second per exosome. To estimate the error in the extracted catalytic rates, we used a jackknife approach, where we fitted the data multiple times after randomly removing a subset of the data.
Fluorescence anisotropy
RNA (GCCCCCCCCGAAAGGGGGGGG-A(21)-4-S-U-A(11)-GCU) for Fluorescence anisotropy measurements was obtained from Dharmacon. The attachment of the 6-(Iodoacetamido)-fluorescein (Sigma-Aldrich) to the thio-uridine (4-S-U) was performed according to the Ramos et al. (22). Dilution series of the inactive exosome (2000, 1000, 500, 250, 125, 60, 30, 15, 10 and 0 nM) or of the neck mutant exosome (80, 60, 40, 20, 10, 5, 2, 1, 0.5 and 0 μM) were mixed with 10 nM of RNA labeled with 6-(Iodoacetamido)-fluorescein. For the competition assays, 20 nM of either 32, 63, 98, or 118As RNA was added to the exosome:fluorescent RNA mixture. In all measurements, buffer (30 mM KPO4 pH 6.9, 100 mM NaCl, 0.005% Triton X-100) was used as a reference. Fluorescence anisotropy was recorded every 5 min using a plate reader (Tecan, Infinite F200; filter linear polarization XP38: excitation at 485 nm and emission at 535 nm). Affinity constants were obtained from the data using in-house written scripts using standard equations (23).
NMR
All NMR samples were in buffer B, based on 100% D2O. NMR spectra were recorded on AVIII‐600 and AVIII‐800 spectrometers with room temperature probe-heads. Methyl TROSY spectra were recorded at 50°C using a carbon chemical shift evolution time of 40 ms. SQ (single quantum) dispersion experiments were recorded at 600 and 800 MHz using a relaxation delay of 50 ms and Car-Purcell-Meiboom-Gill (CPGM) frequencies ranging from 40 to 1000 Hz. Relaxation dispersion data were fitted numerically using in-house written scripts using published equations (24). For the final analysis, two residues (I71 and I85), two magnetic fields (600 and 800 MHz) and three temperatures (308, 315 and 323 K) were fitted together to one intrinsic R2 rate per curve, one exchange rate per temperature and one chemical shift difference per residue. Errors in the extracted parameters were obtained using a Monte Carlo analysis, where the measured data-points were randomly varied around the experimental error. Chemical exchange saturation transfer (CEST) experiments were recorded on an 800 MHz spectrometer at 20°C and using a 400 ms, 5 Hz B1 field, at 42 different carbon offsets that were spaced by 10 Hz. All NMR spectra were processed using the NMRPipe/NMRDraw software suite (25). Figures displaying NMR spectra and molecular structures were produced using NMRview (onemoonscientific.com) and Pymol (pymol.org), respectively.
RESULTS
The exosome interacts tightly with RNA substrate
The first step in an enzymatic cycle is the formation of an enzyme:substrate complex. Here, we used fluorescence anisotropy measurements to determine the affinity between the exosome and an RNA substrate that contains 32 adenines downstream of a stable GC hairpin structure. To visualize the RNA, we introduced a single 4-thiouridine 15 bases downstream of the 3′ end and coupled this base to 6-(Iodoacetamido)-fluorescein. We then added increasing amounts of a catalytically inactive version of the exosome (D182A in Rrp41). Upon substrate:enzyme complex formation, the rotational lifetime of the RNA is changed, from which we extracted an affinity of 11.7 (0.9) nM (Figure 2A) for the interaction between the exosome and the RNA. This indicates that the exosome interacts tightly with substrates and that substrates can thus be recruited to the complex very efficiently.
Figure 2.

(A) Fluorescence anisotropy measurements to determine the affinity between an RNA substrate and the exosome. 32A refers to an RNA that contains a 5′ GC hairpin structure, followed by 32 adenine bases. The extracted error is based on three independent measurements. (B) The interaction strength between RNA substrates and the exosome is independent of the length of the RNA. Shown are fluorescence anisotropy measurements, where 20 nM exosome and 20 nM fluorescently labeled 32A RNA (see above), were complemented with increasing amounts of non-fluorescently labeled RNA. The extracted affinities are very similar for RNA species that contain 32, 63, 92 or 118 adenine bases 3′ to a GC hairpin structure. The errors in the extracted parameters result from independent measurements.
We then asked if the interaction between the exosome and RNA depends on the length of the RNA substrate. To that end we performed fluorescence anisotropy competition experiments where we added increasing amounts of non-fluorescently labeled RNA to preformed exosome:fluorescently labeled RNA complex (Figure 2B). As competitors, we used RNA species that contain 32, 63, 92 or 118 adenines downstream of the stable GC hairpin. In these experiments, the fluorescently labeled RNA is competed away from the exosome, which results in a decrease in the fluorescence anisotropy. We then used the program DynaFit (26) to extract the kD for the competitor and found that all RNA species we tested interact with an affinities around 2.7 (0.9) nM (Figure 2B). Note that the competition experiments yield somewhat lower affinities, which is potentially due to minor interference of the fluorescence label with the binding. These data show that the affinity of the enzyme for the substrate is independent of the length of the substrate and implies that the 3′ end of the RNA is the prime recognition site for the exosome.
The tight exosome substrate interaction provides processivity
The RNA degradation of the exosome is highly processive, where RNA substrates are not released from the enzyme prior to complete degradation (27). Here, we confirm this processivity and show that an RNA substrate that contains a stable GC hairpin followed by 32 adenines is degraded into an RNA species that contains 10 adenines in addition to the hairpin (Figure 3A). This product results from the fact that the stable hairpin prevents entrance of the substrate into the exosome barrel and from the distance between the entrance pore and the active sites that spans 10 bases (27). During the degradation reaction no intermediate products are observed, indicating that the substrate is not released from the enzyme until degradation has been completed.
Figure 3.

(A) Left: Substrate RNA (32A) is processively degraded into product (10A; a GC hairpin with 10 adenine bases) by WT exosome as no intermediate degradation products are detected. Right: a single point mutation in the neck region (Rrp41 R67G) abolishes the processivity as intermediate degradation products appear during the reaction. (B) Fluorescence anisotropy binding curve of the neck mutant exosome (Rrp41 R67G) with the fluorescently labeled 32A RNA. The single point mutation results in a 1000-fold reduction of the affinity between the RNA and the enzyme (compare: Figure 2A).
To shed light on the interactions that are responsible for this processive degradation we introduced a point mutation in the neck region of the exosome complex (R67G in Rrp41). Interestingly, this mutation that, due to the symmetry of the complex removes three positive charges, caused a reduction in the affinity between the RNA and the exosome from 11.7 nM (Figure 2A) to 27 μM (Figure 3B). The removal of the positive charge at the entrance pore thus reduces the affinity between the exosome complex and the substrate RNA by three orders of magnitude. At the same time, the processivity of the degradation reaction has been lost as intermediate degradation products appear during the reaction (Figure 3A, right). Over time, these intermediates decrease in length and disappear as they act as substrates in subsequent rounds of degradation by the exosome complex. These results indicate that the neck region of the exosome is responsible for the tight interaction between the enzyme and the substrate and that this tight interaction results in processive RNA degradation.
The RNA is mobile inside the exosome barrel
Based on RNA degradation experiments that use a substrate that contains a stable 5′ hairpin structure, the distance between the neck region of the exosome and the active sites is 10 nucleotides (Figure 3) (27). The exact path of the RNA between the neck region and the active sites has, however, not been revealed. In crystal structures of the Sulfolobus solfataricus exosome, the substrate RNA is only visible for the 4 bases (bases N1 to N4; where base N1 is the 3′ terminal base; Figure 1) encompassed in the active site as well as the base N10 in the entrance pore (10,27). Except for these bases, no electron density is observed for the RNA inside the chamber. This suggests that parts of the RNA are not well ordered in the barrel of the exosome.
To obtain additional insights into the path of the substrate RNA within the exosome core, we turned to methyl TROSY NMR spectroscopy (28). This NMR method results in high quality proton-carbon NMR spectra (29) that can be used to identify intermolecular interactions (30). To that end, we recorded methyl TROSY NMR spectra of the exosome in the absence and presence of RNA substrate. Methyl groups that come close to the RNA undergo chemical shift perturbations (CSPs) that can report on the path of the substrate.
First, we prepared an exosome complex that contained NMR invisible Rrp41 and a version of Rrp42 that was NMR active in methyl groups of isoleucine, valine and leucine residues (ILV labeling). We recently obtained resonance assignments for these methyl groups and reported that the RNA interacts with residues around the pore region and with the isoleucine 85 of Rrp42 (31) that is close to the active sites of the enzyme. In case the RNA adopts a static structure with one of the three active sites in the exosome barrel, one would expect that resonances of residues close to the RNA split into two, where 2/3 of the original resonance remains and a novel resonance with intensity 1/3 arises. For residue 85, however, we observe that the resonance intensity is reduced by a factor of ∼8, whereas we failed to identify a novel resonance. This indicates that the RNA substrate is not stably bound to a single active site when it is inside the exosome barrel. To determine if other resonances from Rrp42 also undergo line broadening upon RNA interaction, we divided resonance intensities observed in the free exosome by the resonance intensities of the complex with RNA (Figure 4A). Although most resonance intensities are unaffected by the RNA interaction, we noticed that isoleucine 71 and valine 137 are also significantly weakened in the presence of the RNA. Like isoleucine 85, these residues cluster close to the four nucleotides of the 3′ end of the RNA substrate. The peak broadening of isoleucine 85 is more pronounced because it interacts directly with the RNA, whereas isoleucine 71 and valine 137 are sensing the presence of RNA through conformational changes of the loop underneath the active site (32). Interestingly, the line broadening effect is more prominent at higher temperature (50°C) than at lower temperature (20°C) (Figure 4A). Most likely, this temperature dependence is due to transient interactions between the RNA and the enzyme, where the RNA binding-unbinding causes exchange broadening of the methyl groups in the vicinity. The reduction of the line broadening at lower temperatures can be explained by slower motions of the RNA at 20°C than at 50°C. In summary, our NMR titration data suggest that the RNA is mobile inside the barrel of the exosome.
Figure 4.

(A) The methyl group resonance intensities of Rrp42 resonances in the RNA free exosome divided by the corresponding resonance intensity in the presence of RNA. High bars indicate significant line broadening upon RNA binding, notably observed for residues Ile 71, Ile 85 and Val 137 of Rrp42. The line broadening is shown for three different temperatures, 50, 30 and 20°C. Note that a large number of resonances are not affected by the interaction with RNA (below the dashed bar). (B) Methyl TROSY NMR spectrum of the isoleucine region of the exosome that contains NMR active groups in Rrp41 and Rrp42. NMR spectra in the absence (black) and presence (red) of RNA are shown and a number of assignments are indicated. Regions that are highlighted with a dashed box correspond to resonances in Rrp41 that experience large CSPs upon interaction with the RNA substrate. Spectra were recorded at 323 K. (C) Structure of the Sulfolobus solfataricus exosome (PDB: 2C38) (27) superimposed onto the RNA substrate (in red) visible in the structure of the Pyrococcus abyssi exosome (35) (PDB: 2PO1). Assigned residues that show CSPs in the presence of RNA are indicated in blue. Only a single RNA is present per hexameric exosome complex, as the narrow entrance pore does not allow for the recruitment of multiple substrates simultaneously.
To identify residues in Rrp41 that are important for the RNA interaction, we prepared an exosome complex that contained NMR active methyl groups for the isoleucine residues of Rrp41 and Rrp42 (Figure 4B). Resonances from Rrp41 can be identified in a straightforward manner based on spectra that were recorded on an exosome that was only labeled in Rrp42 (Supplementary Figure S2). Addition of RNA to the Rrp41:Rrp42 isoleucine labeled sample resulted in a number of CSPs (Figure 4B) where CSPs in Rrp42 confirmed the data that we obtained from the Rrp42 ILV labeled sample. To identify amino acids in Rrp41 that are affected by the RNA, the NMR resonances need to be assigned to the residues in the complex. A full resonance assignment of the Rrp41 methyl groups was, in our hands, not feasible, as Rrp41 in isolation had a high tendency to aggregate. Nevertheless, we assigned a number of Rrp41 methyl group resonances using a mutational approach, where we mutated Rrp41 isoleucine residues into closely related amino acids (33,34). This ideally results in the disappearance of a single isoleucine resonance from the methyl TROSY spectrum. In that way we identified that isoleucines 101 and 131 in Rrp41 interact with the substrate RNA (Figure 4B). Interestingly, these residues shift rather than broaden upon RNA binding. In addition, we noticed that an additional set of unassigned residues in Rrp41 experiences resonance shifts (boxed regions in Figure 4B). Shifting of resonances in NMR titration experiments takes place when the binding–unbinding process is fast on the NMR chemical shift timescale. This suggests that a number of residues in Rrp41, including isoleucine 101 and 131, interact weaker with the RNA than the residues close to the active sites in Rrp42. This observation is in agreement with the lack of electron density for the nucleotides that come close to the Rrp41 protomer.
In comparison with the structure of the Sulfolobus solfataricus exosome (27), the structure of the Pyrococcus abyssi exosome (35) in complex with RNA shows additional electron density for RNA bases N5 and N7, albeit with low quality. To validate our NMR titration experiments, we superimposed the RNA of the Pyrococcus abyssi complex onto the structure of the Sulfolobus solfataricus exosome (Figure 4C). This reveals that CSPs that we observe in our NMR titration experiments are in some cases further than 5 Å away from the substrate. This is especially true for isoleucine 101 in Rrp41 that is more than 9 Å away from base N7. We can, however, explain the CSPs of isoleucine 101 in Rrp41 with the mobility of the RNA in the exosome barrel, where structural changes in the RNA result in shorter distances between the substrate and isoleucine 101.
Taken together, our NMR data and the lack of electron density of the RNA substrate (10,27,35) point to a high mobility of the substrate RNA inside the barrel of the exosome.
Quantification of the RNA motions inside the exosome barrel
To directly measure motions of the substrate RNA in the barrel of the exosome, we made use of methyl group relaxation dispersion experiments (36,37). In those experiments, the line broadening that is induced by an exchange process can be quantified and exchange rates can be extracted. We focused our analysis on isoleucine residues of Rrp42 because concentrated samples that are only labeled in Rrp42 can be produced (31). In agreement with the line broadening that we observed in isoleucine 85 and isoleucine 71 (Figure 4A), we detected significant dispersion profiles for these residues in the presence of substrate RNA (Figure 5A). Importantly, these dispersion profiles are solely due to the interaction of the enzyme with the substrate, as they were not observed in the absence of RNA (Supplementary Figure S3). In total, we measured dispersion data at three different temperatures (35, 42 and 50°C) and two magnetic field strengths (600 and 800 MHz; Supplementary Figure S3). To extract the underlying exchange parameters, we fitted all data together and assumed that the chemical shift difference was temperature independent. In addition, we assumed that the excited (RNA bound) state had a population of 1/3, as the substrate RNA can only interact with one of the three active sites at a time. Based on that, we extracted exchange rates of 1021 (163), 1615 (230) and 1744 (249) per second at 35, 42 and 50°C, respectively. The extracted chemical shift differences between the free state and the RNA bound state are 0.25 (0.01) and 0.17 (0.02) p.p.m. for isoleucine 85 and 71, respectively. Isoleucine 71 and isoleucine 85 are located close to the active site of the enzyme and the motions that we detect through those methyl groups thus report on the binding–unbinding of the substrate RNA with the active site region of the exosome. We conclude that the RNA moves from one active site to the next one with a frequency between 1000 and 1700 Hz, depending on the temperature. These data reinforce the notion that the RNA is highly mobile in the lumen of the exosome.
Figure 5.

(A) Single quantum relaxation dispersion profiles in the presence of RNA. The profiles for isoleucine 71 and isoleucine 85 are shown at two magnetic field strengths and at 50°C. For clarity, the dispersion profiles at 600 MHz are plotted 5 Hz lower than they actually are. The circles indicate the measurement points, where the error bars have been derived from duplicate measurement points. The drawn lines are the best fit to the data, where both residues at three temperatures were fitted simultaneously to one exchange rate and one carbon chemical shift difference per residue. (B) CEST profiles for isoleucine 85 in the absence (blue) and presence (red) of the RNA substrate. In the presence of RNA, an invisible state 0.25 downfield of the main resonance of isoleucine 85 appears.
To validate the analysis of the relaxation dispersion experiments, we used CEST experiments (38) to identify the resonance frequency of the invisible RNA-bound state of isoleucine 85. The exchange rates that we determined between 35 and 50°C are too fast for efficient CEST to occur. Therefore, we lowered the experimental temperature to 20°C. Unfortunately, at temperatures lower than 20°C, the signal to noise ratio of the NMR spectra drops significantly, which prevented us from measuring at even lower temperatures. In the CEST experiment, we clearly observe the presence of a second and invisible state for isoleucine 85 (Figure 5B). Interestingly, this invisible state is located 0.25 p.p.m. downfield from the resonance frequency of residue 85. This is in excellent agreement with the chemical shift difference extracted from the relaxation dispersion experiments and confirms that the parameters that we extracted from the relaxation dispersion experiments are accurate and reliable.
The exosome exploits all three active sites during catalysis
Our NMR data show that the 3′ end of the RNA substrate is highly mobile inside the lumen of the exosome. Based on the relaxation dispersion experiments, the substrate interacts with the active sites around 1700 times per second at 50°C. This raises the question whether all three active sites are used or potentially even required during the degradation process. To address this, we used two complementary biochemical experiments where we measured the activity of exosome complexes that contained different numbers of active sites.
First, we reconstituted exosome complexes from separately expressed Rrp42 and Rrp41. To vary the number of active sites in the reconstituted complex, we used different mixtures of catalytically active and catalytically inactive (D182A, that does not interfere with the RNA binding) Rrp41. For example, when a mixture of 40% active and 60% inactive Rrp41 is used in the reconstitution process, statistically the following complexes will form: exosome complexes without any active sites (22%), with one active site (43%), with two active sites (29%) and with three active sites (6%). The relation between the average number of active sites and the activity of the complex depends on the mechanism that is used during the degradation process. In case all active sites equally and independently contribute to the reaction, the activity will linearly increase with the average number of active sites (Figure 6A, red curve). Alternatively, in case only one active site is used in the degradation process (e.g. when the substrate stays on a single active site during the degradation process), the activity will level off with increasing average number of active sites (Figure 6A, blue curve). In case all three active sites are essential for efficient catalysis, the activity of the exosome will only reach high levels when there are on average a high number of active sites (Figure 6A, green curve). Experimentally we can distinguish between these three scenarios by measuring the catalytic activity of exosomes that (on average) contain a different number of active sites. Here, we increased the average number of active sites in a stepwise manner from 10% to 100% in steps of 10% (see Methods). Each of these exosome complexes was incubated with substrate RNA and the reaction was quenched at different time-points. In those assays, we choose to use the RNA substrate that contains a 5′ hairpin structure followed by 32 adenine nucleotides. The product of the degradation reaction will then be the hairpin structure with 10 nucleotides, which can be readily detected (see above). Initially, we quantified the levels of substrate and product during the reaction using an Urea PAGE analysis (Supplementary Figure S4), however, we found that quantification using an HPLC approach was more accurate (Supplementary Figure S5) and thus we used this method for all degradation experiments. To obtain degradation rates, we fitted the substrate and product concentrations to a progression curve (Supplementary Figures S4, S5, S6 and methods). In summary, we observe that the number of cleaved nucleotides increases linearly with increasing amounts of active Rrp41 (Figure 6B). These data thus show that all three active sites in the exosome are used during the degradation process. Based on these data, we conclude that one substrate RNA is degraded by three active sites in one exosome. At 50°C, this results in 14.7 (0.6) nucleotide cleavages per second per exosome (Figure 6B).
Figure 6.

(A) Theoretical relationship between the activity and number of active sites inside the exosome complexes, which were reconstituted, with different ratios of active and inactive Rrp41. In case all three active sites in Rrp41 are independently used in the RNA degradation process, the predicted relation between the activity and the percentage of active Rrp41 is shown in red. The blue and green relationships apply to the situation where only one active site is used and where the exosome requires all three sites for full activity, respectively. (B) Experimental relationship between the percentage of active Rrp41 in the exosome and the activity. The blue circles indicate the degradation rates (nucleotides cleaved per second per exosome). The red line is the best fit to the data from which the activity of the fully active exosome is determined to be 14.7 cleavages per exosome at 50°C. The data clearly show that all three sites in the exosome are used in the degradation process. (C) Theoretical relationship between the activity and the discrete number of active sites inside the exosome. The dashed lines are shown for clarity only. See also (A). (D) Experimental relationship between the number of discrete active sites in the exosome complex and the activity. The blue circles refer to the measurements, the red line is the best fit to the data. As in (B), the data show that all three sites in the exosome are used in the degradation process.
In a second and independent approach to determine how many active sites in the exosome play a role in the RNA degradation process, we prepared three different samples with exosome complexes that contained exactly one, two or three active sites. These complexes were obtained by overexpression of untagged Rrp42 with three copies of Rrp41, each of them fused to a different affinity tag (His-tag, MBP-tag or Strep-tag). Importantly, we were not able to detect any subunit exchange between different exosome complexes (Supplementary Figure S7), establishing that the exosome complexes with a discrete number of active sites are extremely stable. Subsequently, three consecutive purification steps were used to obtain complexes that contained exactly one of each Rrp41 affinity tags. During expression of the exosome complex, we used one, two or three catalytically active versions of the differently tagged Rrp41 proteins and were thus able to prepare exosome complexes with a discrete number of active sites. As described before, the activity of these complexes can provide information on the mechanism that is used during RNA degradation (Figure 6C, see above). We then experimentally determined the activity of the exosome complexes that harbored a discrete number of active sites and found that all three active sites are equally involved during the degradation process (Figure 6D). This confirms the experiments that we performed using exosomes with mixed number of active sites. The overall activity of the exosome complexes with a discrete number of active sites appears somewhat lower (9.6 cleavages per second per exosome at 50°C) than the activity of the exosome complexes that contain mixture of active and inactive sites. This is most likely due to a loss in activity of the exosome complex during the long purification protocol that is required for the preparation of the exosome complexes with a discrete number of active sites.
The RNA motions are much faster than the enzymatic turnover rates
Our degradation assays show that the RNA substrate uses all three active sites in the exosome barrel. This is in agreement with the NMR data that show that the substrate rapidly exchanges between the active sites. Interestingly, at 50°C the rate of exchange (1700 s−1) is two orders of magnitude faster than the number of cleavages per second per exosome (∼10 s−1). It is worth noting that these differences are not a peculiarity of this temperature. Indeed, we compared degradation experiments at 20, 35, 50 and 65°C (Figure 7A) and found that the temperature dependence of the degradation rate follows the Eyring relationship (Figure 7B). The temperature dependence of the RNA hopping frequency in the exosome barrel, as we determined using relaxation dispersion experiments, follows a very similar trend, albeit at much higher frequencies. This indicates that the RNA hopping frequency is significantly larger than the degradation rates for biologically relevant temperatures.
Figure 7.

(A) Effect of the temperature on the activity of the exosome. (B) Eyring analysis of the temperature dependence of the degradation rates (lower line) and NMR jumping rates (upper line). The lines follow the same trend, indicating that the substrate jumping rate is much faster than the degradation rate at all biologically relevant temperatures. (C) The number of nucleotides cleaved by the exosome is independent of the length of the RNA. This shows that substrate recruitments or product release are not rate limiting in the reaction. The larger errors for the 118A RNA substrates might result from some inhomogeneity in the exact length of the substrate that resulted from slippage or stalling of the T7 polymerase that was used to prepare this long and highly repetitive sequence.
From the temperature dependence of the activity of the exosome core (Figure 7B) the RNA degradation rate at 75°C (the optimal growth temperature of Sulfolobus solfataricus) can be predicted to be around 65 nucleotides/second. This degradation rate is comparable to the RNA degradation rate that was previously determined for the PNPase enzyme (120 nucleotides/second) (39). In addition, the rate of RNA degradation by the exosome core is in the same order of magnitude as the elongation rate of the archaeal RNA polymerases that was determined to be around 20 nucleotides per second in Methanothermobacter thermautotrophicus (40).
The degradation rate is independent on the RNA length
The catalytic cycle of the exosome includes multiple steps. If substrate binding or product release is very slow, the exosome would require a significant time between finishing the degradation of one substrate and initiating the degradation of the next one. Such a time would reduce the average cleavage frequency that we measured in our biochemical experiments. To probe whether substrate binding or product release are rate limiting in the catalytic cycle of the exosome we performed degradation assays with RNA substrates of increasing length (32, 63, 92 and 118 adenine repeats, that all interact with the exosome with similar affinities, Figure 2B). For short RNA substrates, the exosome would need to reload the substrate significantly more often than for long substrates, which would reduce the overall turnover rate. In our experiments, we designed the substrates such that short (less than ∼10 nucleotides) single stranded RNA stretches are not formed (Supplementary Figure S4A), as these have been shown to be degraded at very low rates (41). Interestingly, in our degradation experiments, we find that the activity of the exosome (number of nucleotides cleaved per second) is very similar for the four RNAs used, showing that the activity of the exosome is largely independent of the length of the substrate (Figure 7C). This shows that substrate binding and product release are not significantly limiting catalytic turnover. The nucleotide cleavage frequencies that we measured thus directly report on the activity of the active sites. This validates our conclusion that the RNA jumping frequencies are much faster than the catalytic cleavage rates.
DISCUSSION
Self‐compartmentalization is a principle that is exploited not only by the exosome, but also by other enzymes, including the proteasome (42). The sequestering of active sites inside a small space prevents degradation of substrates that are not actively targeted to the enzyme. For the archaeal exosome, substrate selection takes place through RNA interacting proteins that dock around the entrance pore of the barrel (9,10,19). In the eukaryotic exosome, these RNA-binding proteins have evolved further and include helicases that are able to unfold RNA species that contain secondary structure (43).
Despite the functional advantages that are related to substrate selection, self-compartmentalization comes at a cost: multiple protomers in a larger complex can only act on a single substrate whereas the same number of monomeric proteins could act on multiple substrates simultaneously. For the Archaeoglobus fulgidus exosome, the activity of the native (Rrp41:Rrp42)3 exosome complex was previously compared to the activity of a version of the complex that only assembles into (Rrp41:Rrp42)1 dimers (41). Interestingly, for long RNA substrates, it was found that the catalytic activity of one (Rrp41:Rrp42)3 hexameric complex is higher than that of three (Rrp41:Rrp42)1 dimers. One (Rrp41:Rrp42)3 exosome that acts on one substrate is thus more efficient than three (Rrp41:Rrp42)1 dimers that act on three substrates simultaneously. For the Archaeoglobus fulgidus exosome complex, the advantages of self‐compartmentalization thus outweigh the disadvantages. The molecular basis that underlies this gain in activity upon assembly of the Rrp41 and Rrp42 protomers into a barrel like quaternary structure remained undetermined.
Here, we addressed the catalytic advantages of oligomerization of the Sulfolobus solfataricus exosome. Two features that appear in the enzyme upon oligomerization of the Rrp41 and Rrp42 proteins into a hexameric barrel are the entrance pore (the neck region) and a lumen that contains a very high concentration of active sites. We find that these two aspects are fundamental to the efficiency of the exosome complex (Figure 8).
Figure 8.
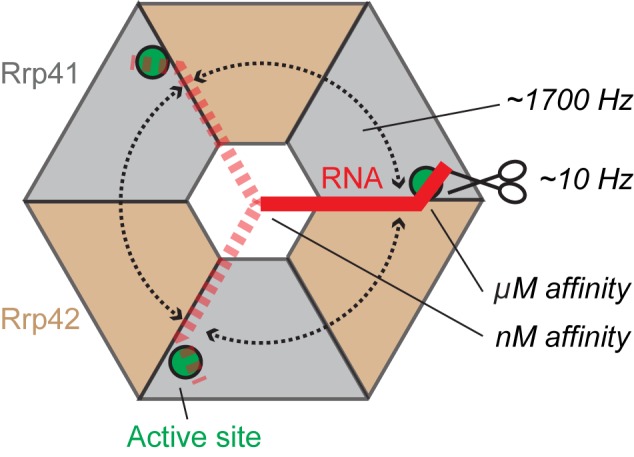
Cartoon that summarizes our findings. The RNA (red, dashed red) enters the exosome barrel (gray, sand) through the neck and can interact with one of three active sites (green circles). The substrate jumps between these three active sites with frequencies of ∼1700 s−1 (rounded dashed arrows). The catalytic cleavage rates are in the order of 10 s−1 (scissor symbol), two orders of magnitude slower than the RNA jumping rate. Only 1 in 100 RNA:exosome encounters results in a catalytic reaction. The exosome:RNA nM affinity interactions through the neck region keep the 3′ end of the RNA in the exosome barrel. The high concentration of active sites and substrate in the barrel of the exosome ensures that the 3′ end of the RNA is always in contact with one of the active sites.
Using binding measurements, we show that the neck region, where the RNA enters the exosome lumen, strongly interacts with unstructured RNA. Interestingly, this interaction involves substrate nucleotides that are located 10 bases upstream of the degradation site. This very strong interaction ensures that substrates can be recruited efficiently, and at the same time prevents that substrates are released from the enzyme complex before complete degradation (Figures 2 and 3). Experimentally we have shown this using an exosome complex that contains a single point mutation in the neck region. This mutation results in a 1000-fold decrease of the enzyme substrate interaction strength (Figure 3B). Importantly, also the processivity that is observed in the WT complex is lost upon weakening the neck-substrate interactions (Figure 3A) as substrates can no longer be retained to the enzyme complex during turnover. The importance of the neck region for RNA degradation appears to be conserved in exosome and exosome-like complexes (Supplementary Figure S1). First, it has been shown that the hexameric structure of the RNAse PH complex is essential for activity as a dimeric form of the enzyme that does not form a neck region is unable to interact with RNA (44). Secondly, mutations of basic residues in the entrance channel of the PNPase complex result in a loss of activity and processivity (45). Finally, mutations within the Rrp41 neck region of the yeast exosome have been shown to be important for the channeling of substrates through the catalytically inactive Exo-9 complex toward the catalytically active Rrp44 subunit (18).
To address the importance of the high concentration of active sites in the lumen of the exosome, we determined the kinetics of the substrate in the proximity of the active sites. Using methyl TROSY NMR methods we show that the 3′ end of the RNA is highly mobile and that it jumps between the active sites with a frequency of around 1700 Hz at 50°C (Figure 5). This high mobility will allow for a rapid dissociation of the reaction products, thereby facilitating turnover. Based on fluorescence anisotropy experiments that use a mutant where the neck interactions are impaired, the affinity between the active sites and the 3′ end of the RNA is in the μM regime (Figure 3B). This affinity is in line with previous ITC measurements that used a very small RNA substrate (46). Based on this μM affinity and the determined exchange rate of the RNA in the barrel it can be concluded that the on-rate of the binding process is fast (in the order of 109 M−1s−1). This fast on-rate is probably a direct consequence of the small volume of the lumen of the exosome and the resulting high substrate concentration. Using a series of RNA degradation experiments (Figures 6 and 7) we show that the motions of the RNA allow that all active sites in the exosome lumen equally participate in RNA degradation. Interestingly, the number of nucleotide cleavage events (∼10 s−1) is two orders of magnitude lower than the number of active-site: substrate encounters (∼1700 s−1). This shows that around 100 encounter complex formation events are required for one cleavage event.
When one assumes that the exosome has a spherical lumen with a diameter of 32 Å, the concentration of active sites in the enclosed volume can be estimated to be around 300 mM whereas the local substrate concentration is one-third of that (one RNA can enter the lumen). Based on the μM affinity of the 3′ end of the RNA for the active sites, the active-site occupancy can be predicted to be essentially 100%. As soon as RNA enters the lumen of the exosome it will thus be bound to one of the three active sites. In that light, the oligomerization of the exosome enhances catalytic efficiency by ensuring that the enzyme interacts with the substrate in a highly efficient manner at all times. In the theoretical case, where the archaeal exosome would only contain a single active site, the RNA would still be in contact with this site for more than 98% of the time. The reduction of the number of active sites in exosome(-like) complexes (6 active sites in RNase PH, 3 active sites in the PNPase and archaeal exosome, one in the plant exosome; Supplementary Figure S1) does not result in a reduction of the catalytic activity; the RNA will always be in full contact with an active site. After oligomerization of the exosome-like complexes into a barrel-like quaternary structure, the diversification of the subunits and the removal of active sites thus posed no catalytic disadvantages. It should be noted that in our degradation experiments (Figure 6) we intentionally removed activity through a single-point mutation that did not alter the interaction with RNA. In that situation, the 3′ end of the RNA partitions between active and inactive sites, which leads to the observed reduction in activity. When the removal of active sites is accompanied by the removal of the substrate interaction, the activity should stay at its maximum, independent of the number of active sites. In that light, it should be noted that the eukaryotic exosome indeed makes no contacts with the substrate RNA as all contact points have been removed (47).
In summary, we show that the oligomerizaiton of the exosome complex into a barrel like structure provides novel catalytic advantages. The basis for this lies in the fact that the enzyme interacts very strongly with the substrate close to base N10, whereas the 3′ end of the RNA close to base N1 remains highly flexible (Figure 8). The evolution of the hexameric exosome complex from single protein chains might have benefited from the increase in catalytic efficiency that is associated with the formation of the quaternary structure.
Supplementary Material
Acknowledgments
We acknowledge all members of the laboratory for discussions. We thank Silke Wiesner for helpful suggestions and Lewis E. Kay (University of Toronto) for sharing the code for the CEST NMR experiment with us.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
M.A.C. acknowledges funding from the IMPRS ‘From Molecules to Organisms’. This work was supported by the Max Planck Society and the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013), ERC grant agreement no. 616052.
Conflict of interest statement. None declared.
REFERENCES
- 1.Houseley J., LaCava J., Tollervey D. RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 2006;7:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- 2.Deutscher M.P., Marshall G.T., Cudny H. RNase PH: an Escherichia coli phosphate-dependent nuclease distinct from polynucleotide phosphorylase. Proc. Natl. Acad. Sci. U.S.A. 1988;85:4710–4714. doi: 10.1073/pnas.85.13.4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishii R., Nureki O., Yokoyama S. Crystal structure of the tRNA processing enzyme RNase PH from Aquifex aeolicus. J. Biol. Chem. 2003;278:32397–32404. doi: 10.1074/jbc.M300639200. [DOI] [PubMed] [Google Scholar]
- 4.Symmons M.F., Jones G.H., Luisi B.F. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Structure. 2000;8:1215–1226. doi: 10.1016/s0969-2126(00)00521-9. [DOI] [PubMed] [Google Scholar]
- 5.Koonin E.V., Wolf Y.I., Aravind L. Prediction of the archaeal exosome and its connections with the proteasome and the translation and transcription machineries by a comparative-genomic approach. Genome Res. 2001;11:240–252. doi: 10.1101/gr.162001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell P., Petfalski E., Shevchenko A., Mann M., Tollervey D. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′–>5′ exoribonucleases. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- 7.Evguenieva-Hackenberg E., Walter P., Hochleitner E., Lottspeich F., Klug G. An exosome-like complex in Sulfolobus solfataricus. EMBO Rep. 2003;4:889–893. doi: 10.1038/sj.embor.embor929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lorentzen E., Walter P., Fribourg S., Evguenieva-Hackenberg E., Klug G., Conti E. The archaeal exosome core is a hexameric ring structure with three catalytic subunits. Nat. Struct. Mol. Biol. 2005;12:575–581. doi: 10.1038/nsmb952. [DOI] [PubMed] [Google Scholar]
- 9.Buttner K., Wenig K., Hopfner K.P. Structural framework for the mechanism of archaeal exosomes in RNA processing. Mol. Cell. 2005;20:461–471. doi: 10.1016/j.molcel.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Lorentzen E., Dziembowski A., Lindner D., Seraphin B., Conti E. RNA channelling by the archaeal exosome. EMBO Rep. 2007;8:470–476. doi: 10.1038/sj.embor.7400945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evguenieva-Hackenberg E., Roppelt V., Finsterseifer P., Klug G. Rrp4 and Csl4 are needed for efficient degradation but not for polyadenylation of synthetic and natural RNA by the archaeal exosome. Biochemistry. 2008;47:13158–13168. doi: 10.1021/bi8012214. [DOI] [PubMed] [Google Scholar]
- 12.Roppelt V., Klug G., Evguenieva-Hackenberg E. The evolutionarily conserved subunits Rrp4 and Csl4 confer different substrate specificities to the archaeal exosome. FEBS Lett. 2010;584:2931–2936. doi: 10.1016/j.febslet.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Chekanova J.A., Shaw R.J., Wills M.A., Belostotsky D.A. Poly(A) tail-dependent exonuclease AtRrp41p from Arabidopsis thaliana rescues 5.8 S rRNA processing and mRNA decay defects of the yeast ski6 mutant and is found in an exosome-sized complex in plant and yeast cells. J. Biol. Chem. 2000;275:33158–33166. doi: 10.1074/jbc.M005493200. [DOI] [PubMed] [Google Scholar]
- 14.Januszyk K., Lima C.D. The eukaryotic RNA exosome. Curr. Opin. Struct. Biol. 2014;24:132–140. doi: 10.1016/j.sbi.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q., Greimann J.C., Lima C.D. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127:1223–1237. doi: 10.1016/j.cell.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 16.Dziembowski A., Lorentzen E., Conti E., Seraphin B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 2007;14:15–22. doi: 10.1038/nsmb1184. [DOI] [PubMed] [Google Scholar]
- 17.Wahle E. Wrong PH for RNA degradation. Nat. Struct. Mol. Biol. 2007;14:5–7. doi: 10.1038/nsmb0107-5. [DOI] [PubMed] [Google Scholar]
- 18.Bonneau F., Basquin J., Ebert J., Lorentzen E., Conti E. The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell. 2009;139:547–559. doi: 10.1016/j.cell.2009.08.042. [DOI] [PubMed] [Google Scholar]
- 19.Walter P., Klein F., Lorentzen E., Ilchmann A., Klug G., Evguenieva-Hackenberg E. Characterization of native and reconstituted exosome complexes from the hyperthermophilic archaeon Sulfolobus solfataricus. Mol. Microbiol. 2006;62:1076–1089. doi: 10.1111/j.1365-2958.2006.05393.x. [DOI] [PubMed] [Google Scholar]
- 20.Mund M., Overbeck J.H., Ullmann J., Sprangers R. LEGO-NMR spectroscopy: a method to visualize individual subunits in large heteromeric complexes. Angew. Chem. Int. Ed. Engl. 2013;52:11401–11405. doi: 10.1002/anie.201304914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easton L.E., Shibata Y., Lukavsky P.J. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA. 2010;16:647–653. doi: 10.1261/rna.1862210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramos A., Varani G. A new method to detect long-range protein-RNA contacts: NMR detection of electron-proton relaxation induced by nitroxide spin-labeled RNA. J. Am. Chem. Soc. 1998;120:10992–10993. [Google Scholar]
- 23.Johnson P.E., Tomme P., Joshi M.D., McIntosh L.P. Interaction of soluble cellooligosaccharides with the N-terminal cellulose-binding domain of Cellulomonas fimi CenC 2. NMR and ultraviolet absorption spectroscopy. Biochemistry. 1996;35:13895–13906. doi: 10.1021/bi961186a. [DOI] [PubMed] [Google Scholar]
- 24.Korzhnev D.M., Kloiber K., Kay L.E. Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: theory and application. J. Am. Chem. Soc. 2004;126:7320–7329. doi: 10.1021/ja049968b. [DOI] [PubMed] [Google Scholar]
- 25.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 26.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 27.Lorentzen E., Conti E. Structural basis of 3′ end RNA recognition and exoribonucleolytic cleavage by an exosome RNase PH core. Mol. Cell. 2005;20:473–481. doi: 10.1016/j.molcel.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Tugarinov V., Hwang P.M., Ollerenshaw J.E., Kay L.E. Cross-correlated relaxation enhanced 1H[bond]13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J. Am. Chem. Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- 29.Sprangers R., Kay L.E. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 30.Wiesner S., Sprangers R. Methyl groups as NMR probes for biomolecular interactions. Curr. Opin. Struct. Biol. 2015;35:60–67. doi: 10.1016/j.sbi.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Audin M.J., Dorn G., Fromm S.A., Reiss K., Schutz S., Vorlander M.K., Sprangers R. The archaeal exosome: identification and quantification of site-specific motions that correlate with cap and RNA binding. Angew. Chem. Int. Ed. Engl. 2013;52:8312–8316. doi: 10.1002/anie.201302811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorentzen E., Conti E. Crystal structure of a 9-subunit archaeal exosome in pre-catalytic states of the phosphorolytic reaction. Archaea. 2012:721869. doi: 10.1155/2012/721869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sprangers R., Gribun A., Hwang P.M., Houry W.A., Kay L.E. Quantitative NMR spectroscopy of supramolecular complexes: dynamic side pores in ClpP are important for product release. Proc. Natl. Acad. Sci. U.S.A. 2005;102:16678–16683. doi: 10.1073/pnas.0507370102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amero C., Asuncion Dura M., Noirclerc-Savoye M., Perollier A., Gallet B., Plevin M.J., Vernet T., Franzetti B., Boisbouvier J. A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies. J. Biomol. NMR. 2011;50:229–236. doi: 10.1007/s10858-011-9513-5. [DOI] [PubMed] [Google Scholar]
- 35.Navarro M.V., Oliveira C.C., Zanchin N.I., Guimaraes B.G. Insights into the mechanism of progressive RNA degradation by the archaeal exosome. J. Biol. Chem. 2008;283:14120–14131. doi: 10.1074/jbc.M801005200. [DOI] [PubMed] [Google Scholar]
- 36.Korzhnev D.M., Kloiber K., Kanelis V., Tugarinov V., Kay L.E. Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme. J. Am. Chem. Soc. 2004;126:3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- 37.Lundstrom P., Vallurupalli P., Religa T.L., Dahlquist F.W., Kay L.E. A single-quantum methyl 13C-relaxation dispersion experiment with improved sensitivity. J. Biomol. NMR. 2007;38:79–88. doi: 10.1007/s10858-007-9149-7. [DOI] [PubMed] [Google Scholar]
- 38.Bouvignies G., Kay L.E. A 2D (1)(3)C-CEST experiment for studying slowly exchanging protein systems using methyl probes: an application to protein folding. J. Biomol. NMR. 2012;53:303–310. doi: 10.1007/s10858-012-9640-7. [DOI] [PubMed] [Google Scholar]
- 39.Fazal F.M., Koslover D.J., Luisi B.F., Block S.M. Direct observation of processive exoribonuclease motion using optical tweezers. Proc. Natl. Acad. Sci. U.S.A. 2015;112:15101–15106. doi: 10.1073/pnas.1514028112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Y., Reeve J.N. Transcription by an archaeal RNA polymerase is slowed but not blocked by an archaeal nucleosome. J. Bacteriol. 2004;186:3492–3498. doi: 10.1128/JB.186.11.3492-3498.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartung S., Niederberger T., Hartung M., Tresch A., Hopfner K.P. Quantitative analysis of processive RNA degradation by the archaeal RNA exosome. Nucleic Acids Res. 2010;38:5166–5176. doi: 10.1093/nar/gkq238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makino D.L., Halbach F., Conti E. The RNA exosome and proteasome: common principles of degradation control. Nat. Rev. Mol. Cell Biol. 2013;14:654–660. doi: 10.1038/nrm3657. [DOI] [PubMed] [Google Scholar]
- 43.Halbach F., Reichelt P., Rode M., Conti E. The yeast ski complex: crystal structure and RNA channeling to the exosome complex. Cell. 2013;154:814–826. doi: 10.1016/j.cell.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 44.Choi J.M., Park E.Y., Kim J.H., Chang S.K., Cho Y. Probing the functional importance of the hexameric ring structure of RNase PH. J. Biol. Chem. 2004;279:755–764. doi: 10.1074/jbc.M309628200. [DOI] [PubMed] [Google Scholar]
- 45.Shi Z., Yang W.Z., Lin-Chao S., Chak K.F., Yuan H.S. Crystal structure of Escherichia coli PNPase: central channel residues are involved in processive RNA degradation. RNA. 2008;14:2361–2371. doi: 10.1261/rna.1244308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oddone A., Lorentzen E., Basquin J., Gasch A., Rybin V., Conti E., Sattler M. Structural and biochemical characterization of the yeast exosome component Rrp40. EMBO Rep. 2007;8:63–69. doi: 10.1038/sj.embor.7400856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Makino D.L., Schuch B., Stegmann E., Baumgartner M., Basquin C., Conti E. RNA degradation paths in a 12-subunit nuclear exosome complex. Nature. 2015;524:54–58. doi: 10.1038/nature14865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.