

Abstract
Type 2 diabetes mellitus is a complex metabolic disease, and cardiovascular disease is a leading complication of diabetes. Epicardial adipose tissue surrounding the heart displays biochemical, thermogenic, and cardioprotective properties. However, the metabolic cross-talk between epicardial fat and the myocardium is largely unknown. This study sought to understand epicardial adipose tissue metabolism from heart failure patients with or without diabetes. We aimed to unravel possible differences in glucose and lipid metabolism between human epicardial and subcutaneous adipocytes and elucidate the potential underlying mechanisms involved in heart failure. Insulin-stimulated [14C]glucose uptake and isoproterenol-stimulated lipolysis were measured in isolated epicardial and subcutaneous adipocytes. The expression of genes involved in glucose and lipid metabolism was analyzed by reverse transcription-polymerase chain reaction in adipocytes. In addition, epicardial and subcutaneous fatty acid composition was analyzed by high-resolution proton nuclear magnetic resonance spectroscopy. The difference between basal and insulin conditions in glucose uptake was significantly decreased (P = 0.006) in epicardial compared with subcutaneous adipocytes. Moreover, a significant (P < 0.001) decrease in the isoproterenol-stimulated lipolysis was also observed when the two fat depots were compared, and it was strongly correlated with lipolysis, lipid storage, and inflammation-related gene expression. Moreover, the fatty acid composition of these tissues was significantly altered by diabetes. These results emphasize potential metabolic differences between both fat depots in the presence of heart failure and highlight epicardial fat as a possible therapeutic target in situ in the cardiac microenvironment.
Keywords: epicardial adipose tissue, heart failure, diabetes, glucose uptake, lipolysis
western lifestyle contributes greatly to the prevalence of overweight and obesity, which are associated with metabolic and cardiovascular disorders, including type 2 diabetes mellitus (DM), hypertension, and coronary heart disease (84). Cardiovascular disease remains the leading cause of death in particular among people with DM (56). In 2014, there were 387 million people with DM, and it is projected to be 592 million by 2035 (81).
The prevalence of heart failure (HF) has been increasing (82), and despite advances in therapy and management, it remains deadly, resulting in high mortality rates that rival those of many cancers (9). Almost two-thirds of HF patients have abnormal glucose homeostasis (74). A marker of DM-impaired metabolism in HF patients is the switch from carbohydrates and fatty acids (FA) as sources of energy to preferentially FA (74).
Adipose tissue (AT) has been identified as a dynamic endocrine organ (31, 83) and metabolic sensor, regulating insulin sensitivity, glucose, and lipid metabolism as well as cardiovascular homeostasis (14). Regional, developmental, structural, and functional variations in fat depots exist (31, 83). Epicardial AT (EAT) is a visceral fat depot around the heart located directly over the myocardium (14). EAT is metabolically active, producing hormones and adipokines and locally modulating cardiac structure and function (31, 83). EAT shares the same microcirculation with the myocardium (15), providing energy directly to cardiomyocytes (83), influencing cardiac angiogenesis and immunity, and providing thermal insulation and mechanical protection to the myocardium (83). When deregulated, EAT may play adverse paracrine roles in cardiac arrhythmias and lipotoxic cardiomyopathies (59) as well as invade the outer region of the adventia in the vascular wall (69). The amount of EAT correlates with insulin resistance, metabolic syndrome (51), and carotid stiffness in hypertensive obese patients (55), contributing to cardiovascular disease (83). Moreover, EAT thickness reflects intra-abdominal visceral (32, 33) and intramyocardial lipid content (49). Thus, EAT volume may play a role in stratification of the cardiometabolic risk and could serve as a therapeutic target (32, 33).
In addition, nonesterified FA profiles are different in EAT compared with subcutaneous adipose tissue (SAT), whereas the saturated FA content is higher and the unsaturated FA content is lower (62).
In EAT, glucose utilization is lower compared with intra-abdominal fat (50, 71), and insulin action is impaired (71), resulting in decreased glucose utilization as a substrate in HF patients. However, the rate of insulin-stimulated glucose uptake in EAT cells from HF patients is unknown.
Furthermore, EAT from HF patients expresses lower levels of fatty acid-binding protein 4 (FABP4) (72) and fat-mobilizing genes (35). In addition, loss of diacylglycerol O-acyltransferase 1 (DGAT1) activity in heart muscle reduces gene expression involved in free FA uptake, such as CD36, reducing toxic lipid accumulation in the heart (47). This indicates that entry of long-chain FA (LCFA) into the heart is tightly coupled to both triglyceride (TG) synthesis and lipolysis (11). Moreover, this is consistent with a model of reciprocal regulation between LCFA uptake by CD36, TG synthesis by DGAT1, and TG lipolysis by ATGL, working to buffer the intracellular availability of LCFA while preventing the accumulation of potentially toxic or physiologically active metabolites (11). However, isoproterenol-stimulated lipolysis in EAT from HF patients has not been studied.
Although there are some studies in EAT from HF patients, the metabolic cross-talk between EAT and the myocardium in these patients is largely unknown, and metabolic studies are scarce. Therefore, the main and novel purpose of this study was to evaluate glucose uptake and lipid metabolism in EAT in the presence and absence of DM and to clarify underlying mechanisms that might be implicated in HF by analyzing the expression of genes that regulate glucose and lipid metabolism and how these impact inflammation.
MATERIALS AND METHODS
AT donors.
This study included 158 subjects with HF; 95 were nondiabetic (NDM; 67 male patients), and 63 had DM (50 male patients). Demographic and clinical characteristics are shown in Table 1, whereas biochemical characteristics are in Table 2. Paired SAT and EAT biopsies were obtained during elective coronary artery bypass grafting [coronary artery disease (CAD)], valvular replacement, or valvuloplasty [non-coronary artery disease (NCAD)] surgery. EAT was extracted from the proximal right artery and SAT from the sternum region. The study was performed after written consent was obtained from participants, and it was approved by the Ethics Committee of the University Hospital of Coimbra. Studies were carried out in accordance with the Declaration of Helsinki.
Table 1.
Demographics and clinical characteristics of the study population
| Nondiabetic Patients | Diabetic Patients | P Value | |
|---|---|---|---|
| No. of patients (%) | 95 (60.1) | 63 (39.9) | |
| Male, % | 70.5 | 79.4 | 0.215 |
| Age, yr | 64.23 ± 1.37 | 69.17 ± 1.05 | 0.058 |
| BMI, kg/m2 | 26.01 ± 0.32 | 27.62 ± 0.36 | 0.001*** |
| Risk factors, % | |||
| Hypertension | 65.4 | 85.7 | 0.004** |
| Dyslipidemia | 60.5 | 79.6 | 0.011* |
| Blood pressure, mm/Hg | |||
| Systolic | 128.7 ± 2.26 | 132.3 ± 2.72 | 0.317 |
| Diastolic | 70.49 ± 1.41 | 73.00 ± 1.61 | 0.257 |
| Medications, % | |||
| Antiplatelet | 63.0 | 79.6 | 0.030* |
| Antiarrhythmic | 12.3 | 14.3 | 0.764 |
| Anticoagulant | 34.6 | 22.4 | 0.092 |
| Antidiabetic | |||
| Insulin | 32.7 | <0.001*** | |
| Biguanide | 34.7 | <0.001*** | |
| α-Glucosidase inhibitor | 2.0 | 0.136 | |
| DPP-4 inhibitor | 12.2 | <0.001*** | |
| DPP-4 inhibitor + biguanide | 28.6 | <0.001*** | |
| Sulfonylurea | 24.5 | <0.001*** | |
| Sulfonylurea + biguanide | 2.0 | 0.218 | |
| β-Blocker | 55.6 | 69.4 | 0.076 |
| Calcium channel blocker | 8.6 | 22.5 | 0.014* |
| ACEI | 49.4 | 53.1 | 0.720 |
| Diuretic | 88.9 | 85.7 | 0.616 |
| Electrolyte, KCl | 71.6 | 65.3 | 0.387 |
| Statin | 55.6 | 77.6 | 0.005** |
| Vasodilator | 12.3 | 22.5 | 0.111 |
| Non-CAD patients | |||
| No. of patients (%) | 45 (47.4) | 20 (31.7) | |
| Male, % | 53.3 | 60.0 | 0.618 |
| Age, yr | 62.00 ± 2.27 | 73.15 ± 1.63 | 0.003** |
| BMI, kg/m2 | 25.47 ± 0.54 | 28.30 ± 0.88 | 0.007** |
| NYHA functional classification of heart failure, % | |||
| I; I–II | 7.9; 2.6 | 11.8; 0 | 0.886; 0.502 |
| II; II–III | 50.0; 18.4 | 58.8; 5.9 | 0.454; 0.169 |
| III; III–IV | 15.8; 5.3 | 17.6; 0 | 0.659; 0.338 |
| IV | 5.9 | 0.002** | |
| CCS functional classification of angina, % | |||
| I; I–II | 13.2; 5.3 | 23.5; 0 | 0.247; 0.338 |
| II; II–III | 52.6; 7.9 | 58.8; 5.9 | 0.618; 0.587 |
| III; III–IV | 13.2; 7.8 | 11.8; 0 | 0.706; 0.169 |
| Operation type, % | |||
| Valvular replacement | 86.8 | 94.1 | 0.317 |
| Valvuloplasty | 52.6 | 35.3 | 0.172 |
| LVEF, % | |||
| <50% | 7.9 | 17.6 | 0.208 |
| ≥50% | 92.1 | 82.4 | 0.208 |
| LVSF, % | |||
| <27% | 10.5 | 17.6 | 0.338 |
| ≥27% | 89.5 | 82.4 | 0.338 |
| Heart rate, beats/min | 72.45 ± 2.49 | 72.41 ± 6.33 | 0.314 |
| CAD patients | |||
| No. of patients (%) | 50 (52.6) | 43 (68.3) | |
| Male, % | 86.0 | 88.4 | 0.734 |
| Age, yr | 66.24 ± 1.58 | 67.33 ± 1.26 | 0.939 |
| BMI, kg/m2 | 26.50 ± 0.34 | 27.30 ± 0.34 | 0.100 |
| NYHA functional classification of heart failure, % | |||
| I; I–II | 20.9; 11.6 | 3.1; 0 | 0.009**; 0.019* |
| II; II–III | 41.9; 11.6 | 62.5; 9.4 | 0.102; 0.675 |
| III; III–IV | 9.3; 4.7 | 25.0; 0 | 0.047*; 0.185 |
| CCS Functional classification of angina, % | |||
| I; I–II | 27.9; 9.3 | 9.4; 3.1 | 0.023*; 0.133 |
| II; II–III | 34.9; 9.3 | 59.4; 6.2 | 0.011*; 0.604 |
| III; III–IV | 13.9; 4.7 | 21.9; 0 | 0.377; 0.185 |
| Operation type, % | |||
| 1-Vessel disease | 25.6 | 12.5 | 0.080 |
| 2-Vessel disease | 18.6 | 12.5 | 0.392 |
| 3-Vessel disease | 48.8 | 68.8 | 0.034* |
| 4-Vessel disease | 2.3 | 6.3 | 0.238 |
| Aneurysm | 7.0 | 3.1 | 0.235 |
| LVEF, % | |||
| <50% | 16.7 | 21.9 | 0.540 |
| ≥50% | 83.3 | 78.1 | 0.540 |
| LVSF, % | |||
| <27% | 12.2 | 25.0 | 0.091 |
| ≥27% | 87.8 | 75.0 | 0.091 |
| Heart rate, beats/min | 71.33 ± 3.86 | 76.38 ± 5.45 | 0.439 |
Quantitative measurements are presented as means ± SE, and qualitative parameters are presented as percentage. BMI, body mass index; ACEI, angiotensin-converting enzyme inhibitor; CAD, coronary artery disease; NYHA, New York Heart Association; CCS, Canadian Cardiovascular Society; DPP-4, dipeptidyl peptidase-4; LVEF, left ventricle ejection fraction; LVSF, left ventricle shortening fraction. For normally distributed data, a parametric t-test was performed, whereas a nonparametric Mann-Whitney test was applied for nonnormally distributed data. P <0.05 was considered significant. For categorical variables, a χ2 test was applied.
P ≤ 0.05;
P ≤ 0.01;
P ≤ 0.001.
Table 2.
Biochemical characteristics of the study population
| Nondiabetic Patients | Diabetic Patients | P Value | |
|---|---|---|---|
| Metabolic biochemical parameters | |||
| Fasting glucose, mg/dl | 96.86 ± 1.78 | 129.0 ± 4.85 | <0.0001*** |
| Fasting insulin, mU/l | 5.40 ± 0.66 | 10.95 ± 2.28 | 0.417 |
| Fasting C-peptide, ng/ml | 2.14 ± 0.09 | 2.11 ± 0.12 | 0.831 |
| HOMA-IR, units | 1.32 ± 0.16 | 3.94 ± 0.88 | 0.013* |
| HOMA-β, units | 64.44 ± 7.97 | 65.32 ± 13.45 | 0.017* |
| QUICKI, units | 0.40 ± 0.01 | 0.38 ± 0.01 | 0.063 |
| Kidney biochemical parameters, mg/dl | |||
| Urea | 21.83 ± 0.88 | 25.58 ± 1.61 | 0.145 |
| Creatinine | 1.07 ± 0.13 | 1.10 ± 0.06 | 0.052 |
| Liver biochemical parameters, U/l | |||
| GOT | 27.47 ± 1.29 | 24.47 ± 1.93 | 0.183 |
| GPT | 28.47 ± 2.42 | 26.33 ± 3.03 | 0.585 |
| GGT | 37.90 ± 3.39 | 38.08 ± 6.29 | 0.334 |
| ALP | 67.36 ± 2.51 | 78.29 ± 4.18 | 0.054 |
| LDH | 212.7 ± 6.83 | 196.8 ± 11.60 | 0.017* |
| Non-CAD patients | |||
| INR, units | 1.40 ± 0.07 | 1.53 ± 0.159 | 0.402 |
| CK, U/l | 134.2 ± 19.90 | 110.2 ± 20.39 | 0.464 |
| CAD patients | |||
| INR, units | 1.26 ± 0.09 | 1.21 ± 0.06 | 0.740 |
| CK, U/l | 113.0 ± 12.99 | 88.71 ± 15.30 | 0.230 |
Data are presented as means ± SE. HOMA-IR, homeostatic model assessment-insulin resistance index; HOMA-β, HOMA-β-cell function; QUICKI, quantitative insulin sensitivity check index; GOT, glutamic oxaloacetic transaminase; GPT, glutamic pyruvic transaminase; GGT, γ-glutamyl transferase; ALP, alkaline phosphatase; LDH, lactate dehydrogenase; INR, international normalized ratio; CK, creatine kinase. For normally distributed data, a parametric t-test was performed, whereas a nonparametric Mann-Whitney test was applied for nonnormally distributed data. P <0.05 was considered significant.
P ≤ 0.05;
P ≤ 0.001.
Blood tests.
Glucose levels were measured with an Accu-Chek Performa glucometer (Roche Diagnostics, Indianapolis, IN). Serum and plasma samples were stored at −80°C for metabolic assays. C-peptide and ultrasensitive insulin ELISA kits were obtained from Mercodia (Uppsala, Sweden).
Chemicals.
Collagenase type II was from Roche (Lisbon, Portugal). d-[U-14C]glucose (250 mCi·mmol−1·l−1) was from Scopus Research (Wageningen, The Netherlands). Human insulin (Actrapid) was kindly supplied by Novo Nordisk (Paço de Arcos, Portugal). N-heptane was from Merck (Whitehouse Station, NJ). Optiphase Hisafe was from Perkin-Elmer (Waltham, MA). Adipocyte lipolysis kits were from Zen Bio, (Research Triangle Park, NC). RNeasy MiniKits were from Qiagen Sciences (Germantown, MD). High Capacity cDNA Reverse Transcriptase kits were from Applied Biosystems (Forest City, CA). PCR primers were designed using Beacon Designer software and synthesized by IDT-Integrated DNA Technologies (BVBA, Leuven, Belgium). SYBR Green Supermix was from Quanta Biosciences (Gaithersburg, MD). All other reagents were from Sigma (St. Louis, MO).
Cell size, glucose uptake, and lipolysis in isolated human adipocytes.
SAT and EAT biopsies were digested with collagenase, and subsequent adipocyte size and weight were measured as reported previously (61).
Insulin-stimulated d-[U-14C]glucose uptake in isolated adipocytes was assessed as reported previously (61). Briefly, surgical subcutaneous and EAT biopsies were immediately cut into small pieces and digested with collagenase type II from Clostridium histolyticum in 6 mM glucose Krebs-Ringer HEPES (KRH) buffer for 60 min at 37°C in a shaking water bath. The resulting cell suspension was isolated from the undigested tissue by filtration through a 250-μm nylon mesh and washed four times in medium without glucose. KRH buffer was prepared with 4% bovine serum albumin (BSA), 140 mM sodium chloride (NaCl), 4.7 mM potassium chloride (KCl), 1.25 mM magnesium sulfate (MgSO4), 1.26 mM calcium chloride (CaCl2), 5.8 mM sodium phosphate (NaH2PO4), 200 nM adenosine deaminase, and 25 mM HEPES, pH 7.4, adjusted with NaOH. Then, the isolated adipocytes were diluted 10 times in KRH buffer without glucose and stimulated or not with human insulin (1,000 μU/ml) for 15 min in a shaking water-bath. Subsequently, 0.86 μM d-[U-14C]glucose was added, and after 45 min, the cell suspension was transferred to prechilled tubes containing silicone oil, allowing the cells to be separated from the buffer by centrifugation. Cell-associated radioactivity was determined by liquid scintillation counting, allowing us to determinate the rate of transmembrane glucose transport, which was calculated according to the following formula: cellular clearance of medium glucose = (counts/min cells × volume)/(counts/min medium × cell number × time) (13).
Lipolysis was also performed as reported previously (60). Briefly, the adipocyte suspension was incubated in the presence or absence of insulin (1,000 μU/ml) in KRH buffer containing 6 mM glucose in a gently shaking water bath at 37°C for 120 min. The medium was supplemented or not with isoproterenol (1 μM) for 120 min. Adipocytes were separated from the medium by centrifugation, and secreted glycerol levels were measured in the extracellular medium using a lipolysis assay kit.
Adipose tissue gene expression.
RNA from SAT and EAT was isolated using the RNeasy MiniKit, and its concentration was determined by OD260 measurement using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Waltham, MA). cDNA was synthesized using the Applied Biosystems High Capacity cDNA Reverse Transcriptase kit. Reverse transcription-polymerase chain reaction was then performed in a Bio-Rad iCycler iQ5. The presence of specific gene products was confirmed with melting curve analysis and electrophoresis to confirm product size. Biopsies were too small to measure corresponding protein expression.
Proton nuclear magnetic resonance spectroscopy.
Forty-nine SAT and 44 EAT biopsies were subjected to high-resolution, magic-angle, spinning-proton nuclear magnetic resonance (HR-MAS 1H-NMR) spectroscopy. Tissues (∼15 mg) were inserted into a zirconium rotor with 10 μl of 50 mM trimethylsilyl propionate in D2O, and NMR analysis performed on a Brucker Avance 800 spectrometer using a 4-mm HR-MAS probe (77). Spectra were processed with NutsPro (NMR Utility Transform Software Professional; Acorn, Livermore, CA), as reported previously (77). The TG composition was calculated as described previously (75). The fractions of unsaturated, saturated, and diunsaturated FA were calculated as reported previously (66). The fraction of monounsaturated FA was calculated as FAmonounsat = FAunsat − FAdiunsat
The fraction of saturated FA determined by NMR agrees with previous analysis obtained by gas chromatography-mass spectrometry (39). However, in the NMR analysis of intact adipose tissue biopsies, the fraction of monounsaturated FA is underestimated, whereas that of diunsaturated FA is slightly overestimated (66).
Statistical analyses.
Statistical tests were performed in SPSS (version 21) with significance at 0.05. Moderate outliers, shown as circles in box-and-whisker plots in all of the figures, lie more than one and a half times the interquartile range (IQR), that is, below Q1 − 1.5 × IQR or above Q3 + 1.5 × IQR. Extreme outliers, shown as stars in box-and-whisker plots in all of the figures, lie more than three times the IQR, that is, below Q1 − 3 × IQR or above Q3 + 3 × IQR. For normality, the Kolmogorov-Smirnov test was performed. To assess the role of tissue type or disease status on quantitative variables, nonparametric two-way ANOVA and multivariate ANOVA tests were performed. The Spearman correlation coefficient was also used. To compare variables related to different stimuli, Wilcoxon tests were used when one stimulus was to be compared with basal, and Friedman tests were used when several stimuli were compared with basal. In the latter case, post hoc Wilcoxon tests were conducted when adequate, with Dunn's test for multiple comparison correction. For lipid metabolism gene expression, we eliminated variables with >10% missing values. The remaining missing values were replaced by the mean value of the corresponding variable. To take into account the correlated values, the expression of individual genes were averaged out (using the mean value) into two variables, the first being lipid storage [variables: DGAT1, FABP4, peroxisome proliferator-activated receptor-γ (PPARG), sterol regulatory element-binding transcription factor 1 (SREBP1), cluster of differentiation 36 (CD36), fatty acid synthase (FASN), lipin-1 (LPIN1), microsomal triglyceride transfer protein large subunit (MTTP), lipoprotein lipase (LPL), stearoyl-CoA desaturase (SCD1)] and the second being lipolysis [variables: hormone-sensitive lipase (HSL), perilipin 1 (PLIN1), aquaporin-7 (AQP7), β1-adrenergic receptor (B1AR), protein kinase A (PKA)]. For NMR data, variables with >10% missing values were eliminated.
RESULTS
Characterization of AT donors.
Of the 95 NDM subjects, 45 had NCAD, and most presented levels II and II–III in the New York Heart Association (NYHA) functional classification of HF, level II in the Canadian Cardiovascular Society (CCS) functional classification of angina, left ventricle ejection fraction (LVEF) ≥50%, and left ventricle shortening fraction (LVSF) ≥27%, as observed in Table 1. The remaining 50 NDM subjects had CAD, and most had levels I and II in the NYHA functional classification, levels I and II in the CCS functional classification of angina, LVEF ≥50%, and LVSF ≥27%. Among the NDM CAD patients, most of them presented three-vessel CAD, as indicated in Table 1.
Of the 63 DM patients, 20 had NCAD, and presented mostly level II in the NYHA functional classification, level II in the CCS functional classification of angina, LVEF ≥50%, and LVSF ≥27%. The remaining 43 NDM patients had CAD and presented mostly level II in the NYHA functional classification, level II in the CCS functional classification of angina, LVEF ≥50%, and LVSF ≥27%, as described in Table 1. Among the DM CAD patients, most of them presented three-vessel CAD.
Moreover, DM subjects exhibited a significantly higher body mass index (BMI) and other comorbidities, namely hypertension and dyslipidemia (Table 1).
DM subjects presented a significantly higher homeostatic model assessment-insulin resistance index, fasting blood glucose, and HOMA-β cell function compared with NDM subjects (Table 2).
Subcutaneous and epicardial adipocyte size and weight in HF patients.
A strong correlation was found between cell weight and cell size [ρ(106) = 0.925, P < 0.001; data not shown]. In fact, the relationship between cell weight and cell size was maintained for both depots and subject groups when they were considered independently. Spearman correlations for each group were SAT + NDM [ρ(26) = 0.929, P < 0.001], SAT + DM [ρ(27) = 0.903, P < 0.001], EAT + NDM [ρ(26) = 0.872, P < 0.001], and EAT + DM [ρ(27) = 0.945, P < 0.001] (Fig. 1, A–C). Cell size was not significantly different (Fig. 1D). The effect of disease status on cell size was nearly significant [F(1, 102) = 3.738, P = 0.056; Fig. 1E], whereas the effect of tissue type on cell size was significant [F(1, 102) = 6.787, P = 0.011; Fig. 1F], indicating that cell size is significantly different between EAT (median = 83.00) and SAT (median = 97.70) (Fig. 1F). Moreover, the interaction effect between tissue type and disease status (DM and NDM) yielded an F ratio of F(1, 102) = 0.004, P = 0.951, indicating that there is no dependence of the variables.
Fig. 1.

Adipocyte size (μm) and weight (μg) in subcutaenous (SAT) and epicardial adipose tissue (EAT) from heart failure (HF) patients with or without diabetes mellitus (DM) (no. of patients is indicated in parentheses). A: scatter plot displaying cell size as a function of the cell weight. Different colors represent each of four groups [SAT + nondiabetic (NDM), SAT + DM, EAT + NDM, and EAT + DM]. B: scatter plot displaying cell size as a function of the cell weight based on disease status. C: scatter plot displaying cell size as a function of the cell weight based on tissue type. D: adipocyte size (μm) in SAT and EAT from HF patients with or without DM. E: adipocyte size based on disease status. F: adipocyte size based on tissue type. G: representative images of hematoxylin and eosin-stained fat depots. Magnification, ×200. Scale bar, 10 μm. Statistical analysis: in A, correlation between cell size and cell weight was evaluated using the Spearman correlation coefficient. SAT + NDM: ρ(26) = 0.929, P < 0.001; SAT + DM: ρ(27) = 0.903, P < 0.001; EAT + NDM: ρ(26) = 0.872, P < 0.001; EAT + DM: ρ(27) = 0.945, P < 0.001. In E and F, cell size was compared between factors (disease status and tissue type) using a nonparametric 2-way ANOVA. P value is as indicated. Moderate outliers, shown as circles in box-and-whisker plots in this and the rest of the figures, lie more than one and a half times the interquartile range (IQR), that is, below Q1 − 1.5 × IQR or above Q3 + 1.5 × IQR. Extreme outliers, shown as stars in box-and-whisker plots in this and the rest of the figures, lie greater than three times the IQR, that is, below Q1 − 3 × IQR or above Q3 + 3 × IQR.
Figure 1G illustrates histological features of SAT and EAT from one representative NCAD NDM patient. Hematoxylin and eosin (H & E) staining illustrates that SAT cells have a larger diameter than EAT cells.
Glucose uptake in SAT and EAT of HF patients.
Insulin-stimulated glucose uptake in isolated SAT and EAT was evaluated in 26 NDM (17 CAD and 9 NCAD) and 27 DM (20 CAD and 7 NCAD) patients. Glucose uptake in each group was significantly elevated compared with basal (no insulin, P < 0.001; Fig. 2A). No differences were observed when comparing the difference between basal and insulin conditions in nondiabetic and diabetic patients [F(1,102) = 0.023, P = 0.880; Fig. 2B]; however, there were differences when comparing tissues [F(1,102) = 7.892, P = 0.006], indicating a significant decrease in glucose uptake in EAT (median = 5.83) compared with SAT (median = 8.06) (Fig. 2C). The interaction effect yielded an F ratio of F(1, 102) = 4.430, P = 0.038, indicating differences between basal and insulin conditions within DM patients between EAT (median = 6.13) and SAT (median = 8.06) and within NDM patients between EAT (median = 4.23) and SAT (median = 8.64).
Fig. 2.
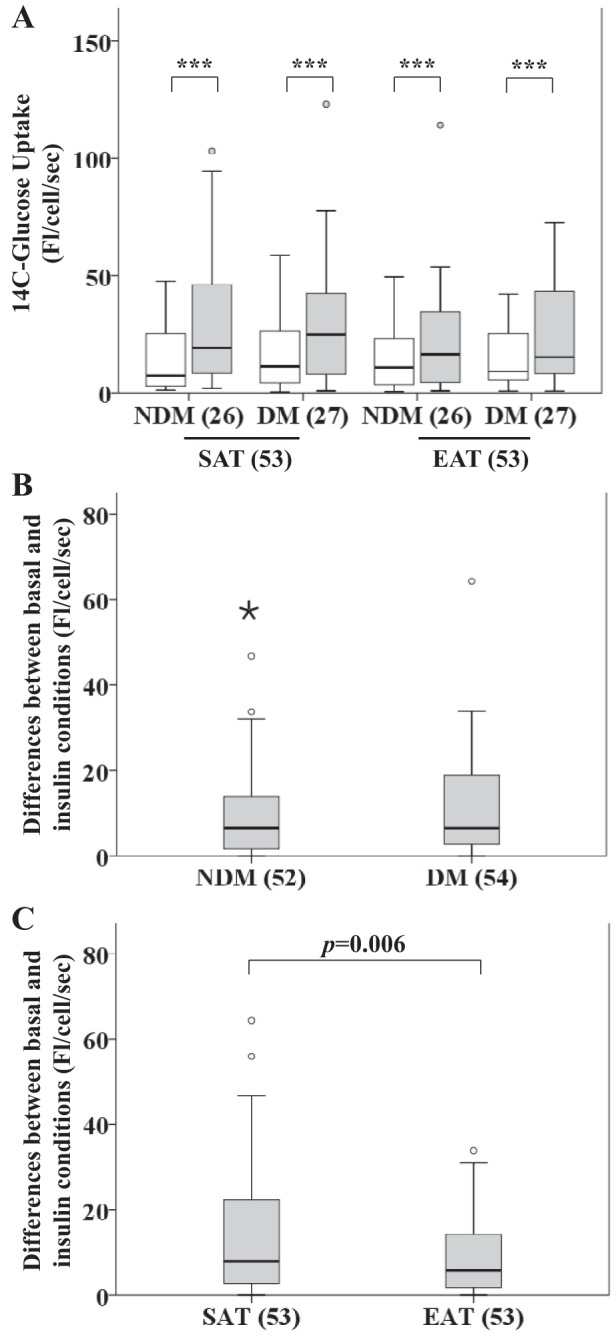
Insulin-stimulated glucose uptake in SAT and EAT from HF patients with or without DM. No. of patients is indicated in parentheses. A: glucose uptake in SAT and EAT from the same patients. B: difference between basal and insulin conditions based on disease status. C: difference between basal and insulin conditions based on tissue type. Statistical analysis: for each tissue type and disease status in A, differences between basal measurements of glucose uptake and corresponding measurements when insulin was used were assessed using Wilcoxon tests. ***P < 0.001. For a better visualization of the results, we excluded a value in the insulin-stimulated condition in the EAT + DM group (200.06). In B, the difference between basal and insulin conditions was compared based on disease status using a nonparametric 2-way ANOVA. For a better visualization of the results, we excluded a value in the DM group (110.83). In C, the difference between basal and insulin conditions was compared based on tissue type using a nonparametric 2-way ANOVA. P value is as indicated. For a better visualization of the results, we excluded a value in the EAT group (110.83). Open bars, basal; gray bars, insulin.
Glucose metabolism gene expression in SAT and EAT from HF patients.
Protein-tyrosine phosphatase 1B (PTP1B), glucose transporter 4 (GLUT4), insulin receptor substrate 1 (IRS1), insulin receptor substrate 2 (IRS2), adiponectin (ADIPOQ), and leptin (LEP) gene expression were compared between disease status and tissue type. The main effect of disease status was not statistically significant, with χ2(6) = 6.494, n = 35, and P = 0.370, whereas effect of tissue type was nearly significant, with χ2(6) = 12.478, n = 35, and P = 0.052 (Fig. 3). Relative expression of the different genes was normalized to β-actin expression levels.
Fig. 3.

Glucose metabolism-related gene expression [Protein-tyrosine phosphatase 1B (PTP1B), glucose transporter 4 (GLUT4), insulin receptor substrate 1 (IRS1), IRS2, adiponectin (ADIPOQ), and leptin (LEP)] in SAT and EAT from HF patients with or without DM. No. of patients is indicated in parentheses. Gene expression was normalized using the reference gene β-actin (ACTB) that was selected based on our previous results demonstrating that it does not change under these conditions. Statistical analysis: glucose metabolism-related gene expression was compared between factors (disease status and tissue type) using a nonparametric 2-way multivariate ANOVA (MANOVA).
Lipolysis in SAT and EAT from HF patients.
Isoproterenol-stimulated lipolysis was evaluated in isolated SAT and EAT cells from 22 NDM (12 CAD and 10 NCAD) and 17 DM (11 CAD and 6 NCAD) patients. For each tissue type and disease status, absolute rates of lipolysis between basal lipolysis and the corresponding treatment were different (P < 0.001; Fig. 4A). In the SAT + NDM group, significant differences were found between basal and insulin (P < 0.05), isoproterenol (P < 0.05), and isoproterenol + insulin (P < 0.05) as well as between insulin and isoproterenol (P < 0.05) and isoproterenol + insulin (P < 0.05) (Fig. 4A). In the SAT + DM, EAT + NDM, and EAT + DM groups, significant differences were found between basal and isoproterenol (P < 0.01) and isoproterenol + insulin (P < 0.01) as well as between insulin and isoproterenol (P < 0.01) and isoproterenol + insulin (P < 0.01) (Fig. 4A). However, at the isoproterenol and insulin concentrations tested we found no significant antilipolytic effect of insulin (Fig. 4A).
Fig. 4.
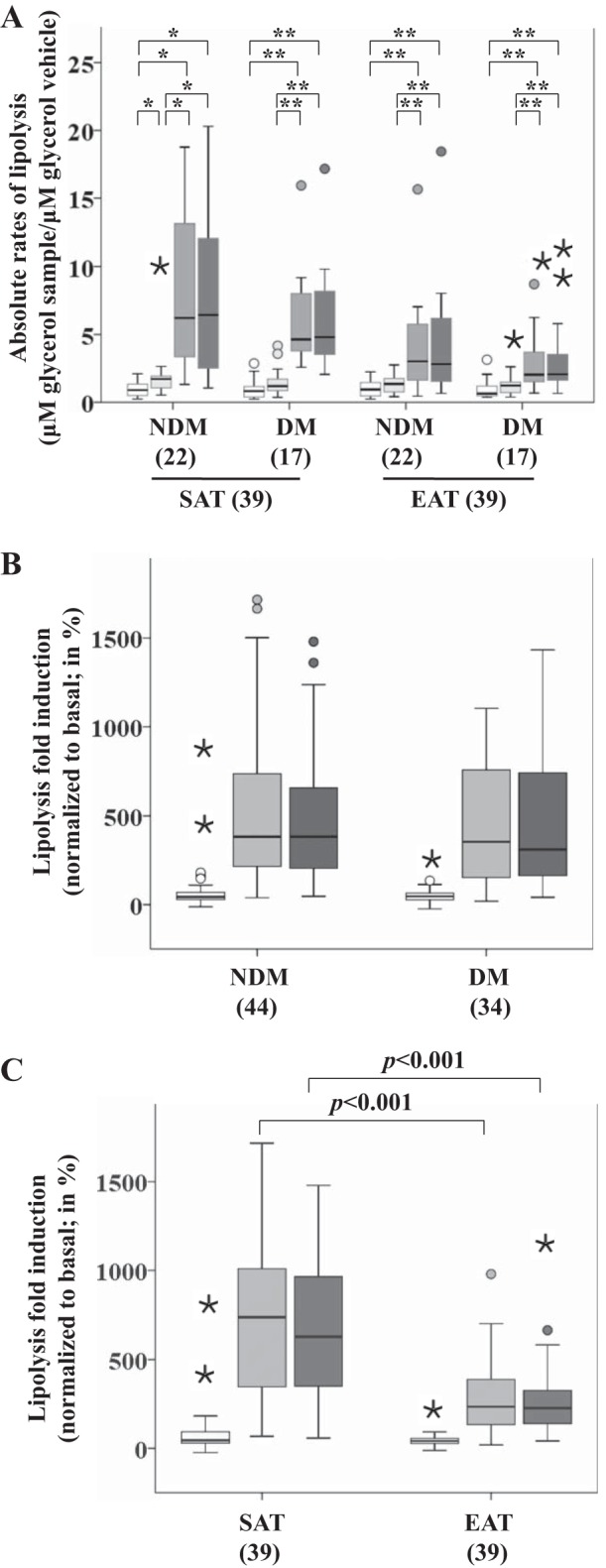
Isoproterenol-stimulated lipolysis in SAT and EAT from HF patients with or without DM. No. of patients is indicated in parentheses. A: absolute rates of lipolysis in each group. B: lipolysis fold induction based on disease status. C: lipolysis fold induction based on tissue type. Statistical analysis: for each tissue type and disease status in A, differences between basal and the measurements corresponding to the different stimuli were done using Friedman tests and subsequent post hoc tests, employing Dunn's correction for multiple comparisons, **P < 0.01, *P < 0.05. The changes in lipolysis in B and C were compared between factors (disease status and tissue type) using a nonparametric 2-way MANOVA. P value is as indicated. Open bars, basal; light gray bars, insulin; gray bars, isoproterenol; dark gray bars, insulin + isoproterenol.
Induced-lipolysis fold changes between disease status were not different (P = 0.910; Fig. 4B); however, significant differences were observed between tissues (P < 0.001; Fig. 4C). Both isoproterenol (P < 0.001) as well as insulin + isoproterenol (P < 0.001) were different when comparing tissues. Isoproterenol-stimulated lipolysis was significantly decreased in EAT (median = 234.76) compared with SAT (median = 737.50). There were no significant differences with insulin (P = 0.093) within tissues.
Lipid storage and lipolysis gene expression in SAT and EAT from HF patients.
We correlated all results from the different genes without applying a correction for multiple comparisons and found moderate to very strong correlations in 74% of the combinations. As described in materials and methods, the different genes were averaged out into two variables: lipid storage and lipolysis. By overlooking the correlation between genes and using univariate analysis (of variance), we obtained the P values reported in Tables 3 and 4. A strong correlation was found between lipid storage and lipolysis variables [ρ(67) = 0.839, P < 0.001; Fig. 5A]. Lipid storage genes were compared between disease status and tissue type, and the main effect was observed in tissue type, F(1, 97) = 9.176, P = 0.003 (Fig. 5C), indicating that the expression of genes involved in lipid storage was significantly different between EAT and SAT. The same did not hold true for lipolysis genes, for which no statistical significance was found, which may be due to the fact that it was possible to average out lipid storage genes for a greater number (n = 101) of cases than for lipolysis variables (n = 74) due to missing values.
Table 3.
Lipid storage gene expressions
| DGAT1 | FABP4 | PPARG | SREBP1 | CD36 | FASN | LPIN1 | MTTP | LPL | SCD1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Tissue type | 0.018 | 0.512 | 0.197 | 0.126 | 0.049 | 0.430 | 0.626 | 0.539 | 0.904 | 0.390 |
| Disease status | 0.010 | 0.744 | 0.837 | 0.063 | 0.276 | 0.001 | 0.804 | 0.564 | 0.018 | 0.003 |
| Interaction | 0.145 | 0.053 | 0.177 | 0.799 | 0.107 | 0.231 | 0.079 | 0.351 | 0.104 | 0.185 |
P values relating to differences between tissues, nondiabetics vs. diabetics, and the interaction are reported. DGAT1, diacylglycerol O-acyltransferase; FABP4, fatty acid-binding protein 4; SREBP1, sterol regulatory element-binding protein 1; CD36, cluster of differentiation 36; FASN, fatty acid synthase; LPIN1, lipin 1; MTTP, microsomal triglyceride transfer protein large subunit; LPL, lipoprotein lipase; SCD1, stearoyl-CoA desaturase. For each gene, an analysis of variance was performed to compare the gene expression between tissue type and disease status and also to account for possible interactions.
Table 4.
Lipolysis gene expressions
| HSL | PLIN1 | AQP7 | B1AR | PKA | |
|---|---|---|---|---|---|
| Tissue type | <0.001 | 0.360 | 0.156 | 0.042 | 0.661 |
| Disease status | 0.019 | 0.229 | 0.910 | 0.696 | 0.174 |
| Interaction | 0.067 | 0.094 | 0.120 | 0.658 | 0.869 |
P values relating to differences between tissues, nondiabetics vs. diabetics, and their interaction are reported. HSL, hormone-sensitive lipase; PLIN1, perilipin 1; AQP7, aquaporin-7; B1AR, β1-adrenergic receptor. For each gene, an analysis of variance was performed to compare the gene expression between tissue type and disease status and also to account for possible interactions.
Fig. 5.

Lipid storage and lipolysis-related gene expression in SAT and EAT from HF patients with or without DM. No. of patients is indicated in parentheses. Different genes were averaged out into 2 variables: lipid storage [diacylglycerol O-acyltranferase 1 (DGAT1), fatty acid-binding protein 4 (FABP4), peroxisome proliferator-activated receptor-γ (PPARG), sterol-regulatory element-binding protein 1 (SREBP1), cluster of differentiation (CD36), fatty acid synthase (FASN), lipin 1 (LPIN1), microsomal triglyceride transfer protein large subunit (MTTP), lipoprotein lipase (LPL), and stearoyl-CoA desaturase 1 (SCD1)] and lipolysis [hromone-sensitive lipase (HSL), perililpin (PLIN1), aquaporin-7 (AQP7), β1-adrenergic receptor (B1AR), and PKA]. Gene expression was normalized using the reference gene ACTB that was selected based on our previous results demonstrating that it does not change under these conditions. A: correlation between lipid storage and lipolysis gene expression. B: lipid storage and lipolysis gene expression based on disease status. C: lipid storage and lipolysis gene expression based on tissue type. Statistical analysis: correlation between lipolysis and lipid storage in A was evaluated using the Spearman correlation coefficient; n = 74, ρ(67) = 0.839, P < 0.001. Lipid storage group in B and C was compared between factors (disease status and tissue type) using a nonparametric 2-way ANOVA. P value is as indicated. Light gray bars, lipid storage; dark gray bars, lipolysis.
Inflammation-related gene expression in SAT and EAT from HF patients.
Inflammation-related gene expression was compared between factors (disease status and tissue type). Disease status effect on inflammation was not statistically significant, with χ2(4) = 4.087, n = 62, and P = 0.394. However, tissue type effect was significant, with χ2(4) = 22.265, n = 62, and P < 0.001. The existence of significant differences between tissues was tested using a post hoc Mann-Whitney U-test for each inflammation-related gene expression: interleukin 6 (IL6; U = 467.0, P = 0.849), interleukin 8 (IL8; U = 297.0, P = 0.010), tumor necrosis factor-α (TNFA; U = 427.0, P = 0.905), and serpin E1 (SERPINE1; U = 239.0, P = 0.001) (Fig. 6).
Fig. 6.

Inflammation-related gene expression [IL8, serpin E1 (SERPINE1), IL6, and TNFA] in SAT and EAT from HF patients with or without DM. No. of patients is indicated in brackets. A, C, E, and G: inflammation-related gene expression based on disease status. B, D, F, and H: inflammation-related gene expression based on tissue type. Gene expression was normalized using the reference gene ACTB, which was selected based on our previous results demonstrating that it does not change under these conditions. Statistical analysis: inflammation-related gene expression was compared between factors (disease status and tissue type) using a nonparametric 2-way MANOVA. P value is as indicated. For a better visualization of the results, we excluded from the SERPINE1 graphs a value in the DM (C) and EAT (D) groups (0.3997).
Fatty acid composition of SAT and EAT from HF patients.
A perfect negative correlation between the saturated and unsaturated FA fractions and between the C16 and C18 fractions was found [ρ(91) = −1.000, P < 0.001].
Monoglyceride (MG), diglyceride (DG), and saturated and C16 fraction measurements were compared between disease status (Fig. 7A) and tissue type (Fig. 7B) and the main effect of disease status was significant (P = 0.009; Fig. 7A). The DG fraction (P = 0.004), the saturated fraction (P = 0.003), and the C16 fraction (P = 0.018) were found to be different for disease status. However, no differences were found for the MG fraction. The effect of tissue type was not significant.
Fig. 7.

Fatty acid composition in SAT and EAT from HF patients, with or without DM, by proton nuclear magnetic resonance spectroscopy. No. of patients is indicated in brackets. A: fatty acid composition based on disease status. B: fatty acid composition based on tissue type. Statistical analysis: monoglyceride (MG), diglyceride (DG), and saturated and C16 fraction measurements were compared between factors (disease status and tissue type) using a nonparametric 2-way MANOVA. P value is as indicated. Black bars, MG; light gray bars, DG; gray bars, saturated fraction; dark gray bars, C16 fraction.
DISCUSSION
Our novel findings show that glucose uptake upon insulin stimulation and lipolysis upon isoproterenol stimulation are decreased in EAT compared with SAT in HF patients. Similarly, lipid storage, lipolysis, and inflammation-related gene expression are significantly decreased in EAT compared with SAT from the same patients. In addition, we found that diabetes alters FA composition in SAT and EAT.
Although insulin-stimulated glucose uptake was significantly increased compared with basal in all groups, a decrease in glucose uptake in EAT compared with SAT cells from the same patient was observed. EAT has lower rates of glucose utilization (64), which is probably due not only to lower hexokinase and phosphofructokinase activities (32) but also to decreased insulin action (34), which may partly explain the lower glucose uptake and antilipolytic actions of insulin stimulation we observed in EAT. Furthermore, insulin concentration for half-maximal suppression of lipolysis in fat is 101 pM in lean but 266 pM in obese subjects, consistent with insulin resistance in obesity (10). We used supraphysiological insulin concentrations (1,000 μU/ml = 6,000 pM), and therefore, the antilipolytic effects of insulin at physiological concentrations should be evaluated further.
In agreement with our findings, GLUT4 gene expression was found to be lower in EAT from CAD patients (72), explaining the decreased insulin-stimulated glucose uptake. However, the uptake of 18-fluorodeoxyglucose measured by positron emission tomography was significantly greater in EAT from patients with atrial fibrillation (AF) vs. non-AF controls (52) when compared with SAT and visceral thoracic fat, which was probably due to the rapid uncoordinated heart rates, increasing cardiac work, and energy requirements in AF patients. Thus, depending on the cardiac pathology, EAT may modulate glucose uptake. Abdominal fat cells from DM patients have decreased insulin action, insulin receptor tyrosine kinase activity, insulin receptor substrate expression, and PI 3-kinase activity, leading to impaired GLUT4 translocation and glucose transport (12). There were no further effects of diabetes on the insulin-induced glucose uptake when the two groups studied were compared, and the fold increase was very small compared with what one expects to find in healthy subjects (68). Alterations in insulin signaling due to the inherent insulin resistance observed in HF patients may have played a key role in the relatively low insulin stimulation effect on glucose uptake, regardless of diabetes status. The fact that we had no healthy control subjects matched for age, sex, or BMI, made it impossible to compare the effect of insulin on glucose uptake in the subjects studied.
Furthermore, we observed a significant decrease in EAT lipolysis, compared with SAT from the same HF patients, regardless of diabetes status. Under physiological conditions of high energy demand, EAT shows high lipogenic and lipolytic activities (50, 64, 83), which suggests that EAT may act as a local energy supply for the adjacent myocardium and/or as a buffer against toxic levels of free FA (50). Larger adipocytes from healthy subjects have increased lipolytic capacity compared with smaller ones (44). Interestingly, and in agreement with previous reports (4, 5), we found epicardial adipocytes to be lighter and smaller than the subcutaneous adipocytes from the same patients as well as a very strong correlation between lipid storage and lipolysis gene expression. Lipid storage gene expression was significantly decreased in EAT compared with SAT. The strong correlation between lipid storage and lipolysis genes suggests that lipolysis gene expression would also be decreased in EAT compared with SAT, and in fact, our results agree with the literature, where EAT from HF patients expresses low levels of fat-mobilizing genes such as PLIN1, LPL, HSL, and ATGL (35). The lower expression of lipolytic genes in epicardial adipocytes translates into a lower lipolysis rate compared with SAT. Others have also observed that HSL gene levels were lower in EAT than in substernal intrathoracic human fat (26). Moreover, protein expression regulating the final steps in hormone signaling during lipolysis was increased in larger adipocytes, explaining the increased lipolytic capacity in larger cells (44). Thus, at least in part, our lipolysis results may be explained by the significantly low HSL gene expression. The low lipid storage and lipolysis gene expression observed in EAT from HF patients could also be due to insulin resistance induced by excess free FA in circulation. Besides the impaired inhibitory effect of insulin on lipolysis, insulin resistance also impairs lipid storage by interfering with pathways involved in uptake, synthesis, and storage of TG in adipocytes (57). In addition, in HSL-deficient mice, compensatory reduction in FA esterification and de novo lipogenesis was observed to counteract the reduced release of free FA into circulation (89). Importantly, the decrease in lipid storage and lipolysis genes observed in EAT could be a protective mechanism against cardiac lipotoxicity, which, ultimately, could lead to heart steatosis and loss of function. Therefore, the heart should be less exposed to the deleterious effects of high FA levels in the bloodstream, which are major contributors to insulin resistance (6) and HF pathogenesis (45). Moreover, the insulin antilipolytic effect is mediated by cGMP-inhibitable phosphodiesterase 3B (16–18, 48, 58), a PI 3-kinase substrate (67) whose phosphorylation leads to HSL phosphorylation and activation, increasing lipolysis (22), mechanisms that are impaired in diabetes and insulin resistance.
Accordingly, diabetic patients with endogenous hypertriglyceridemia are resistant to the antilipolytic action of insulin when compared with weight-matched normolipemic nondiabetic individuals (85).
Other studies have reported mix results regarding the effects of insulin resistance/DM on antilipolysis effects of insulin in human adipose tissue (1, 8, 65, 86). These differences may be due to the different populations studied, type of diabetes, insulin concentration or in the assays performed to evaluate lipid metabolism.
It is known that the cause/effect relationship between different pathologies can work in both directions; for example, HF can lead to metabolic derangements, such as insulin resistance (37), and systemic insulin resistance was reported to be a risk factor for the development of HF (24). Moreover, myocardial insulin resistance in advanced dilated cardiomyopathy can lead to the subsequent alteration in the insulin-signaling cascade, impairing both glucose uptake and lipolysis (73), as we observed in our study. In agreement with our data, other studies have shown that under cardiac pathological conditions, not only is myocardial insulin signaling compromised, impairing insulin-stimulated glucose utilization, but also lipid metabolism (2, 24, 46).
In addition to diabetes and heart disease, the advanced age of the study population may also contribute to peripheral insulin resistance. SAT from middle-aged (40–59 yr), healthy, normal-weight subjects presents a lower insulin binding per cell than younger subjects, which is due mainly to a decreased insulin receptor number (7). Concomitantly, the degree of antilipolysis was 10–20 times smaller in the older subjects (7).
Although we were not able to determine peripheral insulin signaling status in our studied population, they may already present alterations in insulin signaling due to age and HF, with associated effects on lipolysis in response to isoproterenol and insulin. The lack of healthy control subjects matched for age, sex, and BMI prevented us from comparing results with the subjects studied.
Given the importance of inflammation in heart pathophysiology and metabolism, including diabetes (41, 78, 79, 87), we tested the proinflammatory markers IL6, TNFA, IL8, and SERPINE1 gene expression and observed that although there were no differences for IL6 and TNFA, both IL8 and SERPINE1 were significantly decreased in EAT compared with SAT, regardless of disease status, in correlation with the lipolysis results measured in each fat depot. Previous studies demonstrated that lipolysis products induce inflammatory responses by increasing IL8 levels (3, 19, 30). In agreement with our lipolysis results obtained in EAT, the inflammatory markers in EAT were also decreased compared with SAT, perhaps a compensatory mechanism toward cardioprotection. However, other studies have reported IL8 increases in EAT (70, 88), perhaps due to population differences. Previous studies have reported that SERPINE1 was found to be increased in EAT from patients with severe stable CAD (70) and overexpressed in EAT from acute coronary syndrome (43). In our study, SERPINE1 gene expression correlates with the lipolysis results, indicating that increased lipolysis associates with increased inflammation observed in SAT, whereas the decreased lipolysis and inflammatory gene expression observed in EAT may indicate a possible protective mechanism toward the cardiomyocyte. Moreover, depending on the models studied, SERPINE1 may be regulated by insulin and by prescribed medications (25, 43, 53, 76). However, we did not detect any effect of diabetes on inflammatory markers when comparing with nondiabetics with HF. Inflammation is currently recognized as responsible for the development and maintenance of diverse chronic diseases, including diabetes and atherosclerosis (29). It has been demonstrated that IL6 shows cardiodepressive properties (27). In patients with systolic HF, IL6 and TNFα are associated with the functional NYHA class (78). Moreover, patients with left ventricular diastolic dysfunction had significantly higher IL6 and TNFα plasma levels compared with those with normal diastolic function, suggesting a link between low-grade inflammation and the pathogenesis of diastolic dysfunction (21). Furthermore, IL6 and TNFα have been shown to be independent predictors of mortality in HF (20). Recent studies have linked insulin resistance with TNFα and IL6, a measure of proinflammation predictive for DM. Furthermore, increased circulating concentrations of IL6 and TNFα were found in DM and impaired glucose-tolerant subjects (40, 54, 63). The development of cardiac dysfunction is likely to be multifactorial with putative mechanisms, including metabolic disturbances, insulin resistance, myocardial fibrosis, endothelial dysfunction, autonomic dysfunction, and myocyte damage. Proinflammatory cytokines are involved in most if not all of these pathophysiological changes and might be a link between these abnormalities. Thus, due to the large number of factors that are involved in cardiomyopathies, the presence of diabetes probably did not further worsen the inflammatory profile in the collected tissues obtained from our study population, where the results are similar.
In addition, our study population is not naïve for medication and treatments that have anti-inflammatory properties, such as statins, angiotensin blockers, metformin, and pioglitazone (36), and all of these may be masking at least some of the inflammatory effects.
Furthermore, in our study, we observed that DGs and saturated FA were increased in DM patients in both fat depots. The C16 fraction was also decreased in these patients. The TGs stored in fat depots are predominantly saturated FA, palmitic and stearic, which have 16 and 18 carbons, respectively, and are the two most abundant saturated FA in humans (38). These saturated FA are the preferred substrate for the heart and are positively associated with DM (28). Several studies report that saturated FA levels are elevated in obesity and DM and that an excess supply of these molecules can cause diabetic cardiomyopathies (42). In response to chronic high plasma concentration of LCFA, as is the case in DM, the heart is forced to increase FA uptake, and over time this may result in the development of cardiomyopathies (23). Mechanistically, FA can interact with peroxisome proliferator-activated receptors, which upregulate the expression of enzymes necessary for their disposal through mitochondrial β-oxidation but also stimulate FA uptake (23). This can lead to further increases in FA concentration in the cytoplasm of cardiomyocytes (23). Although the onset of diabetic cardiomyopathies results in an increased FA utilization by the heart, the subsequent lipid overload results in an increased production of reactive oxygen species and accumulation of lipid intermediates that over time will introduce structural changes that affect cardiac contractile characteristics (23). Accordingly, we observed that DM patients presented an increase in DGs and saturated FA content in both adipose fat depots studied, whereas the C16 fraction was decreased in these tissues, compared with NDM patients. Thus, the DM-induced metabolic changes lead to an excessive accumulation of saturated FA in EAT and SAT. Since C16 FA are metabolized by the heart, leading us to believe that both fat depots release more C16 FA into the bloodstream and will be metabolized and accumulated in the heart of diabetics, resulting in the development of diabetic cardiomyopathy.
In conclusion, although more studies are required to clarify the underlying molecular mechanisms regulating EAT glucose and lipid metabolism in HF patients, our results identify important new markers of glucose and lipid metabolism that are deregulated in EAT in HF patients. In fact, our study indicates that glucose uptake, lipolysis, lipid storage, and inflammation-related gene expression are significantly decreased in EAT compared with SAT in HF patients.
Metabolic alterations could modify energy requirements in the heart and, consequently, contribute to cardiac disease. In our study population, we do not have alterations at the level of the difference between basal and insulin conditions in glucose uptake or isoproterenol-stimulated lipolysis in the presence or absence of diabetes. However, diabetes influenced the adipocyte size and FA composition in both fat depots. Since there are no data available comparing sternal SAT and abdominal SAT, the potential differences between sternal and abdominal subcutaneous fat may explain why we did not observe differences between diabetic and nondiabetic HF patients.
EAT volume may play a role in the stratification of the cardiometabolic risk and serve as a therapeutic target (32, 33), and our data highlight the important metabolic diversity between SAT and EAT in HF patients, with or without diabetes, bringing to light EAT as a potential therapeutic target against altered glucose and lipid metabolism in these patients.
Limitations of the study.
The available amounts of EAT and SAT biopsies collected from these patients was insufficient to perform all of the experiments in the same sample. Ongoing studies will evaluate posttranslational modifications of proteins as well as enzymatic activities and insulin signaling. In addition, we were unable to obtain EAT biopsy samples from healthy subjects.
GRANTS
This work wa supported by a SPD/GIFT award, the European Foundation for the Study of Diabetes, SFRH/BPD/26837/2006, PTDC/SAU-OSM/104124/2008, EXCL/DTP-PIC/0069/2012, Strategic Project 2015-UID/NEU/04539/2013 funded by FEDER through Operational Programme Competitiveness Factors-COMPETE, and by FCT-Foundation for Science and Technology. E. Carvalho was partially supported by National Institutes of Health Grants P30-AG-028718 and RO1-AG-033761.
DISCLOSURES
The authors declare that they do not have any conflicts of interest. The funding agency had no role in the manuscript content or the decision to publish.
AUTHOR CONTRIBUTIONS
A.B., M.A., and E.C. conception and design of research; A.B., A.F., S.C., D.E., I.J., R.A.C., M.A., and E.C. performed experiments; M.L. and M.P. analyzed data; A.B., I.J., R.A.C, M.A., and E.C. interpreted results of experiments; A.B., M.L., and M.P. prepared figures; A.B. drafted manuscript; A.B. and E.C. edited and revised manuscript; M.A. and E.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We kindly thank Novo Nordisk for the human insulin, Actrapid, and Dr. L. Carvalho for the H & E image of SAT and EAT.
REFERENCES
- 1.Arner P, Engfeldt P, Skarfors E, Lithell H, Bolinder J. Insulin receptor binding and metabolic effects of insulin in human subcutaneous adipose tissue in untreated non-insulin dependent diabetes mellitus. Ups J Med Sci 92: 47–58, 1987. [DOI] [PubMed] [Google Scholar]
- 2.Aroor AR, Mandavia CH, Sowers JR. Insulin resistance and heart failure: molecular mechanisms. Heart Fail Clin 8: 609–617, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aung HH, Lame MW, Gohil K, An CI, Wilson DW, Rutledge JC. Induction of ATF3 gene network by triglyceride-rich lipoprotein lipolysis products increases vascular apoptosis and inflammation. Arterioscler Thromb Vasc Biol 33: 2088–2096, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bambace C, Sepe A, Zoico E, Telesca M, Olioso D, Venturi S, Rossi A, Corzato F, Faccioli S, Cominacini L, Santini F, Zamboni M. Inflammatory profile in subcutaneous and epicardial adipose tissue in men with and without diabetes. Heart Vessels 29: 42–48, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Bambace C, Telesca M, Zoico E, Sepe A, Olioso D, Rossi A, Corzato F, Di Francesco V, Mazzucco A, Santini F, Zamboni M. Adiponectin gene expression and adipocyte diameter: a comparison between epicardial and subcutaneous adipose tissue in men. Cardiovasc Pathol 20: e153–e156, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 18: 139–143, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolinder J, Ostman J, Arner P. Influence of aging on insulin receptor binding and metabolic effects of insulin on human adipose tissue. Diabetes 32: 959–964, 1983. [DOI] [PubMed] [Google Scholar]
- 8.Bolinder J, Ostman J, Arner P. Postreceptor defects causing insulin resistance in normoinsulinemic non-insulin-dependent diabetes mellitus. Diabetes 31: 911–916, 1982. [DOI] [PubMed] [Google Scholar]
- 9.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol 8: 30–41, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell PJ, Carlson MG, Nurjhan N. Fat metabolism in human obesity. Am J Physiol Endocrinol Metab 266: E600–E605, 1994. [DOI] [PubMed] [Google Scholar]
- 11.Carley AN, Bi J, Wang X, Banke NH, Dyck JR, O'Donnell JM, Lewandowski ED. Multiphasic triacylglycerol dynamics in the intact heart during acute in vivo overexpression of CD36. J Lipid Res 54: 97–106, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho E, Eliasson B, Wesslau C, Smith U. Impaired phosphorylation and insulin-stimulated translocation to the plasma membrane of protein kinase B/Akt in adipocytes from Type II diabetic subjects. Diabetologia 43: 1107–1115, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Carvalho E, Jansson PA, Nagaev I, Wenthzel AM, Smith U. Insulin resistance with low cellular IRS-1 expression is also associated with low GLUT4 expression and impaired insulin-stimulated glucose transport. FASEB J 15: 1101–1103, 2001. [PubMed] [Google Scholar]
- 14.Cherian S, Lopaschuk GD, Carvalho E. Cellular cross-talk between epicardial adipose tissue and myocardium in relation to the pathogenesis of cardiovascular disease. Am J Physiol Endocrinol Metab 303: E937–E949, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Corradi D, Maestri R, Callegari S, Pastori P, Goldoni M, Luong TV, Bordi C. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc Pathol 13: 313–316, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Degerman E, Belfrage P, Manganiello VC. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J Biol Chem 272: 6823–6826, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Degerman E, Manganiello VC, Newman AH, Rice KC, Belfrage P. Purification, properties and polyclonal antibodies for the particulate, low Km cAMP phosphodiesterase from bovine adipose tissue. Second Messengers Phosphoproteins 12: 171–182, 1988. [PubMed] [Google Scholar]
- 18.Degerman E, Smith CJ, Tornqvist H, Vasta V, Belfrage P, Manganiello VC. Evidence that insulin and isoprenaline activate the cGMP-inhibited low-Km cAMP phosphodiesterase in rat fat cells by phosphorylation. Proc Natl Acad Sci USA 87: 533–537, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.den Hartigh LJ, Altman R, Norman JE, Rutledge JC. Postprandial VLDL lipolysis products increase monocyte adhesion and lipid droplet formation via activation of ERK2 and NFκB. Am J Physiol Heart Circ Physiol 306: H109–H120, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103: 2055–2059, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Dinh W, Futh R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, Bansemir L, Bufe A, Barroso MC, Lankisch M. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol 8: 58, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiPilato LM, Ahmad F, Harms M, Seale P, Manganiello V, Birnbaum MJ. The Role of PDE3B Phosphorylation in the Inhibition of Lipolysis by Insulin. Mol Cell Biol 35: 2752–2760, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dirkx E, Schwenk RW, Glatz JF, Luiken JJ, van Eys GJ. High fat diet induced diabetic cardiomyopathy. Prostaglandins Leukot Essent Fatty Acids 85: 219–225, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Dirkx E, van Eys GJ, Schwenk RW, Steinbusch LK, Hoebers N, Coumans WA, Peters T, Janssen BJ, Brans B, Vogg AT, Neumann D, Glatz JF, Luiken JJ. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J Mol Cell Cardiol 76: 208–217, 2014. [DOI] [PubMed] [Google Scholar]
- 25.Fain JN, Madan AK. Insulin enhances vascular endothelial growth factor, interleukin-8, and plasminogen activator inhibitor 1 but not interleukin-6 release by human adipocytes. Metabolism 54: 220–226, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Fain JN, Sacks HS, Bahouth SW, Tichansky DS, Madan AK, Cheema PS. Human epicardial adipokine messenger RNAs: comparisons of their expression in substernal, subcutaneous, and omental fat. Metabolism 59: 1379–1386, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257: 387–389, 1992. [DOI] [PubMed] [Google Scholar]
- 28.Forouhi NG, Koulman A, Sharp SJ, Imamura F, Kroger J, Schulze MB, Crowe FL, Huerta JM, Guevara M, Beulens JW, van Woudenbergh GJ, Wang L, Summerhill K, Griffin JL, Feskens EJ, Amiano P, Boeing H, Clavel-Chapelon F, Dartois L, Fagherazzi G, Franks PW, Gonzalez C, Jakobsen MU, Kaaks R, Key TJ, Khaw KT, Kuhn T, Mattiello A, Nilsson PM, Overvad K, Pala V, Palli D, Quiros JR, Rolandsson O, Roswall N, Sacerdote C, Sánchez MJ, Slimani N, Spijkerman AM, Tjonneland A, Tormo MJ, Tumino R, van der ADL, van der Schouw YT, Langenberg C, Riboli E, Wareham NJ. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident type 2 diabetes: the EPIC-InterAct case-cohort study. Lancet Diabetes Endocrinol 2: 810–818, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freitas Lima LC, Braga VA, do Socorro de França Silva M, Cruz J C, Sousa Santos SH, de Oliveira Monteiro MM, Balarini CM. Adipokines, diabetes and atherosclerosis: an inflammatory association. Front Physiol 6: 304, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grisouard J, Bouillet E, Timper K, Radimerski T, Dembinski K, Frey DM, Peterli R, Zulewski H, Keller U, Muller B, Christ-Crain M. Both inflammatory and classical lipolytic pathways are involved in lipopolysaccharide-induced lipolysis in human adipocytes. Innate Immun 18: 25–34, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Hassan M, Latif N, Yacoub M. Adipose tissue: friend or foe? Nat Rev Cardiol 9: 689–702, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Iacobellis G, Bianco AC. Epicardial adipose tissue: emerging physiological, pathophysiological and clinical features. Trends Endocrinol Metab 22: 450–457, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iacobellis G, Malavazos AE, Corsi MM. Epicardial fat: from the biomolecular aspects to the clinical practice. Int J Biochem Cell Biol 43: 1651–1654, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Iacobellis G, Sharma AM. Epicardial adipose tissue as new cardio-metabolic risk marker and potential therapeutic target in the metabolic syndrome. Curr Pharm Des 13: 2180–2184, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Jaffer I, Riederer M, Shah P, Peters P, Quehenberger F, Wood A, Scharnagl H, Marz W, Kostner KM, Kostner GM. Expression of fat mobilizing genes in human epicardial adipose tissue. Atherosclerosis 220: 122–127, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Jialal I, Devaraj S. Anti-inflammatory Strategies to Prevent Diabetic Cardiovascular Disease. Clin Pharmacol Ther 98: 121–123, 2015. [DOI] [PubMed] [Google Scholar]
- 37.Jiménez-Amilburu V, Jong-Raadsen S, Bakkers J, Spaink HP, Marín-Juez R. GLUT12 deficiency during early development results in heart failure and a diabetic phenotype in zebrafish. J Endocrinol 224: 1–15, 2015. [DOI] [PubMed] [Google Scholar]
- 38.Johnson EJ, Schaefer EJ. Potential role of dietary n-3 fatty acids in the prevention of dementia and macular degeneration. Am J Clin Nutr 83: 1494S–1498S, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Jones OA, Spurgeon DJ, Svendsen C, Griffin JL. A metabolomics based approach to assessing the toxicity of the polyaromatic hydrocarbon pyrene to the earthworm Lumbricus rubellus. Chemosphere 71: 601–609, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Kado S, Nagase T, Nagata N. Circulating levels of interleukin-6, its soluble receptor and interleukin-6/interleukin-6 receptor complexes in patients with type 2 diabetes mellitus. Acta Diabetol 36: 67–72, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Kanda T, Takahashi T. Interleukin-6 and cardiovascular diseases. Jpn Heart J 45: 183–193, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, Kita T, Kimura T, Ono K. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res 116: 279–288, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Langheim S, Dreas L, Veschini L, Maisano F, Foglieni C, Ferrarello S, Sinagra G, Zingone B, Alfieri O, Ferrero E, Maseri A, Ruotolo G. Increased expression and secretion of resistin in epicardial adipose tissue of patients with acute coronary syndrome. Am J Physiol Heart Circ Physiol 298: H746–H753, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Laurencikiene J, Skurk T, Kulyte A, Heden P, Astrom G, Sjolin E, Ryden M, Hauner H, Arner P. Regulation of lipolysis in small and large fat cells of the same subject. J Clin Endocrinol Metab 96: E2045–E2049, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Leichman JG, Aguilar D, King TM, Vlada A, Reyes M, Taegtmeyer H. Association of plasma free fatty acids and left ventricular diastolic function in patients with clinically severe obesity. Am J Clin Nutr 84: 336–341, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y, Chen T, Wang Y, Huang Y, Li CY, Cao C, Zhang Y, Hu X, Xiao RP. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation 131: 795–804, 2015. [DOI] [PubMed] [Google Scholar]
- 47.Liu L, Yu S, Khan RS, Ables GP, Bharadwaj KG, Hu Y, Huggins LA, Eriksson JW, Buckett LK, Turnbull AV, Ginsberg HN, Blaner WS, Huang LS, Goldberg IJ. DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. J Lipid Res 52: 732–744, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lonnroth P, Smith U. The antilipolytic effect of insulin in human adipocytes requires activation of the phosphodiesterase. Biochem Biophys Res Commun 141: 1157–1161, 1986. [DOI] [PubMed] [Google Scholar]
- 49.Malavazos AE, Di Leo G, Secchi F, Lupo EN, Dogliotti G, Coman C, Morricone L, Corsi MM, Sardanelli F, Iacobellis G. Relation of echocardiographic epicardial fat thickness and myocardial fat. Am J Cardiol 105: 1831–1835, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Marchington JM, Mattacks CA, Pond CM. Adipose tissue in the mammalian heart and pericardium: structure, foetal development and biochemical properties. Comp Biochem Physiol B 94: 225–232, 1989. [DOI] [PubMed] [Google Scholar]
- 51.Mazur A, Ostanski M, Telega G, Malecka-Tendera E. Is epicardial fat tissue a marker of metabolic syndrome in obese children? Atherosclerosis 211: 596–600, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Mazurek T, Kiliszek M, Kobylecka M, Skubisz-Gluchowska J, Kochman J, Filipiak K, Krolicki L, Opolski G. Relation of proinflammatory activity of epicardial adipose tissue to the occurrence of atrial fibrillation. Am J Cardiol 113: 1505–1508, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Mottillo EP, Shen XJ, Granneman JG. beta3-adrenergic receptor induction of adipocyte inflammation requires lipolytic activation of stress kinases p38 and JNK. Biochim Biophys Acta 1801: 1048–1055, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muller S, Martin S, Koenig W, Hanifi-Moghaddam P, Rathmann W, Haastert B, Giani G, Illig T, Thorand B, Kolb H. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia 45: 805–812, 2002. [DOI] [PubMed] [Google Scholar]
- 55.Natale F, Tedesco MA, Mocerino R, de Simone V, Di Marco GM, Aronne L, Credendino M, Siniscalchi C, Calabrò P, Cotrufo M, Calabrò R. Visceral adiposity and arterial stiffness: echocardiographic epicardial fat thickness reflects, better than waist circumference, carotid arterial stiffness in a large population of hypertensives. Eur J Echocardiogr 10: 549–555, 2009. [DOI] [PubMed] [Google Scholar]
- 56.Nichols M, Townsend N, Scarborough P, Rayner M. Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J 35: 2950–2959, 2014. [DOI] [PubMed] [Google Scholar]
- 57.Nielsen TS, Jessen N, Jørgensen JO, Møller N, Lund S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 52: R199–R222, 2014. [DOI] [PubMed] [Google Scholar]
- 58.Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3-kinase. J Biol Chem 269: 3563–3567, 1994. [PubMed] [Google Scholar]
- 59.Payne GA, Kohr MC, Tune JD. Epicardial perivascular adipose tissue as a therapeutic target in obesity-related coronary artery disease. Br J Pharmacol 165: 659–669, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira MJ, Palming J, Rizell M, Aureliano M, Carvalho E, Svensson MK, Eriksson JW. The immunosuppressive agents rapamycin, cyclosporin A and tacrolimus increase lipolysis, inhibit lipid storage and alter expression of genes involved in lipid metabolism in human adipose tissue. Mol Cell Endocrinol 365: 260–269, 2013. [DOI] [PubMed] [Google Scholar]
- 61.Pereira MJ, Palming J, Rizell M, Aureliano M, Carvalho E, Svensson MK, Eriksson JW. mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol Cell Endocrinol 355: 96–105, 2012. [DOI] [PubMed] [Google Scholar]
- 62.Pezeshkian M, Noori M, Najjarpour-Jabbari H, Abolfathi A, Darabi M, Darabi M, Shaaker M, Shahmohammadi G. Fatty acid composition of epicardial and subcutaneous human adipose tissue. Metab Syndr Relat Disord 7: 125–131, 2009. [DOI] [PubMed] [Google Scholar]
- 63.Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci 67: 291–300, 2000. [DOI] [PubMed] [Google Scholar]
- 64.Rabkin SW. Epicardial fat: properties, function and relationship to obesity. Obes Rev 8: 253–261, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Reckless JP, Galton DJ. Catecholamine receptor sensitivity and the regulation of lipolysis in adult diabetes. Diabetologia 12: 351–358, 1976. [DOI] [PubMed] [Google Scholar]
- 66.Ren J, Dimitrov I, Sherry AD, Malloy CR. Composition of adipose tissue and marrow fat in humans by 1H NMR at 7 Tesla. J Lipid Res 49: 2055–2062, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rondinone CM, Carvalho E, Rahn T, Manganiello VC, Degerman E, Smith UP. Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J Biol Chem 275: 10093–10098, 2000. [DOI] [PubMed] [Google Scholar]
- 68.Rondinone CM, Carvalho E, Wesslau C, Smith UP. Impaired glucose transport and protein kinase B activation by insulin, but not okadaic acid, in adipocytes from subjects with Type II diabetes mellitus. Diabetologia 42: 819–825, 1999. [DOI] [PubMed] [Google Scholar]
- 69.Sacks HS, Fain JN. Human epicardial adipose tissue: a review. Am Heart J 153: 907–917, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Sacks HS, Fain JN, Cheema P, Bahouth SW, Garrett E, Wolf RY, Wolford D, Samaha J. Depot-specific overexpression of proinflammatory, redox, endothelial cell, and angiogenic genes in epicardial fat adjacent to severe stable coronary atherosclerosis. Metab Syndr Relat Disord 9: 433–439, 2011. [DOI] [PubMed] [Google Scholar]
- 71.Salgado-Somoza A, Teijeira-Fernandez E, Fernandez AL, Gonzalez-Juanatey JR, Eiras S. Changes in lipid transport-involved proteins of epicardial adipose tissue associated with coronary artery disease. Atherosclerosis 224: 492–499, 2012. [DOI] [PubMed] [Google Scholar]
- 72.Salgado-Somoza A, Teijeira-Fernandez E, Rubio J, Couso E, Gonzalez-Juanatey JR, Eiras S. Coronary artery disease is associated with higher epicardial retinol-binding protein 4 (RBP4) and lower glucose transporter (GLUT) 4 levels in epicardial and subcutaneous adipose tissue. Clin Endocrinol 76: 51–58, 2012. [DOI] [PubMed] [Google Scholar]
- 73.Shah A, Shannon RP. Insulin resistance in dilated cardiomyopathy. Rev Cardiovasc Med 4, Suppl 6: S50–S57, 2003. [PubMed] [Google Scholar]
- 74.Stratmann B, Tschoepe D. Sweet heart - contributions of metabolism in the development of heart failure in diabetes mellitus. Exp Clin Endocrinol Diabetes 116, Suppl 1: S40–S45, 2008. [DOI] [PubMed] [Google Scholar]
- 75.Strobel K, van den Hoff J, Pietzsch J. Localized proton magnetic resonance spectroscopy of lipids in adipose tissue at high spatial resolution in mice in vivo. J Lipid Res 49: 473–480, 2008. [DOI] [PubMed] [Google Scholar]
- 76.Tanaka Y, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, Sakaguchi M, Sugimoto T, Koya D, Haneda M, Kashiwagi A, Maegawa H, Uzu T. Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int 79: 871–882, 2011. [DOI] [PubMed] [Google Scholar]
- 77.Tavares LC, Jarak I, Nogueira FN, Oliveira PJ, Carvalho RA. Metabolic evaluations of cancer metabolism by NMR-based stable isotope tracer methodologies. Eur J Clin Invest 45, Suppl 1: 37–43, 2015. [DOI] [PubMed] [Google Scholar]
- 78.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 27: 1201–1206, 1996. [DOI] [PubMed] [Google Scholar]
- 79.Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, Mabuchi N, Sawaki M, Kinoshita M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol 31: 391–398, 1998. [DOI] [PubMed] [Google Scholar]
- 80.van den Borst B, Schols AM, de Theije C, Boots AW, Köhler SE, Goossens GH, Gosker HR. Characterization of the inflammatory and metabolic profile of adipose tissue in a mouse model of chronic hypoxia. J Appl Physiol 114: 1619–1628, 2013. [DOI] [PubMed] [Google Scholar]
- 81.Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract 94: 311–321, 2011. [DOI] [PubMed] [Google Scholar]
- 82.WRITING GROUP MEMBERS, Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121: e46–e215, 2010. [DOI] [PubMed] [Google Scholar]
- 83.Wronska A, Kmiec Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol (Oxf) 205: 194–208, 2012. [DOI] [PubMed] [Google Scholar]
- 84.Xu A, Vanhoutte PM. Adiponectin and adipocyte fatty acid binding protein in the pathogenesis of cardiovascular disease. Am J Physiol Heart Circ Physiol 302: H1231–H1240, 2012. [DOI] [PubMed] [Google Scholar]
- 85.Yki-Jarvinen H, Taskinen MR. Interrelationships among insulin's antilipolytic and glucoregulatory effects and plasma triglycerides in nondiabetic and diabetic patients with endogenous hypertriglyceridemia. Diabetes 37: 1271–1278, 1988. [DOI] [PubMed] [Google Scholar]
- 86.Yu ZW, Jansson PA, Posner BI, Smith U, Eriksson JW. Peroxovanadate and insulin action in adipocytes from NIDDM patients. Evidence against a primary defect in tyrosine phosphorylation. Diabetologia 40: 1197–1203, 1997. [DOI] [PubMed] [Google Scholar]
- 87.Yudkin JS. Adipose tissue, insulin action and vascular disease: inflammatory signals. Int J Obes Relat Metab Disord 27, Suppl 3: S25–S28, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Zdychova J, Kralova Lesna I, Maluskova J, Janousek L, Cahova M, Kazdova L. Comparison of gene expression of epicardial and visceral adipocytes with regard to the differentiation stage. Neuro Endocrinol Lett 33, Suppl 2: 93–97, 2012. [PubMed] [Google Scholar]
- 89.Zimmermann R, Haemmerle G, Wagner EM, Strauss JG, Kratky D, Zechner R. Decreased fatty acid esterification compensates for the reduced lipolytic activity in hormone-sensitive lipase-deficient white adipose tissue. J Lipid Res 44: 2089–2099, 2003. [DOI] [PubMed] [Google Scholar]