


Abstract
Trans-lesion synthesis polymerases, like DNA Polymerase-η (Pol-η), are essential for cell survival. Pol-η bypasses ultraviolet-induced DNA damages via a two-metal-ion mechanism that assures DNA strand elongation, with formation of the leaving group pyrophosphate (PPi). Recent structural and kinetics studies have shown that Pol-η function depends on the highly flexible and conserved Arg61 and, intriguingly, on a transient third ion resolved at the catalytic site, as lately observed in other nucleic acid-processing metalloenzymes. How these conserved structural features facilitate DNA replication, however, is still poorly understood. Through extended molecular dynamics and free energy simulations, we unravel a highly cooperative and dynamic mechanism for DNA elongation and repair, which is here described by an equilibrium ensemble of structures that connect the reactants to the products in Pol-η catalysis. We reveal that specific conformations of Arg61 help facilitate the recruitment of the incoming base and favor the proper formation of a pre-reactive complex in Pol-η for efficient DNA editing. Also, we show that a third transient metal ion, which acts concertedly with Arg61, serves as an exit shuttle for the leaving PPi. Finally, we discuss how this effective and cooperative mechanism for DNA repair may be shared by other DNA-repairing polymerases.
INTRODUCTION
The DNA replication machinery guarantees the correct genetic inheritance to the new cells generation. However, DNA alterations, caused by endogenous and/or exogenous agents, represent a major obstacle for DNA replication. For example, daily sunlight exposure can cause the formation of ultraviolet (UV)-induced covalent bonds between adjacent pyrimidine bases within the double helix, resulting in cyclobutane pyrimidine dimers (CPDs). These are one of the major forms of DNA damage (1–6), which are not handled by most replicative polymerases, causing the stall of the replication fork (7,8). One way to restore the DNA replication machinery is to bypass those hurt regions via the so-called translesion synthesis (TLS) process, during which specialized translesion polymerases are capable to extend the damaged DNA. Once base pairing has been restored beyond the lesion, the replicative polymerase regains the control of DNA replication (9–11).
One of those specialized TLS polymerases is human DNA Polymerase-η (Pol-η), member of the Y-family polymerases. Pol-η extends the damaged DNA primer strand in the presence of UV-induced CPDs. It inserts bases opposite these DNA defects, ensuring correct DNA replication (1–6). However, Pol-η bypasses also DNA crosslinking damages generated by anticancer drugs, such as cisplatin, allowing cancer cells to survive and proliferate (12–16). Pol-η's function is also critical for DNA elongation in the somatic hypermutation mechanism, which is a programmed base substitution in the variable regions of immunoglobulin genes (17,18). For these reasons, Pol-η is a promising target to treat skin carcinogenesis (19,20) and overcome cancer drug resistance (21,22).
Like other Y-family members (8), Pol-η contains a two-metal-ion catalytic site, where MgA and MgB are coordinated by the so-called DED-motif (Figure 1). Recent time-resolved X-ray structures of Pol-η in complex with a double-strand DNA (dsDNA) and a 2′-deoxyadenosine triphosphate (dATP) have revealed the enzymatic structural evolution during catalysis (1). The enzymatic reaction of Pol-η catalyzes the two-metal-aided formation of a new bond between the 3′-OH of the primer DNA strand and the α-phosphate of dATP, with the concomitant cleavage of the phosphodiester bond between the α- and β-phosphates of dATP (23–26). Consequently, the primer DNA strand is extended by one nucleotide, with departure of the leaving group pyrophosphate (PPi, Figure 1) (27–29).
Figure 1.

Human polymerase-η structural and catalytic features. (A) Graphic of the ternary polymerase-η/dsDNA/dATP complex (1,2). Protein in graphic representation, dsDNA in ribbon representation; orange spheres indicate the two Mg2+ ions. The following domains are depicted: palm (yellow), thumb (blue), fingers (cyan) and little finger (red). On the right, scheme and close view of the catalytic site of Pol-η during DNA synthesis: (B and D) Reactant state with DNA and dNTP; (C and E) Product state with DNA and PPi. The different rotamer conformations of Arg61, from reactants to products, are highlighted.
Importantly, these recent structural data have revealed the significant flexibility of the conserved and catalytic Arg61 (2,30,31) and, intriguingly, the binding of a third transient magnesium ion (MgC) at the catalytic site of Pol-η (1). In fact, Arg61 shows conformational transitions during catalysis, passing from the reactants to the products. While in the reactant state, Arg61 forms cation–π interactions with the incoming base and hydrogen bonds with the phosphate groups of the incoming dATP. Then, in the products, Arg61 loses its stacking interaction with the incoming base, forming with it a single H-bond. A third Arg61 conformation was also detected in the products, where Arg61 forms two H-bonds with the dATP and the apical oxygen of the template base, respectively (Figure 2A) (1). A similar conformation of Arg61 was resolved in more recent crystal structures of Pol-η in complex with a dsDNA presenting a mispair condition, formed by a non-reactive dGTP paired with a templating thymine, in the product state (18). In addition to the highly flexible Arg61, the third transient MgC has been observed only in the product state during Pol-η catalysis (2). Taken together, these novel experimental results on Pol-η provide a comprehensive set of data for a thorough investigation of the mechanistic role of Arg61 and MgC to catalyze Pol-η function. In addition, other studies have recently revealed that DNA polymerases often contain multiple metal ions at the catalytic site, together with a highly flexible positively charged residue, like an arginine or a lysine (1,25,32). How do these conserved structural features contribute to DNA replication?
Figure 2.

Pre-reactive state on the move. (A) Image showing the diverse Arg61 conformations detected in wt-RS system. Collective variables (CV1 and CV2) used to monitor conformational changes are shown. (B) Different Arg61 conformations for wt-RS system (CV2 versus time) correlated with fluctuations of d-newbond (purple). (C) Frequency distribution of d-newbond length as a function of the different Arg61 conformations. (D) Average internuclear MgA-MgB distance in the different Arg61 conformations.
Through a series of extensive classical molecular dynamics (MD) and free energy simulations, we unravel a highly cooperative mechanism, which is here described by an equilibrium ensemble of structures that connect the reactants to the products in Pol-η catalysis (1). This dynamic mechanism clarifies how Arg61 and multiple metal ions jointly catalyze DNA editing, and allows decoding the conformational transitions disclosed by the recent experimental data on Pol-η. We found that Arg61 initially adopts conformations that favor the recruitment of the incoming base and the proper formation of a pre-reactive complex. Then, in the products, Arg61 changes its conformation to act concertedly with a third transient metal ion to facilitate the exit of the leaving PPi after DNA elongation. Thanks to the conservation of a common catalytic structural motif, we discuss how this efficient and cooperative mechanism for DNA replication could likely be extended to other DNA polymerases.
MATERIALS AND METHODS
Structural models
We considered three different systems for the reactant and two for the product state (1), namely: (i) wild-type reactant state (wt-RS), which is based on the recent time-resolved X-ray structures of the ternary complex (PDB ID: 4ECS) (1) formed by human Pol-η in complex with double-strand DNA (dsDNA) and 2′-deoxyadenosine triphosphate (dATP). In this structure, the Arg61 side chain points toward the dATP α-phosphate, adopting the A-conf (85% occupancy in the X-ray); (ii) Arg61Ala reactant state (mut-RS), which is based on the same X-ray structure used for the wt-RS system, with the catalytic Arg61 mutated into an alanine residue, as studied experimentally (2); (iii) wild-type T:G mispaired reactant state (mp-RS), which is modeled on the recent structure of Pol-η complexed with dsDNA and an incoming non-reactive dATP, which was substituted with a standard dGTP, to form a dGTP:T mispair (PDB ID: 3MR2) (2,18). In this system, Arg61 adopts a conformation defined as C-conf (50% occupancy in the X-ray), with a third H-bond that differentiates it from the wt-RS conformation (see Supplementary Figure S1, mp-RS); (iv) wild-type product state (3M-PS), based on the X-ray structure of the enzyme in complex with dsDNA and three magnesium ions (MgA, MgB and MgC) (1). This model depicts the enzyme structure after completion of the nucleotidyl-transfer reaction and consequent products formation (PDB ID: 4ECW) (1). The Arg61 side chain points toward the templating thymine, adopting the C-conf (60% occupancy in the X-ray) (1) (Figure 2). MgC coordinates three non-bridging oxygen atoms of the γ-phosphate, while three water molecules complete its octahedral coordination; (v) MgA,B, product state (2M-PS), which is based on the same X-ray structure used to construct the model 3M-PS, with MgC removed.
Molecular dynamics (MD) simulations
The all-atom AMBER/parm99SB-ILDN (33) force field was adopted for the Pol-η in complex with dsDNA, whereas dATP, dGTP and PPi were treated with the general amber force field (34). The atomic charges were derived by fitting the electrostatic potential according to the Merz-Singh-Kollman scheme, also known as the RESP fitting procedure (Supplementary Table S1). The length of all covalent bonds, including hydrogen atoms, was set using the LINCS algorithm, allowing a time-integration step of 2 fs. All simulations were performed using Namd 2.9 code (35). Long-range electrostatic interactions were calculated with the particle mesh Ewald method with a real space cutoff of 12 Å. Periodic boundary conditions in the three directions of Cartesian space were applied. Constant temperature (310 K) was imposed using Langevin dynamics (36) with a damping coefficient of 1 ps. A constant pressure of 1 atm was maintained with Langevin-Piston dynamics with a 200 fs decay period and a 50 fs time constant. The metal active site was treated with a flexible non-bonded approach based on the ‘atoms in molecules’ partitioning scheme of the DFT-BLYP electronic density of the active site (Supplementary Table S1) (37). We could thus consider the charge-transfer interactions between Mg2+ ions and their ligands, permitting possible structural rearrangements at the active site during the MD simulations. All the simulated systems were hydrated using TIP3P (38) water molecules. A total of 7 Mg2+ ions were added to each system to reach a final concentration of ∼1 mM, while Na+ and Cl− ions were added to neutralize the total charge. The size of the final systems was approximately 113 × 97 × 88 Å, with ∼28 000 water molecules, resulting in a total number of ∼70 000 atoms each.
We adopted the following simulation protocol: the systems were minimized using a steepest-descent algorithm and then slowly heated up to 310 K in 10 ns for a total of 2000 steps. The first 50 ns of production run are considered as the equilibration phase. Approximately ∼1 μs of MD simulations were collected in the NPT ensemble for each of the five systems, resulting in a total of ∼5 μs of dynamics. Coordinates of the systems were collected every 5 ps, for a total of ∼200 000 frames for each run. Statistics were collected considering the equilibrated trajectories only, thus discarding the first ∼100 ns of simulation for all the systems.
Free energy estimation
We used well-tempered metadynamics (39) to characterize Arg61 flexibility and estimate the free-energy landscape associated with its rearrangement. The free energy was determined as a function of two selected collective variable (CVs), which identify A-, B- and C-conf as observed during unbiased MD. The CVs are (Figure 2): (i) CV1, which is the distance between the centers of mass of Arg61 heavy atoms (Cζ, Nη1 and Nη2) and the heavy atoms of the incoming adenine, or the guanine in the dGTP:T pair detected in the mp-RS system; (ii) CV2, which is the distance between the two centers of mass of Arg61 heavy atoms (Cζ, Nη1 and Nη2) and α- and β-phosphate groups of the incoming dATP (Pα, Pβ and the O atom between them). In particular, based on the crystallographic evidence and our classical MD simulations, when ∼2.00 Å < CV1 < ∼4.40 Å and ∼1.50 Å < CV2 < ∼4.50 Å, Arg61 is considered in A-conf, while different values of CVs refer to B- (∼2.00 Å < CV1 < ∼5.50 Å and ∼5.00 Å < CV2 < ∼7.00 Å) or C-conf (∼3.00 Å < CV1 < ∼6.00 Å and ∼7.00 Å < CV2 < ∼12.00 Å).
The fictitious temperature associated with the CVs was set to 930 K while the Gaussian function deposition rate was set to 1 ps. The initial hills height and width were set to 0.05 kcal mol−1 and 0.01 kcal mol−1, respectively. The well-tempered simulations were carried out until their convergence (∼100 ns of each of them), i.e. the progressive stabilization of the energetic minima on the free-energy surface (Supplementary Figure S2). All other parameters correspond to those used for the plain MD simulations, described above.
Well-tempered metadynamics was also used to explore the PPi unbinding process in the presence of MgC using system (4). In this case, we considered a single CV (CVPPi) that describes the difference between the bound and unbound state of PPi during its departure. This CV is the distance between the centers of mass of the PPi atoms and the αC of the DED-motif (Supplementary Figure S2). In this case, we applied a fictitious CV temperature of 4650 K. The Gaussian function deposition rate was set to1 ps while the initial hills height and width were set to 0.05 kcal mol−1 and 0.01 kcal mol−1, respectively.
RESULTS
Pre-reactive configurations in Pol-η
Through two independent MD simulations (∼500 ns) of the wild-type reactant wt-RS complex, formed by Pol-η and its substrates, dsDNA and dATP, we examined the evolution over time of key structural features of the catalytic pocket. Arg61 stochastically adopted different rotamer conformations along the trajectories (Figure 2B). During the first ∼60 ns of production run, Arg61 conserved its crystallographic A-conf pose (Figure 2B). In this conformation, its guanidine group forms a bi-dentate H-bond interaction with the α/β-phosphate groups of dATP. Subsequently, Arg61 changed its side-chain orientation, adopting the B-conf pose, in which it forms its characteristic H-bond with the N7 atom of the incoming dATP. This conformational change was well described by the distance CV2 (Figure 2A), which increased from ∼4.00 Å to ∼5.00 (Figure 2B). Furthermore, Arg61 occasionally assumed a C-conf pose, which is characterized by an additional H-bond formed by its guanidine group with the apical oxygen of the 5′-terminal thymine of the template strand (2). Each B- and C-conf lasted for a short time, ranging from ∼10 to ∼22 ns, before Arg61 returned to the native A-conf (Figure 2B).
We then monitored the alignment of the substrates dsDNA and the incoming dATP to form the in-line conformation, which preludes the catalytic SN2-like phosphoryl-transfer reaction in Pol-η. Notably, the 3′-end sugar pucker remains in a stable C3′-endo conformation for the entire simulations, in agreement with the X-ray data (1). We also examined the d-newbond variation, i.e. the distance between the two reactive groups, the 3′-OH of the terminal base of the DNA, and the α-phosphorus of the incoming nucleotide (Figure 2A). We found that d-newbond had a mean value of 3.35 ± 0.07 Å when Arg61 adopted A-conf, which agreed well with the X-ray value of d-newbond (3.30 Å) (1). However, when Arg61 was in B- or C-conf during the simulations, d-newbond slightly increased to a longer stable value of 3.52 ± 0.07 Å and 3.65 ± 0.05 Å, respectively. This suggests a conformational state that is likely less prone to promote catalysis. Three Gaussians fit the frequency distribution of d-newbond when Arg61 assumes A-conf, B-conf and C-conf, pointing out the elongation of d-newbond when Arg61 moved away from A-conf (Figure 2C). In particular, the greater the distance between the Arg61 guanidine group and the phosphate tail of the incoming base, the longer the length of d-newbond (A-conf > B-conf > C-conf), as confirmed by confidence interval analysis (see Supporting Text). Arg61 fluctuations seemed to affect d-newbond only, while the surrounding structural environment maintained its native geometry throughout the whole trajectory.
Concomitantly, the distance between MgA and MgB became shorter (3.51 ± 0.07 Å) when Arg61 assumed A-conf, in fine agreement with the X-ray structure of the pre-reactive state (1). This inter-metal distance became larger (3.65 ± 0.06 Å) when Arg61 switched from A- to B-conf and further increased (3.75 ± 0.06 Å) when Arg61 adopted C-conf. Three Gaussian curves indicate the frequency distribution of the inter-metal length (MgA-MgB) with respect to the three different Arg61 conformations (Figure 2D), highlighting the plasticity of the catalytic pockets in aiding catalysis, as for those enzymes that use a bi(tri)-metal core to catalyze phosphoryl-transfer reactions (25,26,37,40,41). See further analysis in Supplementary Figures S3–6 and Supplementary Table S2.
A mutant model system mut-RS (Arg61Ala) of the ternary Pol-η/dsDNA/dATP complex was also simulated for ∼1 μs. The enzyme well maintained its overall structural framework and the catalytic metal ions conserved their typical octahedral coordination geometry. However, the only substrate showed a slight rearrangement already in the early stages of the simulation. After only ∼50 ns of production run, we observed a conformational change of the adenine base of the incoming dATP, which was buckled with respect to the 2′-deoexyribose. Because of this buckling, the incoming dATP lost the H-bonds established with the complementary 5′-thymine (inter-base H-bonds). Indeed, the double H-bond interaction expected for a dATP:T pairing (according to Watson–Crick's base-pairing model) is present in only ∼11.00% of the collected frames of the mut-RS simulations and ∼20.00% of the wt-RS system frame. Hence, the lack of Arg61 compromises the base interaction pattern, which becomes unstable. As a consequence, the incoming dATP lost its native position. This motion is captured well by the dATP RMSD value, which increases from 0.33 ± 0.09 Å in the wt-RS to 0.89 ± 0.26 Å in mut-RS (Supplementary Figure S3). This motion is also reflected by an increased fluctuation of d-newbond, which was 3.44 ± 0.14 Å.
The mispaired model mp-RS shows a non-Watson–Crick base-pairing dGTP:T in the dsDNA, with the formation of a ternary Pol-η/dsDNA/dGTP complex (18). Notably, C-conf is here characterized by an H-bond pattern that exists in the mp-RS crystal only, due to the mispairing dGTP:T condition (Supplementary Figure S1). We ran two simulations (∼250 ns, each) using both A- and C-conf Arg61 conformations as a starting pose. Interestingly, in the A-conf system, Arg61 suddenly rotated around its φ dihedral angle (Cβ-Cγ-Cδ-Nϵ) readopting its native C-conf (18), which then remained stable. The fast A-to-C-conf switching is captured well by CV2, which was ∼3.50 Å (A-conf) in the first 5 ns of simulation. Then, CV2 rapidly increased to a value of ∼10.00 Å (C-conf) during the remaining part of the simulation. We also simulated a system in which Arg61 starts in C-conf. As expected, C-conf was stably maintained throughout the entire simulated timescale and no Arg61 conformational changes were observed (Supplementary Figure S6). In fact, the three H-bonds characteristic of C-conf displayed high stability with an average length of 2.63 ± 0.19 Å. Here, d-newbond shows a value of 3.23 ± 0.07 Å, indicating a perfect alignment of the reactive groups. The limited simulated timescale of this system did not capture the large conformational changes in the catalytic pocket, which are displayed by X-ray structures of this system (18) and which likely explain its lower catalytic efficiency (see ‘Discussion’ section).
Energetics of Arg61 motions in pre-reactive states
We calculated the free energy surfaces (FESs) of the Arg61 A↔B/A↔C interconversions for the wt-RS and mp-RS systems (Figure 3), as a function of CV1 and CV2 (see ‘Materials and Methods’ section). The FES related to the wt-RS system was characterized by three low free-energy minima. The deepest minimum corresponded to the X-ray conformation (1), in which Arg61 was detected in A-conf (CV1 = ∼4.00 Å and CV2 = ∼2.00 Å). This reflects our unbiased MD simulations, in which Arg61 was most likely to be in this conformation. Nearby this absolute minimum, a second free-energy basin revealed a variant form of A-conf (CV1 = ∼4.00 Å, CV2 = ∼4.00 Å), which was frequently observed in wt-RS unbiased simulations. In this variant, only one of the bi-dentate H-bond interactions with the phosphate groups (characteristic of A-conf) was formed. Then, a less stable minimum was found for Arg61 in B-conf (CV1 = ∼2.20 Å, CV2 = ∼7.00 Å), while a third relative minimum (CV1 = ∼3.80 Å and CV2 = ∼9.50 Å) identified Arg61 in C-conf. The overall relative stability of the three rotamers for Arg61 (A-conf > B-conf > C-conf) corroborated our findings from unbiased MD (see Supplementary Table S3).
Figure 3.
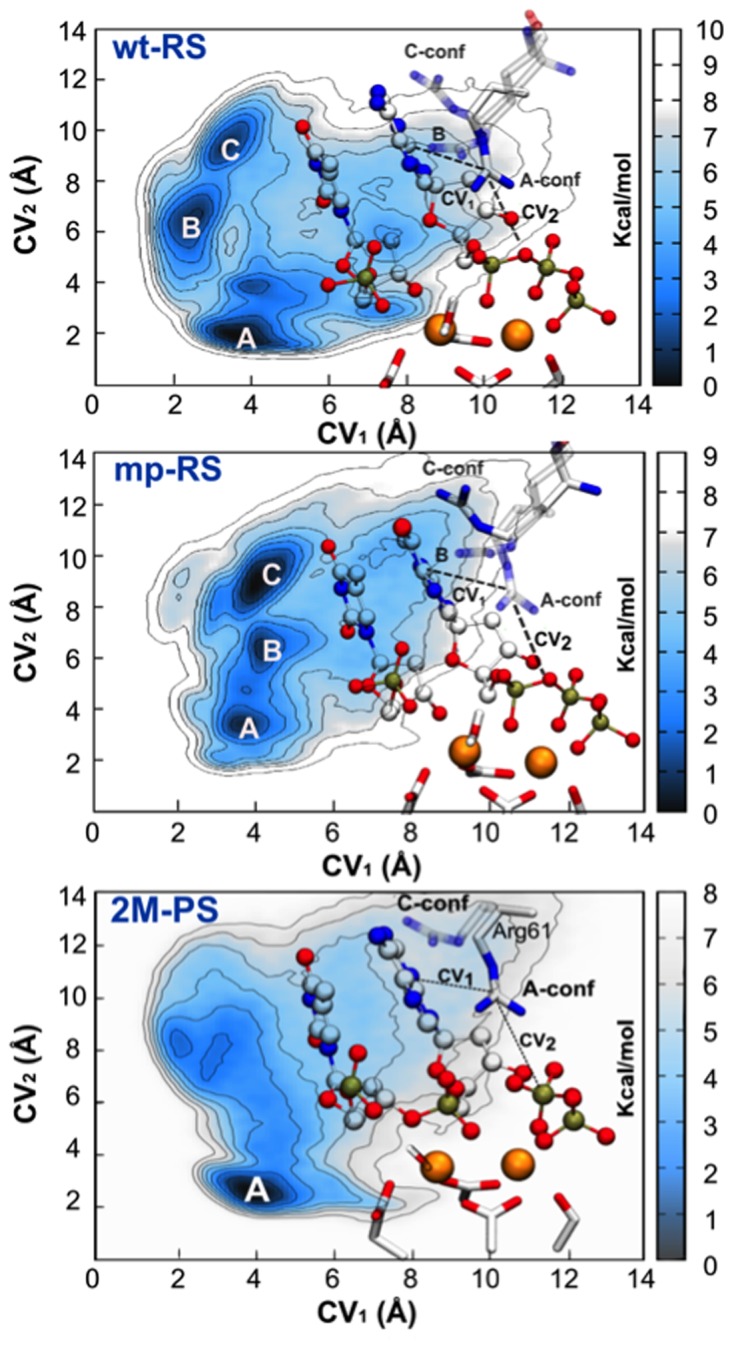
Free energy simulations of Arg61 motions. (wt-RS) Free energy surface (FES) of wt-RS system showing three main energetic basins for different Arg61 conformations (A-, B- and C-conf). (mp-RS) FES of mp-RS system characterized by three minima. The deepest one corresponds to C-conf. Nomenclature is maintained as for wt-RS. CVs are also depicted (see ‘Materials and Methods’ section). (2M-PS) Free energy landscape of 2M-PS system. CVs are also depicted (see ‘Materials and Methods’ section). Isolines are every 1.00 kcal mol-1 for each FES.
The free-energy landscape for the mp-RS system was quite different, with the basin of Arg61 in A-conf (CV1 = ∼3.80 Å, CV2 = ∼3.00 Å), which was then no longer the absolute minimum of the FES (Figure 3). As expected, C-conf was now the deepest free-energy minimum (CV1 = ∼4.00 Å and CV2 = ∼9.50 Å). This explains the high population of this configuration during the unbiased MD simulations of mp-RS. Arg61 in B-conf (CV1 = ∼4.00 Å, CV2 = ∼7.00 Å) was found in a basin of ∼3.50–4.00 kcal mol−1 higher than in C-conf. A variant form of the A-conf (CV1 = ∼4.00 Å, CV2 = ∼4.30 Å) was also found ∼6.00 kcal mol−1 higher in free energy than C-conf. In this latter case, Arg61 H-bonds only one non-bridging oxygen atom of the Pα of the dGTP, rather than forming the favored bifurcated interaction as observed for the wt-RS system. As a result, these less stable Arg61 states are rarely detected in unbiased MD (see Supplementary Table S3).
Post-reactive configurations in Pol-η
We collected ∼1 μs of simulation time for two configurations of the product system, where the enzyme is characterized by Pol-η in complex with dsDNA, now extended by one nucleotide with respect to the reactants, and the free PPi leaving group (1). Namely, we simulated 3M-PS and 2M-PS, which respectively have three and two Mg2+ ions in the catalytic pockets (Figure 1). Notably, in 3M-PS, Arg61 steadily maintained C-conf, with no Arg61 A↔C/A↔B interconversions. Instead, in 2M-PS, after removing MgC, Arg61 immediately changed its rotamer conformation from C- to A-conf (t = ∼20 ns), while the overall protein, MgA and MgB ions maintained their general structure and coordination with respect to the X-ray structure (1). Hence, in 2M-PS, Arg61 adopted different conformations along the MD runs, as in the pre-reactive state. In fact, Arg61 assumed all the known conformations with preference for the C-conf, which appeared at t = ∼0 ns, ∼280 ns, ∼422 ns, ∼475 ns, ∼710 ns, ∼940 ns (Supplementary Figure S7).
Crucially, we observed a new transient Mg2+ ion that spontaneously reached, from the water bulk, the negative potential well near the catalytic pocket, filling up the vacancy originally created by the removal of MgC, between the PPi and the α-phosphate of the new 3′-end (Figure 4, and Supplementary Figure S8). This unprompted event rebuilt a structural framework that resembled the original 3M-PS system (RMSD = 1.87 Å versus 3M-PS) (1). Then, further transient rearrangements of Arg61, DED-motif, MgB and PPi occurred during these MD runs, after a new third Mg2+ ion reached the catalytic pocket. In fact, for ∼430 ns (following third Mg2+ ion binding) the geometry coordination of MgB was slightly altered (Figure 4). As a consequence, Asp13 and Glu118 increased their distance from MgB, passing from 2.00 ± 0.30 Å to 3.51 ± 0.20 Å, respectively. Concomitantly, the PPi leaving group interacted more closely with MgC, with an average distance of 2.10 ± 0.21 Å. This overall structural rearrangement allowed Arg61 to switch from C-conf to A-conf, forming bifurcated H-bond interactions with the newly formed adduct (MgB–MgC–PPi, Figure 4). This orchestrated motion, which involved MgB, MgC, PPi, Arg61, Arg55 and Tyr52, suggests a step-wise leaving mechanism of the PPi from the catalytic pocket, which is an essential part of the catalytic turnover for Pol-η's function. As a consequence, the distance between the centers of mass of the PPi and the DED-motif increased from ∼3.50 Å to ∼7.20 Å. However, after these ∼430 ns of unbiased MD, this metastable intermediate was likely unable to overcome the energetic barrier to complete the leaving group release mechanism (see below). Consequently, the third metal left the cavity (t = ∼800 ns) and the system fell back into the closest energetic minimum, restoring the original 2M-PS structure, which was maintained well for the remainder of the simulations (Figure 4).
Figure 4.

Third Mg ion binding and release mechanisms. (A) Post-reactive system in which Arg61 adopts C-conf. (B) At t = ∼530 ns, a third Mg2+ metal stably binds the catalytic pocket, forming the 3M-PS system, that matches the X-Ray structure 4ECW (1). This snapshot presages PPi departure, a mechanism investigated by using enhanced sampling methods. In the upper-right corner is reported an indicative free energy profile of the step-wise PPi leaving mechanism. Importantly, to favor PPi release, Arg61 switches from C- to A-conf. (C) MgC-PPi distance monitored along the simulated timescale. Blue line indicates the distance of the third Mg2+ from the pyrophosphate (PPi) center of mass. Orange line indicates the distance between third ion and center of mass of PPi detected in the crystallographic structure 4ECW (1).
Energetics of Arg61 motions in post-reactive configurations and PPi-releasing mechanism
In 2M-PS, the FES showed one deep minimum in which Arg61 adopted the A-conf (CV1 = ∼4.00 Å and CV2 = ∼3.00 Å). However, the energy required to switch from A-conf to C-conf was only ∼3.50 kcal mol−1, which explains the spontaneous Arg61 A↔C interconversion detected in our unbiased MD simulations of 2M-PS (Figure 3 and Supplementary Table S3).
We also investigated the full PPi-releasing mechanism, starting from the 3M-PS system. We used a new CV defined as CVPPi (i.e. the distance between the centers of mass of PPi and the αC of DED-motif and Met14 as a CV, Supplementary Figure S2). Our unbiased MD simulations showed a metastable conformation presaging a possible leaving mechanism of the PPi complexed with MgB and MgC. We thus considered PPi bound to the catalytic pocket when CVPPi ≤ ∼9.50 Å, while the MgB–MgC–PPi was completely unbound when CVPPi ≥ ∼9.50 Å.
Starting our simulations from the crystallographic PPi conformation still bound to the catalytic site (Figure 4B, CVPPi = ∼7.50 Å), a two-step mechanism allowed the release of the PPi, complexed with MgB and MgC, from the Pol-η catalytic site. First, the system reached a metastable intermediate, characterized by the partial unbound state of the MgB–MgC–PPi complex, as suggested in the unbiased MD. During this state, Arg61 passed from C-conf to A-conf and concertedly interacted (together with Arg55 and Tyr52) with the newly formed complex MgB–MgC–PPi, which underwent conformational changes that determined its detachment of ∼3.70 Å, with respect to its native conformation. A free energy contribution of ∼5 kcal mol−1 was required to overcome the first barrier of this two-step releasing mechanism. A second step then occurred, in which Arg61 rotated around its φ dihedral angle, readopting its native C-conf, to aid the final release of the product adduct MgB–MgC–PPi, which was completely disengaged from the DED-motif in Pol-η catalytic site. For this second step, a barrier of ∼4 kcal mol−1 had to be overcome. The unbinding of the PPi from the catalytic site required the build-up of a given amount of biasing potential, which provided a representation of the energy landscape along the dissociation pathway (Figure 4).
DISCUSSION
Recent structural data of the human DNA polymerase-η (Pol-η) have highlighted the significant structural flexibility of the conserved Arg61 and, intriguingly, the binding of a transient third ion resolved at the catalytic site (1). Here, extended MD and free energy simulations of several systems of Pol-η in different pre- and post-reactive conditions demonstrate how the third metal ion cooperate with different Arg61 configurations to facilitate DNA replication.
During the ∼1 μs of MD simulations of the wild-type reactant system wt-RS, we observed three different Arg61 conformations (A-, B- and C-conf), which agreed with the crystallographic evidence (1,2). A-conf was the most sampled conformation, found in the 54.10% of the total frames, while B-conf and C-conf were found in 29.80 and 16.10% of the MD trajectory, respectively (A-conf occupancy of 85% in the 4ECS X-ray structure (1)). Importantly, free energy simulations confirmed A-conf as the most stable Arg61 conformation in wt-RS, located in the deepest energetic minimum of the free-energy surface (Figure 3).
In addition, we found that d-newbond, which specifies the length of the forming bond during catalysis (Figure 2), was shorter (3.35 ± 0.07 Å) when Arg61 assumed A-conf and progressively increased when Arg61 adopted B- (3.52 ± 0.07 Å) or C-conf (3.65 ± 0.05 Å, see Confidence Interval analysis in Supplementary Text). Concomitantly, d-newbond fluctuations correlated with the inter-metal distance (MgA-MgB). Indeed, only when Arg61 adopted A-conf, MgA-MgB length was stably maintained around 3.51 ± 0.07 Å, in fine agreement with the crystallographic evidence (1) and other several other studies (25,26,37,40–43), highlighting the general flexibility of the catalytic metal ions, passing from a pre-reactive to a post-reactive configuration. Also, we found that an increase in d-newbond length is correlated with an increase in inter-metal distance, with respect to the different Arg61 conformations (Figure 2).
Interestingly, the Arg61 conformations along our MD simulations, in both pre- and post-reactive states, match well with two distinct sets of X-ray structures of Pol-η, resolved in complex with damaged DNA (15,44). Zhao et al. (15) have solved a ternary complex formed by Pol-η, a non-hydrolysable incoming nucleotide and a dsDNA containing a lesion caused by cisplatin (45). In the crystallographic pre-reactive state (PDB ID: 4DL4) (15), Arg61 adopts both A- and C-conf (see Supplementary Figure S9). In this crystal, A-conf has an occupancy value of 60%, while C-conf has a lower occupancy of 40%. Here, d-newbond is 3.35 Å. On the other hand, in the post-insertion complex (PDB ID: 4DL6) (15), Arg61 only adopts C-conf, with a longer d-newbond, corresponding to 3.80 Å. Our results also match well with the X-ray structures solved by Patra et al. (44), which report a ternary complex made by Pol-η, a non-hydrolysable incoming nucleotide and a dsDNA affected by the lesion 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxoG) (46–48). Also here, when Arg61 adopts A-conf-like conformations (PDBid 4O3O) (44), d-newbond has a value of 3.13 Å. On the other hand, in a second X-ray structure (PDB ID: 4O3Q) (44), Arg61 assumes C-conf-like conformations and d-newbond increases to a value of 3.71 Å (see Supplementary Figure S9).
In the mutated system mut-RS (Arg61Ala), which was found experimentally to be from 2- to 6-fold less efficient in polymerization (in both normal and damaged DNA) than the wild-type form (2,49), we found a less stable dATP conformation, which was buckled. This caused instability of the Watson–Crick's H-bond interaction pattern (Supplementary Figure S5). As a result, d-newbond fluctuations in mut-RS increase, which is reflected in a wider Gaussian distribution function (Figure 2). Overall, these data suggest that Arg61 is necessary to correctly guide the incoming base into the catalytic site (via B- and C-conf) and to promote (via A-conf) the phosphoryl-transfer reaction, catalyzing the efficient incorporation of the incoming nucleotide into the dsDNA during polymerization. Besides, these results help further rationalizing the outcome of recent MD simulations describing a complex network of local hydrogen-bond interactions, involving Arg61, within the catalytic site of Pol-η. These local interactions thermodynamically favor the selection and binding of specific deoxyribose nucleotides triphosphates in the presence of cyclobutane thymine−thymine dimers (TTDs, a UV-induced DNA damage). As such, they likely contribute to the fidelity and overall efficiency of Pol-η (4,50).
In the mispaired system mp-RS (18), we found that Arg61 was highly stable, never spontaneously leaving its starting C-conf. Even when Arg61 was manually placed into a starting A-conf, the key residue spontaneously reassumed C-conf in the very first ns of unbiased MD (Supplementary Figure S6). This can be explained by the presence of an additional H-bond, which is only formed in C-conf with the dGTP incoming base (Supplementary Figure S1). The preference of C-conf suggests a lower reactivity of this system. This agrees well with the kinetics experiments reporting a drop in Pol-η efficiency (kcat/KM), from 47.80 μM−1 min−1 for the canonical dATP:dT pairing to only 1.70 μM−1 min−1 for this specific dGTP misincorporation (15). Along the same lines, a recent paper of Patra et al. (44) reports kinetics and structural data of Pol-η incorporating different nucleotides (dATP, dTTP, dCTP and dGTP), in presence of the 8-oxoG lesion. The dGTP:8-oxoG incorporation had a low efficiency value, equal to 0.016 ± 0.003 μM−1 s−1. This can be compared with the value of 1.0 ± 0.16 μM−1 s−1 associated with the canonical dCTP:dG incorporation. Notably, Arg61 is in A-conf only in those X-ray structures obtained for the control systems (native base pairing), while Arg61 is in C-conf in those structures having non-native base pairings (see Supplementary Figure S9). Hence, a lower polymerization efficiency for misincorporation may be due to the lack of A-conf formation in pre-reactive states, as observed in our mp-RS dynamics. Free energy simulations of mp-RS also showed the triple-H-bonded C-conf as the deepest energetic basin, while A-conf and B-conf were found in two less stable minima on the FES (Figure 3, mp-RS). This likely prevents the formation of a proper pre-reactive state, as found in wt-RS.
During the MD simulations of the post-reactive two-metal-ion system 2M-PS, we found that C-conf remained the most populated state, being present for ∼62.00% of the simulation time. This suggests that the product formation, where the incoming base is covalently bound to the substrate primer strand, favors C-conf over A-conf. While the latter seems a specific conformation needed in the pre-reactive state, the preference for C-conf in the post-reactive state seems necessary to allow binding of the third ion, as observed experimentally (1,8).
In fact, during the unbiased MD simulations of 2M-PS, a third Mg2+ moved spontaneously to the catalytic pocket at t = ∼370 ns, reconstituting the three-metal-ion system 3M-PS system (Figure 4). The unprompted entry of this transient third ion initially destabilized the pocket environment, with formation of a metastable MgB–MgC–PPi adduct which seemed to evolve toward the exit of the PPi leaving group. However, this metastable intermediate lasted for ∼430 ns. After this time, the third ion spontaneously left the pocket and the leaving MgB–MgC–PPi adduct fell slowly back into its original position, restoring the canonical post-reactive coordination of the catalytic site of Pol-η (1).
Intriguingly, this orchestrated structural motion suggests a putative leaving mechanism of the PPi group that implies a key role of MgC as an exit shuttle, together with MgB and the chelating Arg61, Arg55 and Tyr52. This possible leaving pathway was further suggested by our free energy simulations, which returned an estimated energy barrier of ∼8 kcal mol−1 overcome via a two-step mechanism. First, in 3M-PS, the overall system escaped from the crystallographic post-reactive state, containing three metal ions in the pocket and located at CVPPi = ∼7.50 Å. This first step slowly evolved into a metastable state, in which the MgB–MgC–PPi group was partially unbound, while Arg61 was in A-conf. The second step led to the final exit of the complexed PPi leaving group. Concomitantly, Arg61 rotated along its φ dihedral angle, accompanying the leaving group release (Figure 4).
Using the computed free energy values for the leaving PPi departure and the transition-state theory (51), we could roughly estimate that the unbinding time of the PPi is around ∼100 μs (for further details see Supplementary Text). Experimental results indicate that the Y-family polymerases are low-processivity enzymes (with a rate constant of nucleotide incorporation that often is around ∼1.00–3.00 s−1) (2,44,49). Thus, the leaving PPi departure seems a rather fast step, compared to the time requested for the overall process. For this reason, our results agree with the proposal that the leaving PPi departure is not the rate-limiting step of the overall process (3,44,52,53). The relaxation step of the whole ternary complex, after PPi release, seems more likely to be the rate-limiting step of the polymerization process catalyzed by human Pol-η, as already proposed for the structurally similar Y-family members Dpo4 and Pol-κ (52,53).
Extension to other DNA polymerases
A structural superimposition between Pol-η, Pol-ι (54) and Dpo4 (24), that also are Y-family DNA polymerases (8,11,55,56), highlights the key structural elements shared by these enzymes (Figure 5). Importantly, the conserved Lys77 in the crystallographic pre-reactive Pol-ι complex points down in an A-conf-like conformation, while Lys56 in the crystallographic post-reactive Dpo4 complex points up in a C-conf-like conformation. These different conformations of this positively charged residue agree with our MD simulation findings in Pol-η, where Arg61 adopted mostly A-conf in the reactants and mostly C-conf in the products. Notably, when Arg61 is mutated into a Lys residue in human Pol-η (Supplementary Figure S10), the enzyme retains a slightly lower activity (49). Besides, the presence of a positively charged residue in a similar position of the catalytic site is found in other Y-family polymerases, including Escherichia coli Pol-IV, E. coli Pol-V and human enzymes like Pol-κ and Rev1 (57–63), which also catalyze a two-metal-aided (Mg2+) phosphoryl-transfer reaction to process DNA (25). This suggests its key role in recruiting and processing the (d)NTP/(d)NMP for DNA/RNA elongation/digestion in normal (64–66) and deranged conditions (8,18,29,30,67–70). As for the third transient metal ion, this emerging and still puzzling aspect of two-metal-ion catalysis has been recently reported in several other nucleic-acid-processing enzymes (25) including, recently, DNA polymerase β (32), suggesting a dynamic and functional rearrangement of a third additional metal ion in an extended neighborhood of the two-metal-ion catalytic pocket.
Figure 5.

Structural superimposition of different Y-family members. (A) X-ray structure (PDB ID: 2ALZ) of a pre-reactive configuration of the human Pol-ι enzyme. Lys77 points ‘down’ like Arg61 in A-Conf in Pol-η, establishing H-bond interactions with the phosphate groups of the incoming base. (B) X-ray structure (PDB ID: 2AGO) of a post-reactive configuration in the Sulfolobus solfataricus Dpo4 enzyme. Here, Lys56 adopts an ‘up’ conformation, similar to the C-conf detected in the products of Pol-η catalysis. (C) Superimposition of the three different catalytic sites (Pol-η, Pol-ι and Dpo4).
In conclusion, our results greatly enrich the interpretation of existing structural data on Pol-η and reveal an effective and cooperative mechanism for enzymatic repair of DNA operated by DNA-repairing polymerases. This is based on conformational transitions of a key positively charged residue coupled with a transient third metal ion at the catalytic site of Pol-η, which ultimately acts as an exit shuttle for the leaving PPi departure. Based on the conservation of these key structural features, this efficient mechanism detected in Pol-η may be shared by several DNA polymerases and other binuclear metalloenzymes for nucleic acid processing (25), with relevance for the de-novo design of enzymes for DNA processing or the structure-based discovery of small molecules for anti-cancer therapy that targets DNA polymerases in cancer cells (71–76).
Supplementary Material
Acknowledgments
We thank PRACE for HPC computing time. We thank Grace Fox for proofreading and copyediting the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Italian Association for Cancer Research (AIRC) [‘MFAG n. 14140’]. Funding for open access charge: Istituto italiano di Tecnologia; Italian Association for Cancer Research (AIRC).
Conflict of interest statement. None declared.
REFERENCES
- 1.Nakamura T., Zhao Y., Yamagata Y., Hua Y.J., Yang W. Watching DNA polymerase eta make a phosphodiester bond. Nature. 2012;487:196–201. doi: 10.1038/nature11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biertumpfel C., Zhao Y., Kondo Y., Ramon-Maiques S., Gregory M., Lee J.Y., Masutani C., Lehmann A.R., Hanaoka F., Yang W. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patra A., Banerjee S., Johnson Salyard T.L., Malik C.K., Christov P.P., Rizzo C.J., Stone M.P., Egli M. Structural basis for error-free bypass of the 5-N-Methylformamidopyrimidine-dG lesion by human DNA polymerase eta and Sulfolobus solfataricus P2 polymerase IV. J. Am. Chem. Soc. 2015;137:7011–7014. doi: 10.1021/jacs.5b02701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ucisik M.N., Hammes-Schiffer S. Relative binding free energies of adenine and guanine to damaged and undamaged DNA in human DNA polymerase eta: clues for fidelity and overall efficiency. J. Am. Chem. Soc. 2015;137:13240–13243. doi: 10.1021/jacs.5b08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCulloch S.D., Kokoska R.J., Masutani C., Iwai S., Hanaoka F., Kunkel T.A. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- 6.McCulloch S.D., Kunkel T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruet-Hennequart S., Gallagher K., Sokol A.M., Villalan S., Prendergast A.M., Carty M.P. DNA polymerase eta, a key protein in translesion synthesis in human cells. Subcell. Biochem. 2010;50:189–209. doi: 10.1007/978-90-481-3471-7_10. [DOI] [PubMed] [Google Scholar]
- 8.Yang W. An overview of Y-Family DNA polymerases and a case study of human DNA polymerase eta. Biochemistry. 2014;53:2793–2803. doi: 10.1021/bi500019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodgate R. Evolution of the two-step model for UV-mutagenesis. Mutat. Res. 2001;485:83–92. doi: 10.1016/s0921-8777(00)00076-8. [DOI] [PubMed] [Google Scholar]
- 10.Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reissner T., Chaney S., Friedberg E.C., Wang Z.G., Carell T., et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals (vol 28, pg 383, 2009) EMBO J. 2009;28:992–992. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sale J.E., Lehmann A.R., Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell. Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruet-Hennequart S., Villalan S., Kaczmarczyk A., O'Meara E., Sokol A.M., Carty M.P. Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle. 2009;8:3039–3050. [PubMed] [Google Scholar]
- 13.Ummat A., Rechkoblit O., Jain R., Roy Choudhury J., Johnson R.E., Silverstein T.D., Buku A., Lone S., Prakash L., Prakash S., et al. Structural basis for cisplatin DNA damage tolerance by human polymerase eta during cancer chemotherapy. Nat. Struct. Mol. Biol. 2012;19:628–632. doi: 10.1038/nsmb.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaisman A., Masutani C., Hanaoka F., Chaney S.G. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase eta. Biochemistry. 2000;39:4575–4580. doi: 10.1021/bi000130k. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y., Biertumpfel C., Gregory M.T., Hua Y.J., Hanaoka F., Yang W. Structural basis of human DNA polymerase eta-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. U.S.A. 2012;109:7269–7274. doi: 10.1073/pnas.1202681109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parsons J.L., Nicolay N.H., Sharma R.A. Biological and therapeutic relevance of nonreplicative DNA polymerases to cancer. Antioxid. Redox Signal. 2013;18:851–873. doi: 10.1089/ars.2011.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts S.A., Gordenin D.A. Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer. 2014;14:786–800. doi: 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y., Gregory M.T., Biertumpfel C., Hua Y.J., Hanaoka F., Yang W. Mechanism of somatic hypermutation at the WA motif by human DNA polymerase eta. Proc. Natl. Acad. Sci. U.S.A. 2013;110:8146–8151. doi: 10.1073/pnas.1303126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Q., Clark A.B., McCulloch S.D., Yuan T., Bronson R.T., Kunkel T.A., Kucherlapati R. Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res. 2006;66:87–94. doi: 10.1158/0008-5472.CAN-05-1862. [DOI] [PubMed] [Google Scholar]
- 20.Kulaksiz G., Reardon J.T., Sancar A. Xeroderma pigmentosum complementation group E protein (XPE/DDB2): purification of various complexes of XPE and analyses of their damaged DNA binding and putative DNA repair properties. Mol. Cell. Biol. 2005;25:9784–9792. doi: 10.1128/MCB.25.22.9784-9792.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaney S.G., Campbell S.L., Bassett E., Wu Y. Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit. Rev. Oncol. Hematol. 2005;53:3–11. doi: 10.1016/j.critrevonc.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Tomicic M.T., Aasland D., Naumann S.C., Meise R., Barckhausen C., Kaina B., Christmann M. Translesion polymerase eta is upregulated by cancer therapeutics and confers anticancer drug resistance. Cancer Res. 2014;74:5585–5596. doi: 10.1158/0008-5472.CAN-14-0953. [DOI] [PubMed] [Google Scholar]
- 23.Yang W., Lee J.Y., Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 24.Ling H., Boudsocq F., Woodgate R., Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 25.Palermo G., Cavalli A., Klein M.L., Alfonso-Prieto M., Dal Peraro M., De Vivo M. Catalytic metal ions and enzymatic processing of DNA and RNA. Acc. Chem. Res. 2015;48:220–228. doi: 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- 26.De Vivo M., Dal Peraro M., Klein M.L. Phosphodiester cleavage in ribonuclease H occurs via an associative two-metal-aided catalytic mechanism. J. Am. Chem. Soc. 2008;130:10955–10962. doi: 10.1021/ja8005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L.H., Yu X.Y., Hu P., Broyde S., Zhang Y.K. A water-mediated and substrate-assisted catalytic mechanism for Sulfolobus solfataricus DNA polymerase IV. J. Am. Chem. Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lior-Hoffmann L., Wang L.H., Wang S.L., Geacintov N.E., Broyde S., Zhang Y.K. Preferred WMSA catalytic mechanism of the nucleotidyl transfer reaction in human DNA polymerase kappa elucidates error-free bypass of a bulky DNA lesion. Nucleic Acids Res. 2012;40:9193–9205. doi: 10.1093/nar/gks653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golosov A.A., Warren J.J., Beese L.S., Karplus M. The mechanism of the translocation step in DNA replication by DNA polymerase I: a computer simulation analysis. Structure. 2010;18:83–93. doi: 10.1016/j.str.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergoglio V., Boyer A.S., Walsh E., Naim V., Legube G., Lee M.Y., Rey L., Rosselli F., Cazaux C., Eckert K.A., et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell. Biol. 2013;201:395–408. doi: 10.1083/jcb.201207066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su Y., Patra A., Harp J.M., Egli M., Guengerich F.P. Roles of residues Arg-61 and Gln-38 of human DNA polymerase eta in bypass of deoxyguanosine and 7, 8-Dihydro-8-oxo-2′-deoxyguanosine. J. Biol. Chem. 2015;290:15921–15933. doi: 10.1074/jbc.M115.653691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perera L., Freudenthal B.D., Beard W.A., Shock D.D., Pedersen L.G., Wilson S.H. Requirement for transient metal ions revealed through computational analysis for DNA polymerase going in reverse. Proc. Natl. Acad. Sci. U.S.A. 2015;112:E5228–E5236. doi: 10.1073/pnas.1511207112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 35.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grest G.S., Kremer K. Molecular-dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A. 1986;33:3628–3631. doi: 10.1103/physreva.33.3628. [DOI] [PubMed] [Google Scholar]
- 37.Dal Peraro M., Spiegel K., Lamoureux G., De Vivo M., DeGrado W.F., Klein M.L. Modeling the charge distribution at metal sites in proteins for molecular dynamics simulations. J. Struct. Biol. 2007;157:444–453. doi: 10.1016/j.jsb.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 39.Barducci A., Bussi G., Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 40.Ho M.H., De Vivo M., Dal Peraro M., Klein M.L. Understanding the effect of magnesium ion concentration on the catalytic activity of ribonuclease H through computation: does a third metal binding site modulate endonuclease catalysis. J. Am. Chem. Soc. 2010;132:13702–13712. doi: 10.1021/ja102933y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palermo G., Stenta M., Cavalli A., Dal Peraro M., De Vivo M. Molecular simulations highlight the role of metals in catalysis and inhibition of Type II topoisomerase. J. Am. Chem. Soc. 2013;9:857–862. doi: 10.1021/ct300691u. [DOI] [PubMed] [Google Scholar]
- 42.De Vivo M., Ensing B., Dal Peraro M., Gomez G.A., Christianson D.W., Klein M.L. Proton shuttles and phosphatase activity in soluble epoxide hydrolase. J. Am. Chem. Soc. 2007;129:387–394. doi: 10.1021/ja066150c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vidossich P., Magistrato A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules. 2014;4:616–645. doi: 10.3390/biom4030616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patra A., Nagy L.D., Zhang Q., Su Y., Muller L., Guengerich F.P., Egli M. Kinetics, structure, and mechanism of 8-Oxo-7, 8-dihydro-2′-deoxyguanosine bypass by human DNA polymerase eta. J. Biol. Chem. 2014;289:16867–16882. doi: 10.1074/jbc.M114.551820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wheate N.J., Walker S., Craig G.E., Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 46.Degan P., Shigenaga M.K., Park E.M., Alperin P.E., Ames B.N. Immunoaffinity isolation of urinary 8-Hydroxy-2′-deoxyguanosine and 8-hydroxyguanine and quantitation of 8-hydroxy-2′-deoxyguanosine in DNA by polyclonal antibodies. Carcinogenesis. 1991;12:865–871. doi: 10.1093/carcin/12.5.865. [DOI] [PubMed] [Google Scholar]
- 47.Shimoda R., Nagashima M., Sakamoto M., Yamaguchi N., Hirohashi S., Yokota J., Kasai H. Increased formation of oxidative DNA-damage, 8-hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res. 1994;54:3171–3172. [PubMed] [Google Scholar]
- 48.Fraga C.G., Motchnik P.A., Shigenaga M.K., Helbock H.J., Jacob R.A., Ames B.N. Ascorbic-acid protects against endogenous oxidative DNA damage in human sperm. Proc. Natl. Acad. Sci. U.S.A. 1991;88:11003–11006. doi: 10.1073/pnas.88.24.11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katafuchi A., Sassa A., Niimi N., Gruz P., Fujimoto H., Masutani C., Hanaoka F., Ohta T., Nohmi T. Critical amino acids in human DNA polymerases eta and kappa involved in erroneous incorporation of oxidized nucleotides. Nucleic Acids Res. 2010;38:859–867. doi: 10.1093/nar/gkp1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ucisik M.N., Hammes-Schiffer S. Comparative molecular dynamics studies of human DNA polymerase eta. J. Chem. Inf. Model. 2015;55:2672–2681. doi: 10.1021/acs.jcim.5b00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laidler K., King C. Development of transition-state theory. J. Phys. Chem. 1983;15:2657–2664. [Google Scholar]
- 52.Zhao L., Pence M.G., Eoff R.L., Yuan S., Fercu C.A., Guengerich F.P. Elucidation of kinetic mechanisms of human translesion DNA polymerase kappa using tryptophan mutants. FEBS J. 2014;281:4394–4410. doi: 10.1111/febs.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beckman J.W., Wang Q., Guengerich F.P. Kinetic analysis of correct nucleotide insertion by a Y-family DNA polymerase reveals conformational changes both prior to and following phosphodiester bond formation as detected by tryptophan fluorescence. J. Biol. Chem. 2008;283:36711–36723. doi: 10.1074/jbc.M806785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nair D.T., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Human DNA polymerase iota incorporates dCTP opposite template G via a G.C + Hoogsteen base pair. Structure. 2005;13:1569–1577. doi: 10.1016/j.str.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 55.Maxwell B.A., Suo Z.C. Recent insight into the kinetic mechanisms and conformational dynamics of Y-family DNA polymerases. Biochemistry. 2014;53:2804–2814. doi: 10.1021/bi5000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pata J.D. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta. 2010;1804:1124–1135. doi: 10.1016/j.bbapap.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bunting K.A., Roe S.M., Pearl L.H. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang Q., Karata K., Woodgate R., Cox M.M., Goodman M.F. The active form of DNA polymerase V is UmuD’(2)C-RecA-ATP. Nature. 2009;460:359–363. doi: 10.1038/nature08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uljon S.N., Johnson R.E., Edwards T.A., Prakash S., Prakash L., Aggarwal A.K. Crystal structure of the catalytic core of human DNA polymerase kappa. Structure. 2004;12:1395–1404. doi: 10.1016/j.str.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 60.Lone S., Townson S.A., Uljon S.N., Johnson R.E., Brahma A., Nair D.T., Prakash S., Prakash L., Aggarwal A.K. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 61.Liu Y., Yang Y., Tang T.S., Zhang H., Wang Z., Friedberg E., Yang W., Guo C. Variants of mouse DNA polymerase kappa reveal a mechanism of efficient and accurate translesion synthesis past a benzo[a]pyrene dG adduct. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1789–1794. doi: 10.1073/pnas.1324168111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nair D.T., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- 63.Swan M.K., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Structure of the human Rev1-DNA-dNTP ternary complex. J. Mol. Biol. 2009;390:699–709. doi: 10.1016/j.jmb.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reich S., Guilligay D., Pflug A., Malet H., Berger I., Crepin T., Hart D., Lunardi T., Nanao M., Ruigrok R.W.H., et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature. 2014;516:361–366. doi: 10.1038/nature14009. [DOI] [PubMed] [Google Scholar]
- 65.Balzarini J., Das K., Bernatchez J.A., Martinez S.E., Ngure M., Keane S., Ford A., Maguire N., Mullins N., John J., et al. Alpha-carboxy nucleoside phosphonates as universal nucleoside triphosphate mimics. Proc. Natl. Acad. Sci. U.S.A. 2015;112:3475–3480. doi: 10.1073/pnas.1420233112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Appleby T.C., Perry J.K., Murakami E., Barauskas O., Feng J., Cho A., Fox D., Wetmore D.R., McGrath M.E., Ray A.S., et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science. 2015;347:771–775. doi: 10.1126/science.1259210. [DOI] [PubMed] [Google Scholar]
- 67.Vaisman A., Ling H., Woodgate R., Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Broughton B.C., Cordonnier A., Kleijer W.J., Jaspers N.G., Fawcett H., Raams A., Garritsen V.H., Stary A., Avril M.F., Boudsocq F., et al. Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc. Natl. Acad. Sci. U.S.A. 2002;99:815–820. doi: 10.1073/pnas.022473899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Broyde S., Wang L., Rechkoblit O., Geacintov N.E., Patel D.J. Lesion processing: high-fidelity versus lesion-bypass DNA polymerases. Trends Biochem. Sci. 2008;33:209–219. doi: 10.1016/j.tibs.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franklin M.C., Wang J., Steitz T.A. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 71.Nilforoushan A., Furrer A., Wyss L.A., van Loon B., Sturla S.J. Nucleotides with altered hydrogen bonding capacities impede human DNA polymerase eta by reducing synthesis in the presence of the major cisplatin DNA adduct. J. Am. Chem. Soc. 2015;137:4728–4734. doi: 10.1021/ja512547g. [DOI] [PubMed] [Google Scholar]
- 72.De Vivo M. Bridging quantum mechanics and structure-based drug design. Front. Biosci. (Landmark Ed) 2011;16:1619–1633. doi: 10.2741/3809. [DOI] [PubMed] [Google Scholar]
- 73.Lodola A., De Vivo M. The increasing role of QM/MM in drug discovery. Adv. Protein Chem. Struct. Biol. 2012;87:337–362. doi: 10.1016/B978-0-12-398312-1.00011-1. [DOI] [PubMed] [Google Scholar]
- 74.Srivastava A.K., Han C., Zhao R., Cui T., Dai Y., Mao C., Zhao W., Zhang X., Yu J., Wang Q.E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 2015;112:4411–4416. doi: 10.1073/pnas.1421365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelley M.R., Fishel M.L. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med. Chem. 2008;8:417–425. doi: 10.2174/187152008784220294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chou K.M. DNA polymerase eta and chemotherapeutic agents. Antioxid. Redox Signal. 2011;14:2521–2529. doi: 10.1089/ars.2010.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.