

Abstract
GPR120 (free fatty acid receptor-4) is a G protein-coupled receptor for medium- and long-chain unsaturated fatty acids, including ω-3 fatty acids. Recent studies have shown GPR120 to play cardinal roles in metabolic disorders via modulation of gut hormone secretion and insulin sensitivity and to exert anti-inflammatory effects in macrophages and adipose tissues. However, information on anti-inflammatory role of GPR120 at the level of intestinal epithelium is very limited. Current studies demonstrated differential levels of GPR120 mRNA and protein along the length of the human, mouse, and rat intestine and delineated distinct anti-inflammatory responses following GPR120 activation in model human intestinal epithelial Caco-2 cells, but not in model mouse intestinal epithelial endocrine cell line STC-1. In Caco-2 cells, GPR120 was internalized, bound to β-arrestin-2, and attenuated NF-κB activation in response to 30-min exposure to the agonists GW9508, TUG-891, or docosahexaenoic acid. These effects were abrogated in response to small interfering RNA silencing of β-arrestin-2. Treatment of STC-1 cells with these agonists did not induce receptor internalization and had no effects on NF-κB activation, although treatment with the agonists GW9508 or TUG-891 for 6 h augmented the synthesis and secretion of the gut hormone glucagon-like peptide-1 in this cell line. Our studies for the first time demonstrated a GPR120-mediated novel anti-inflammatory pathway in specific intestinal epithelial cell types that could be of therapeutic relevance to intestinal inflammatory disorders.
Keywords: FFAR4, Caco-2, STC-1, NF-κB, GLP-1
gpr120, also known as free fatty acid (FFA) receptor-4 (FFAR4), is a G protein-coupled receptor for medium- and long-chain unsaturated fatty acids (FAs) (10). Various ω-3 or ω-6 polyunsaturated FAs, including docosahexaenoic acid (DHA) C22:6 (n-3) and eicosapentaenoic acid C20:5 (n-3), activate GPR120 in the micromolar concentration range (14). Although its ligand profiles are similar to those of GPR40 (FFAR1), the amino acid sequence homology between FFAR4 and FFAR1 is only 10% (14). GPR120 has been shown to be widely expressed in various tissue and cell types, including intestinal tissue, adipose tissue, macrophages, and pancreas (14, 18). The diverse tissue distribution of GPR120 suggests that the G protein-coupled receptor has multiple functions in the homeostatic regulation of systemic metabolism and inflammation (14). Indeed, GPR120 agonism by various ligands, more particularly by the ω-3 FAs, has been shown to result in varied potentially beneficial biological effects, including stimulation of secretion of glucagon-like peptide-1 (GLP-1) from enteroendocrine L cells, inhibition of ghrelin secretion, stimulation of glucose uptake by adipocytes, promotion of pancreatic β-cell survival (28), as well as augmentation of autophagy in macrophages (26). Ample studies have shown GPR120 to play cardinal roles in metabolic disorders via modulation of gut hormone secretion, food preference, insulin secretion, and glucose homeostasis (14, 18, 20, 21, 25, 28). GPR120 is highly expressed in the enteroendocrine L cells located in the lower small intestine and colon, and, therefore, earlier studies on GPR120 have focused on its potential ability to stimulate GLP-1 (28). A recent study has shown that GPR120 is highly expressed also in enteroendocrine K cells of the upper small intestine and has a critical role in the secretion of another incretin gastric inhibitory peptide after fat ingestion (15). Induction of GLP-1 secretion following GPR120 activation has been shown to require Gαq/11 coupling (18, 28). GPR120 also activates mitogen-activated protein kinases pathway via Gαq/11 coupling (18, 19).
On the other hand, GPR120 has been shown in macrophages and adipocytes to exert Gαq/11-independent anti-inflammatory effects via inhibition of NF-κB activation that involves binding of GPR120 to β-arrestin-2, followed by internalization of the complex (23). These anti-inflammatory signaling cascades have been suggested to underlie the insulin-sensitizing and anti-diabetic effects of ω-3 FAs in macrophages and adipocytes (18, 21). However, despite abundant intestinal expression levels of GPR120, information on anti-inflammatory role of GPR120 at the level of intestinal epithelium, which could be of key importance in the pathophysiology of intestinal inflammatory disorders, is very limited. In this report, we have systematically examined the expression levels of GPR120 along the length of the intestine in various mammalian species and also elucidated a GPR120-mediated anti-inflammatory pathway that has not been reported earlier in intestinal epithelial cells.
MATERIALS AND METHODS
Chemicals and antibodies.
GPR120 agonists GW9508 and TUG-891 were obtained from Sigma Aldrich (St. Louis, MO) and Tocris (Bristol, UK), respectively. Anti-GPR120 antibody (rabbit monoclonal) was obtained from Novus Biologicals (Littleton, CO), anti-β-arrestin-2 and anti-GLP-1 antibodies were from Santa Cruz Biotechnology (Dallas, TX), and anti-TAB1 (TAK1 binding protein 1) and anti-TAK1 (transforming growth factor-β-activated kinase 1) antibodies were from Cell Signaling (Beverly, MA).
Cell lines and cell culture.
Human intestinal Caco-2 cells obtained from American Type Culture Collection (ATCC) (Manassas, VA) were maintained in Eagle's minimum essential medium with 4.5 g/l glucose, 50 kU/l penicillin, 5 mg/l streptomycin, and 20% fetal bovine serum. STC-1 cells, a mouse intestinal endocrine cell line (10, 17), also obtained from ATCC, were maintained in Dulbecco's modified Eagle's medium high glucose media (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum. For experiments, confluent cells in cell culture flasks were trypsinized and seeded into 6-, 12-, or 24-well plates at a cell density of 2 × 104 cells/ml. At 60–70% confluency, cells were used for treatments. Cells were serum-starved overnight before treatments and also during the treatments.
Animal and human intestinal mucosal tissues.
All of the animal studies were approved by the Animal Care Committee of the University of Illinois at Chicago and Jesse Brown Veteran Affairs Medical Center. C57BL/6J mice (male, 8 wk old) and Sprague-Dawley rats (male, 6 wk old) were obtained from Jackson Laboratories (Bar Harbor, ME) and Harlan Laboratories (Madison, WI), respectively. Animals with normal chow diet were killed after 2 wk of maintenance; the whole intestine was removed and divided into jejunum, ileum, and proximal and distal colon. Mucosa from each region were scrapped and stored at −80°C until used for RNA extraction or preparing tissue lysates. The human studies were approved by the Human Investigation Committee of the Jesse Brown VA Medical Center and the Institutional Review Board of the University of Illinois at Chicago. Small intestine and colon from healthy adult organ donors (primarily trauma victims) were obtained immediately after harvest of transplantation organs. The small intestine was divided into two equal parts after one-third of the middle portion was discarded. The first half was considered to be jejunum, and the second half to be ileum. After the cecum was discarded, the remaining large intestine was divided equally into proximal and distal colon. The mucosa was scraped from the seromuscular layer of these segments and stored at −80°C.
RNA extraction and real-time RT-PCR.
The total RNA from mouse and rat intestinal mucosa was prepared using RNeasy Mini Kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. An equal amount of RNA for each sample was reverse-transcribed and amplified in a one-step reaction using Brilliant SYBR Green QRT-PCR master mix kit (Stratagene, La Jolla, CA) and using Mx 3000 (Stratagene, Santa Clara, CA). The gene-specific primers for mouse and rat GPR120 (5′-3′), respectively, were as follows: forward, CTGGGGCTCATCTTTGTCGT, reverse, ACGACGAGCACTAGAGGGAT; forward, CCACCGTTCTGGGACTCATC, reverse, ACCACGAGCACTAGAGGGAT. The quantitation of the amplification was expressed as a ratio of 2ΔCt-GPR120/2ΔCt-GAPDH, where ΔCt-GPR120 and ΔCt-GAPDH represent the difference between the threshold cycle of amplification of GPR120 and GAPDH.
Immunoblotting.
Lysates were prepared by homogenizing scrapped intestinal mucosal tissues in 1× RIPA buffer (Cell Signaling Technology, Danvers, MA) containing 1× protease inhibitor cocktail (Roche, Indianapolis, IN). Proteins in the mouse, rat, or human intestinal mucosal lysates were separated by SDS-PAGE on an 8% gel. Proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA) and probed with the anti-GPR120 antibody (Novus Biologicals, catalog no. NBP1-00858), possessing immunoreactivity toward human, mouse, and rat antigens. Immunoreactive bands were visualized using the ECL detection kit (Bio-Rad).
Coimmunoprecipitation.
Control cells and cells treated with GPR120 agonists were washed in cold PBS and lysed in a lysis buffer [20 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 30 mM NaF, 2 mM sodium pyrophosphate, and 1× protease inhibitor cocktail (Roche, Indianapolis, IN)]. The cell lysates were precleared with protein A/G plus-agarose (Santa Cruz) and then incubated with the antibody used for immunoprecipitation (anti-β-arrestin-2, Santa Cruz, catalog no. sc-13140, or anti-TAK1, Cell Signaling, catalog no. 5206S) at 4°C for 16 h, followed by incubation with protein A/G plus-agarose for 5 h. Parallel control experiments were performed by incubating the precleared cell lysate with normal rabbit IgG, followed by incubation with protein A/G plus-agarose. The agarose beads were collected by centrifugation, washed four times with lysis buffer, and heated to 95°C for 5 min after adding Laemmli buffer. The resulting immunoprecipitates were separated by SDS-PAGE and probed with the respective probing antibodies (anti-GPR120, anti-TAB1, Cell Signaling, catalog no. 3226S) in immunoblotting.
Cell surface biotinylation.
Cell surface biotinylation was performed using sulfo-NHS-SS-biotin (Thermo Scientific, Rockford, IL) (0.5 g/l) in borate buffer (in mmol/l: 154 NaCl, 7.2 KCl, 1.8 CaCl2, 10 H3BO3, pH 9.0), as previously described by our laboratory (4). Labeling was allowed to proceed for 60 min at 4°C to prevent endocytosis and internalization of antigens. After pull-down of biotinylated antigens with streptavidin agarose, biotinylated proteins were released by boiling in Laemmli buffer containing dithiothreitol, subjected to SDS-PAGE, and then probed with anti-GPR120 antibody. The surface GPR120 was compared with intracellular and total cellular GPR120, as determined by immunoblotting of the solubilized cell extract.
NF-κB activity.
NF-κB activation (basal or TNF-α induced) in response to GPR120 agonists was measured by NF-κB-luciferase reporter assay, as described earlier by our laboratory (5). Briefly, Caco-2 cells were transfected with p-NF-κB-Luc (Clontech, Mountain View, CA) and β-galactosidase expression vector using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). The former plasmid contains NF-κB binding consensus element upstream of luciferase reporter gene. Twenty-four hours after transfection, cells were treated with GPR120 agonists in the presence or absence of TNF-α (10 ng/ml) for another 24 h. Luciferase and β-galactosidase assays were performed according to the manufacturer's instructions. NF-κB activity was expressed in terms of relative luciferase activity normalized to β-galactosidase activity.
Preparation of nuclear extract and measurement of p65.
Nuclear extracts from control and agonists treated Caco-2/STC-1 cells were prepared using the nuclear extraction kit from Active Motif (Carlsbad, CA) following the manufacturer's protocol as described earlier by our laboratory (3). NF-κB activation was assessed by measuring levels of nuclear p65 by immunoblotting with anti-p65 antibody (Santa Cruz Biotechnology).
siRNA silencing of β-arrestin-2.
Small interfering RNA (siRNA) for human β-arrestin-2 (SI 02776928) and scrambled siRNA were obtained commercially from Qiagen, and transfection of cells (1 × 105 cells per well in six-well plates) was performed following the manufacturer's protocol. Forty-eight hours after transfection, cells were treated with GPR120 agonists for 30 min. Effects on GPR120 internalization and on NF-κB activation in response to β-arrestin-2 silencing, respectively, were assessed by cell surface biotinylation and by measuring levels of nuclear p65 by immunoblotting with anti-p65 antibody. The extent of β-arrestin-2 silencing was assessed by measuring its levels in the cell lysates by immunoblotting with anti-β-arrestin-2 antibody.
Immunofluorescence measurements of GLP-1.
STC-1 cells seeded on 12-well Transwell inserts at a density of 5,000 cells/well and grown to 60% confluency were treated with GPR120 agonists (GW9508, 50 μM or TUG-891, 10 μM) for 6 h. For immunostaining, the cells were washed with 1× PBS containing 1 mM CaCl2, pH 7.4, and then fixed with 2% paraformaldehyde at room temperature for 30 min. Fixed cells were permeabilized using 0.1% Triton X-100 in PBS and blocked in 5% normal goat serum for 2 h. Monolayers were then incubated with anti-GLP-1 (Santa Cruz, catalog no. sc-80602, 1:75 dilution) and anti-GPR120 (Novus Biologicals, catalog no. NBP1-00858, 1:75 dilution) antibodies for 2 h, followed by three washes for 5 min with 1× PBS containing CaCl2 and saponin. After washing, cells were finally incubated in Alexa Fluor 568-conjugated goat anti-mouse (for GLP-1) and Alexa Fluor 488-conjugated goat anti-rabbit (for GPR120) secondary antibodies (Invitrogen) at 1:100 dilution for 60 min at room temperature. Inserts were carefully removed from the Transwell and mounted on glass slides using Slowfade gold antifade reagent (Invitrogen). Images were obtained on Carl Zeiss LSM 510 META laser scanning confocal microscope equipped with a ×63 water-immersion objective. Beams of 488 nm and 534 nm from an Ag/Kr laser and 361 nm from a UV laser were used for excitation. LP505 and LP585 filters were used for detecting green and red fluorescence emissions, respectively. Multitracking function was used to sequentially scan two different fluorochromes to avoid any bleed-through among these fluorescent dyes.
Measurement of secreted and cellular levels of GLP-1 by ELISA.
STC-1 cells plated in six-well plates and grown to 60% confluency were treated with GPR120 agonists (GW9508, 50 μM or TUG-891, 10 μM) for 6 h. Cells were harvested following collection of the spent media, and protein concentrations were determined. Protein (20 μg) in a volume of 10 μl total lysate and 10 μl spent media were used to measure cellular and secreted GLP-1 levels, respectively, by ELISA (GLP-1 ELISA Kit, Immuno-biological Laboratories, Minneapolis, MN, catalog no. 27784) following the manufacturer's instructions. Results are expressed as picograms GLP-1 per milligram protein.
Statistical analyses.
The data presented are means ± SE of three to four independent experiments. Difference between controls vs. various treatments was analyzed using one-way ANOVA, with Dunnett's multiple-comparison tests for repeated comparisons to the control. Differences were considered significant at P ≤ 0.05.
RESULTS
GPR120 is differentially expressed along the length of mammalian intestine.
Expression of GPR120 in the intestine (13, 22) and in model intestinal epithelial cell lines (13, 17, 19) has previously been reported. However, distribution of GPR120 mRNA and protein along the length of the intestine in different mammalian species has not been examined. Therefore, we examined the relative expression of GPR120 along the length of human, rat, and mouse intestine. As shown in Fig. 1A, GPR120 mRNA levels in mouse and rat were more in the large intestine (cecum, proximal and distal colon) than in the small intestine (jejunum and ileum). Protein levels of GPR120 were also consistently more in the colonic region than in the small intestine in all of the three mammalian species tested (Fig. 1B). Consistent with earlier reports, our present data also showed GPR120 expression in STC-1 cells, a model endocrine cell line of intestinal epithelium (13, 17), as well as in absorptive enterocyte-like Caco-2 cells (19). Our data, however, showed substantially higher GPR120 protein level in STC-1 cells compared with its level in Caco-2 cells (Fig. 1C). Agonist stimulation of GPR120 in adipocytes and macrophages has previously been shown to elicit anti-inflammatory pathway via inhibition of NF-κB (21). Therefore, we next sought to examine potential anti-inflammatory effects of GPR120 activation in both model intestinal epithelial and endocrine cells.
Fig. 1.

GPR120 expression in mammalian intestinal epithelium. A: total RNA extracted from mucosal scrappings of different regions of mouse and rat intestine was utilized to measure GPR120 mRNA by real-time RT-PCR and normalized with GAPDH mRNA. B, left: lysates prepared from mucosal scrappings of human, mouse, and rat intestinal regions were subjected to SDS-PAGE and probed with GPR120 antibody in immunoblotting. Right: respective densitometric analysis of the band intensities. C, top: Caco-2 and STC-1 cell lysates were subjected to SDS-PAGE and probed with GPR120 antibody. Bottom: densitometric analysis of the band intensities. *Significant difference, P ≤ 0.05. B and C show representative blots from 3–4 independent experiments. Values are means ± SE. Jej, jejunum; Ile, ileum; PC, proximal colon; DC, distal colon.
Agonist stimulation internalizes GPR120 via interaction with β-arrestin-2 in Caco-2, but not in STC-1 cells.
Thus far, all anti-inflammatory effects of GPR120 activation in macrophages appeared to be mediated via GPR120-β-arrestin-2 signaling (13, 28). In this pathway, ligand stimulation has been shown to increase affinity of GPR120 intracellular domain to scaffold protein β-arrestin-2. GPR120 binding to β-arrestin-2 follows internalization of the GPR120-β-arrestin-2 complex and inhibition of proinflammatory responses such as NF-κB activation (18, 21). Therefore, we used synthetic GPR120 agonist GW9508 (13, 28) and the ω-3 FA DHA as natural agonist to stimulate GPR120 and measured GPR120-β-arrestin-2 interaction by coimmunoprecipitation and GPR120 internalization by cell-surface biotinylation in both Caco-2 and STC-1 cells. In Caco-2 cells, both GW9508 and DHA increased GPR120-β-arrestin-2 interactions (Fig. 2, top) that resulted in GPR120 internalization, as shown by decreased levels of GPR120 at the cell surface and corresponding increase in the intracellular levels (Fig. 2, bottom). It should be noted that marginal basal levels of GPR120-β-arrestin-2 interaction and internalization of GPR120 also occur in the absence of the agonists. On the other hand, in enteroendocrine STC-1 cells, GPR120 activation by GW9508 or DHA did not trigger GPR120-β-arrestin-2 interaction (Fig. 3, top) and GPR120 internalization (Fig. 3, bottom) compared with unstimulated cells. Specificity of GPR120-β-arrestin-2 interaction in immunoprecipitation experiments was confirmed by performing appropriate negative controls, where incubation of the cell lysates with normal rabbit IgG, instead of anti-β-arrestin-2 antibody, failed to immunoprecipitate GPR120 (not shown). These results suggest that, similar to what has been reported in macrophages, agonists stimulation triggers GPR120 to bind to β-arrestin-2 in intestinal epithelial Caco-2 cells, but not in enteroendocrine STC-1 cells. We further confirmed GPR120 binding to β-arrestin-2 in Caco-2 cells, and not in STC-1 cells, in response to treatments with TUG-891, a recently reported GPR120-specific agonist (11, 12). As shown in Fig. 4, TUG-891 augments GPR120-β-arrestin-2 coimmunoprecipitation in Caco-2 (A), but not in STC-1 cells (B).
Fig. 2.

Agonist stimulation internalizes GPR120 via interaction with β-arrestin-2 in Caco-2 cells. Top left: lysates of control or agonist [50 μM GW9508 or 50 μM docosahexaenoic acid (DHA)]-treated (30 min) Caco-2 cells, containing equal amounts of proteins, were used to immunoprecipitate (IP) GPR120 with anti-β-arrestin-2 antibody. IPs were subjected to SDS-PAGE and probed with anti-GPR120 antibody in immunoblotting. After being stripped with 0.2 N NaOH, blots were reprobed with anti-β-arrestin-2 antibody. Representative blot of 3 independent experiments is shown. Top right: densitometric analysis of band intensities of GPR120/β-arrestin-2 in different groups is shown. Values are means ± SE; n = 3. *Different from control, P ≤ 0.05. Bottom left: monolayers of Caco-2. Ten-day postplating were treated with 50 μM GW9508 or 50 μM DHA for 30 min, and cell surface levels of GPR120 were measured by cell-surface biotinylation. Representative blot of 3 independent experiments is shown. Bottom right: densitometric analysis of band intensities of apical GPR120/total GPR120 in different groups is shown. Values are means ± SE; n = 3. *Different from control, P ≤ 0.05. WB, Western blot.
Fig. 3.

Agonist stimulation does not trigger β-arrestin-2-mediated internalization of GPR120 in STC-1 cells. Top left: lysates of control or agonist (50 μM GW9508 or 50 μM DHA)-treated (30 min) STC-1 cells, containing equal amounts of proteins, were used to IP GPR120 with anti-β-arrestin-2 antibody. IPs were subjected to SDS-PAGE and probed with anti-GPR120 antibody in immunoblotting. After being stripped with 0.2 N NaOH, blots were reprobed with anti-β-arrestin-2 antibody. Representative blot of 3 independent experiments is shown. Top right: densitometric analysis of band intensities of GPR120/β-arrestin-2 in different groups is shown. Values are means ± SE; n = 3. Bottom left: STC-1 cells (7-day postplating) were treated with 50 μM GW9508 or 50 μM DHA for 30 min, and cell surface levels of GPR120 were measured by cell-surface biotinylation. Representative blot of 3 independent experiments is shown. Bottom right: densitometric analysis of band intensities of apical GPR120/total GPR120 in different groups is shown. Values are means ± SE; n = 3.
Fig. 4.

TUG-891 stimulation enhances GPR120-β-arrestin-2 interaction in Caco-2, but not in STC-1 cells. Top: lysates of control or 10 μM TUG-891-treated Caco-2 (A) or STC-1 (B) cells, containing equal amounts of proteins, were used to IP GPR120 with anti-β-arrestin-2 antibody. IPs were subjected to SDS-PAGE and probed with anti-GPR120 antibody in immunoblotting. After being stripped with 0.2 N NaOH, blots were reprobed with anti-β-arrestin-2 antibody. Representative blots of 3 independent experiments are shown. Bottom: densitometric analysis of band intensities of GPR120/β-arrestin-2 in different groups is shown. Values are means ± SE; n = 3. *Different from control, P ≤ 0.05.
Agonist stimulation of GPR120 in Caco-2 cells increases β-arrestin-2-TAB1 interaction and attenuates TAB1 binding to TAK1.
Previous studies in monocytic RAW 264.7 cells showed that β-arrestin-2-mediated GPR120 internalization caused association of β-arrestin-2 with TAB1 that in turn blocked TAB1 association with TAK1 and thus inhibited TAK1-induced inflammatory signaling (21). Since agonists stimulation of GPR120 in Caco-2 cells induced its β-arrestin-2-mediated internalization, we utilized coimmunoprecipitation studies to investigate the downstream effects on β-arrestin-2 association with TAB1 and TAB1 interaction with TAK1. Our results showed that GPR120 stimulation by GW9508 or TUG-891 increased β-arrestin-2 interaction with TAB1 (Fig. 5A). On the other hand, our results further showed that GPR120 stimulation attenuated basal or TNF-α-induced association of TAB1 with TAK1 (Fig. 5B). Specificity of β-arrestin-2-TAB1 and TAK1-TAB1 interactions in immunoprecipitation experiments was confirmed by performing appropriate negative controls, where incubation of the cell lysates with normal rabbit IgG, instead of anti-β-arrestin-2 or anti-TAK1 antibodies, failed to immunoprecipitate TAB1 (not shown). Taken together, our results suggest that signaling events downstream of GPR120 internalization in Caco-2 cells are similar to those reported earlier in macrophages and monocytes (21). Since agonist treatments did not induce GPR120 internalization or association with β-arrestin-2 in STC-1 cells, we did not attempt to examine these downstream effects in this cell line.
Fig. 5.

Agonist stimulation of GPR120 in Caco-2 cells increases β-arrestin-2-TAB1 (TAK1 binding protein 1) interaction and attenuates TAB1 binding to TAK1 (transforming growth factor-β-activated kinase 1). A, left: lysates of control or agonist (50 μM GW9508 or 10 μM TUG-891)-treated (30 min) Caco-2 cells, containing equal amounts of proteins, were used to IP TAB1 with anti-β-arrestin-2 antibody. IPs were subjected to SDS-PAGE and probed with anti-TAB1 antibody in immunoblotting. After being stripped with 0.2 N NaOH, blots were reprobed with anti-β-arrestin-2 antibody. Left: densitometric analysis of band intensities of TAB1/β-arrestin-2 in different groups is shown. Values are means ± SE; n = 3. *Different from control, P ≤ 0.05. B, right: Caco-2 cells with or without pretreatments with TNF-α (10 ng/ml) for 6 h were treated with the agonists [GW9508 (50 μM) or TUG-891 (10 μM)] in presence or absence of TNF-α (10 ng/ml) for 30 min. Cell lysates containing equal amounts of proteins were used to IP TAB1 with anti-TAK1 antibody. IPs were subjected to SDS-PAGE and probed with anti-TAB1 antibody in immunoblotting. After being stripped with 0.2 N NaOH, blots were reprobed with anti-TAK1 antibody. Left: densitometric analysis of band intensities of TAB1/TAK1 in different groups is shown. Values are means ± SE; n = 3. *Difference between groups: control vs. TNF-α or agonists, TNF-α vs. agonists, P ≤ 0.05.
Agonist stimulation of GPR120 in Caco-2 cells inhibits NF-κB activation.
Earlier reports in monocytic RAW 264.7 cells showed that blocking TAB1 association with TAK1 resulted in anti-inflammatory effects via inhibition of Toll-like receptor 4 or TNF-α-induced NF-κB activation (21, 23). Since agonist stimulation-induced GPR120 signaling in Caco-2 cells also attenuated TAB1-TAK1 interactions, we examined whether these effects led to inhibition of NF-κB activation. We used the NF-κB transcription reporter vector p-NF-κB-Luc for transfection of Caco-2 cells. This vector contains NF-κB consensus sequence located upstream of the firefly luciferase reporter gene. When the plasmid is transfected into an appropriate cell line, expression of the reporter gene can be induced by adding a stimulus that activates the NF-κB pathway. As shown in Fig. 6A, treatment of Caco-2 cells with either GW9508 or TUG-891 significantly inhibited basal as well as TNF-α-induced NF-κB activity.
Fig. 6.

Agonist stimulation of GPR120 inhibits NF-κB activation in Caco-2 and not in STC-1 cells. A: Caco-2 cells transfected with p-NF-κB-Luc reporter plasmid and β-galactosidase expression vector were pretreated with TNF-α (10 ng/ml) for 6 h, followed by treatments with GW9508 (50 μM) or TUG-891 (10 μM) in presence or absence of TNF-α (10 ng/ml) for another 24 h. Cells were harvested; luciferase activities were measured and normalized with the respective β-galactosidase activities. Values are means ± SE; n = 3. *Different from control, P ≤ 0.05. B, top: nuclear extracts were prepared from control or agonist (50 μM GW9508 or 10 μM TUG-891)-treated (6 h) STC-1 cells. Nuclear extracts were subjected to SDS-PAGE and probed with anti-p-65 antibody in immunoblotting. After being stripped with 0.2 N NaOH, blots were reprobed with anti-lamin antibody as loading control. Densitometric analysis of band intensities of p65/lamin in different groups is shown. Values are means ± SE; n = 3.
Although agonist treatments did not induce GPR120 internalization or association with β-arrestin-2 in STC-1 cells, it was logical to examine whether the agonists still could modulate NF-κB activation in this cell line via other mechanisms. Because of very low transfection efficiency of STC-1 cells, however, we did not use NF-κB transcription reporter vector p-NF-κB-Luc and instead examined nuclear p65 level to assess the effects of GPR120 agonists on basal or TNF-α-induced NF-κB activation. As shown in Fig. 6B, treatments of STC-1 cells with GW9508 or TUG-891 for 6 h had no effects on basal NF-κB activity, as there were no changes in the nuclear p65 levels. Furthermore, we did not observe increased nuclear p65 in response to TNF-α in this cell line (results not shown), and, therefore, it was not possible to compare the effects of GPR120 agonists on TNF-α-induced NF-κB activation.
β-Arrestin-2 knockdown abrogates GPR120 stimulation-dependent inhibition of NF-κB in Caco-2 cells.
To further confirm the occurrence of a β-arrestin-2-dependent anti-inflammatory pathway in Caco-2 cells, β-arrestin-2 was knocked down by siRNA, and NF-κB activation was assessed by measuring nuclear levels of p65. As shown in Fig. 7, β-arrestin-2-specific siRNA substantially knocked down its expression (A); GPR120 was internalized in response to GW9508 in control cells but not in β-arrestin-2-deficient cells (B). Furthermore, there was a significant decrease in nuclear p65 level following GW9508 treatment of control siRNA transfected cells. However, GW9508 treatment did not alter nuclear p65 level in cells with siRNA knockdown of β-arrestin-2 (Fig. 7C).
Fig. 7.
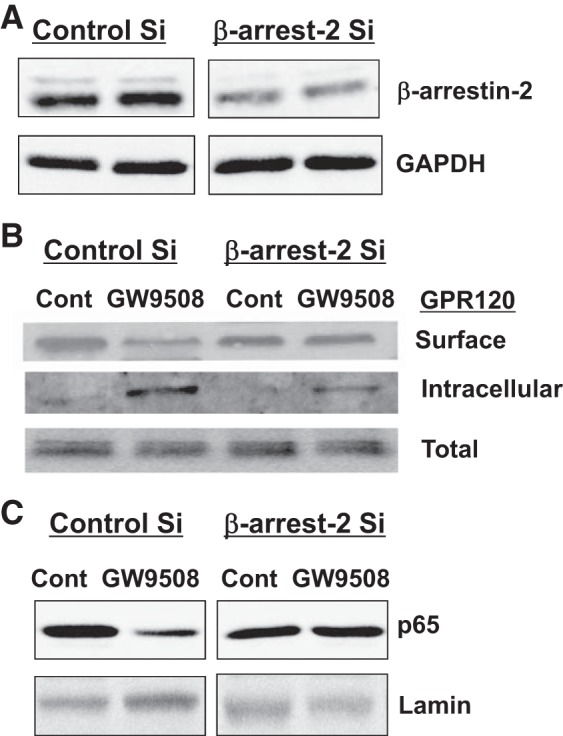
β-Arrestin-2 knockdown abrogates agonist stimulation-dependent internalization of GPR120 and inhibition of NF-κB. Caco-2 cells were transfected with control-scrambled small interfering (siRNA) (Si) or β-arrestin-2 siRNA, as described in materials and methods. Forty-eight hours after transfection, cells were used either to measure β-arrestin-2 levels by immunoblotting with anti-β-arrestin-2 antibody (A), or to measure surface GPR120 by cell surface biotinylation (B), or to prepare nuclear extracts and measure p65 levels by WB (C). Representative blots of 3 independent experiments are shown in A–C.
GPR120 stimulation in STC-1 cells increases GLP-1 level.
Previous studies have shown that agonist stimulation of GPR120 in STC-1 cells leads to increased GLP-1 secretion (10, 28). Our present studies examined whether GPR120 stimulation could, as shown in macrophages (21, 23), elicit an anti-inflammatory pathway in different in vitro models of intestinal epithelial cell lines. However, our results showed β-arrestin-2-dependent anti-inflammatory responses in Caco-2, but not in STC-1 cells, a model of enteroendocrine cells. Therefore, we sought to verify whether GPR120 activation in this cell line still leads to increased GLP-1 level. Our results, as measured by immunofluorescence, showed that treatment of STC-1 cells with GW9508 or TUG-891 increased GLP-1 immunofluorescence compared with control (Fig. 8), although these agonists had no effects on GPR120 immunofluorescence. We next used ELISA to quantitatively measure GLP-1 levels in STC-1 cells and those secreted into the cell culture media in response to treatments with GW9508 and TUG-891. As shown in Fig. 9, both agonists significantly increased cellular and secreted levels of GLP-1, with TUG-891 showing a greater effect.
Fig. 8.

Glucagon-like peptide-1 (GLP-1) expression in STC-1 cells in response to GPR120 agonists. Immunofluorescent staining was performed in control or agonist-treated [50 μM GW9508 (A) or 10 μM TUG-891 (B), 6 h] STC-1 cells grown on six-well Transwell inserts. GLP-1 is shown in red, and GPR120 in green. Images were obtained on Carl Zeiss LSM 510 META laser scanning confocal microscope equipped with a ×63 water-immersion objective. Representative images from 3 different experiments are shown.
Fig. 9.

GPR120 agonists increase cellular and secreted levels of GLP-1 in STC-1 cells. GLP-1 assay was performed by ELISA, as described in materials and methods, in agonist-treated (50 μM GW9508 or 10 μM TUG-891, 6 h) STC-1 cells and in the spent media. Values are means ± SE in pg GLP-1/mg protein; n = 3. *Different from control, P ≤ 0.05.
DISCUSSION
In this report, we show differential effects of activation of the free FA receptor GPR120 (also known as FFAR4) in human intestinal Caco-2 and mouse intestinal STC-1 cells, two model cell lines representing two different cell types of the mammalian intestinal epithelial layer. GPR120 is expressed in diverse tissues with abundant expression in the intestine (10, 13, 28). The columnar epithelium that lines the inner surface of the intestine consists of several distinct cell types that are rapidly and continually renewed by intestinal stem cells that reside near the base of the crypts of Lieberkuhn (24). The absorptive enterocytes/colonocytes comprise the highest population, followed by the secretory goblet cells, whereas enteroendocrine cells, scattered as individual cells throughout the epithelium, comprise only 1% of the total cells of the epithelium (16, 24). Our results on the relative levels of GPR120 mRNA and protein along the length of the intestine of human, mouse, and rat showed similar expression patterns, with higher expression in the large intestine compared with ileum and jejunum. This could presumably be due to higher population of L-type enteroendocrine cells in the colonic epithelium compared with that in the upper segments of the intestine (7, 8). However, since colon also harbors the highest population of bacteria belonging to diverse species, higher expression of GPR120 in the colon could also be due to the availability of specific bacterial metabolites that could induce signaling pathways via activation of GPR120 (7).
GPR120 is activated by unsaturated medium- to long-chain FFAs (10, 14) and has a critical role in various physiological processes, such as incretin hormone secretion, food preference, insulin signaling, and adipogenesis (14, 18, 28). GPR120 was first identified as a FFAR regulating GLP-1 secretion in mice in response to dietary FAs. Unsaturated long-chain FFAs, especially α-linolenic acid, showed a dose-dependent stimulation of GLP-1 secretion in the mouse enteroendocrine cell line STC-1 (10). Transfection with GPR120-specific siRNA, but not GPR40-specific siRNA, significantly reduced α-linolenic acid-induced GLP-1 secretion, indicating that the FFA-induced GLP-1 secretion in STC-1 cells is mediated through GPR120 (10).
Later studies established the critical importance of the GPR120 as a ω-3 FA receptor in mediating potent anti-inflammatory effects both in vitro and in vivo in proinflammatory CD11c+ macrophages and monocytic RAW 264.7 cells (21). Importantly, the observed anti-inflammatory effects were shown to be completely abrogated by siRNA-mediated knockdown of GPR120. Elucidation of the role of GPR120 in mediating potent anti-inflammatory effects established a new route toward understanding the pathogenesis of multiple inflammatory diseases. Therefore, in view of its abundant expression in the intestine, more particularly in the large intestine, we hypothesized that GPR120 could play a pivotal role to confer potent anti-inflammatory effects in intestinal inflammatory disorders. Since intestinal epithelial cells are of critical importance in the pathophysiology of inflammatory bowel diseases (IBD), including Crohn's disease and ulcerative colitis, we sought to investigate GPR120 activation-dependent anti-inflammatory pathways in intestinal epithelial cells utilizing in vitro models of human enterocyte-like Caco-2 and mouse endocrine cell line STC-1. Earlier studies reported GPR120 expression in both STC-1 (13, 17) and Caco-2 (19) cells. Our present studies for the first time demonstrated that ligand activation of GPR120 in intestinal epithelial Caco-2 cells exerted anti-inflammatory effects via inhibition of NF-κB activation, similar to the effects shown earlier in monocytes and macrophages (21). Interestingly, this anti-inflammatory response was not observed in response to GPR120 activation in STC-1 cells, an epithelial endocrine cell line in which GPR120 activation has been shown earlier to stimulate GLP-1 secretion. Our present studies also demonstrated increased levels of GLP-1 compared with control in response to treatments with the GPR120 agonists GW9508 and TUG-891. It should be emphasized, however, that absence of this anti-inflammatory response in STC-1 cells do not necessarily represent a condition in vivo, as this could be due to loss of certain factors/elements in STC-1 cells that are expressed in L cells in vivo and are pivotal in mediating this anti-inflammatory response.
GPR120 has primarily been shown to mediate its effects either via coupling to Gαq proteins or via G-protein-independent and β-arrestin-2-mediated pathways (18, 28). G protein-coupled pathway is involved in GLP-1 secretion (18, 28). On the other hand, stimulation of GPR120 by the ω-3 FA DHA or by the synthetic agonist GW9508 in monocytic RAW 264.7 cells has been shown to increase affinity of GPR120 intracellular domain to the scaffold protein β-arrestin-2 (21). GPR120 binding to β-arrestin-2 follows internalization of the GPR120-β-arrestin-2 complex and inhibition of proinflammatory responses such as NF-κB activation. Our studies also demonstrated β-arrestin-2 dependence of the anti-inflammatory activity of GPR120 in Caco-2 cells. Similar to earlier reports (21), abrogation of β-arrestin-2 signaling via selective RNA interference was shown to attenuate GW9508-mediated anti-inflammatory effects in Caco-2 cells, suggesting that, on binding to GPR120, the ligands exert their effects via β-arrestin-2 signaling. It should be noted that the dual specificity of the synthetic agonist GW9508 for GPR120 as well as for GPR40, another FFAR with high affinity for medium- and long-chain FFAs (28), could present as a confounding variable in the interpretation of results in studies using GW9508 as a result of off-target effects at the other receptor. However, this discrepancy seemed to be ruled out in our studies based on the earlier reports showing that Caco-2 cells do express GPR120, but not GPR40 (19). Furthermore, we also confirmed the role of GPR120 in parallel studies by using TUG891, a ligand with greater selectivity and potency to GPR120 than GPR40 (11), which was commercially available to us only during the later part of this study.
The distinct mechanisms of GPR120 action via coupling of the receptor either to Gαq or to β-arrestin-2 might account for the divergence in the reported functions of this receptor in different tissues. Our studies further suggested that GPR120 stimulation could induce distinct signaling pathways, not only in different tissue types, but also in different cell types of the same tissue, such as intestinal epithelium. Thus cell type-specific effects of GPR120 agonism are of critical importance in understanding how pharmaceutical agents that target a distinct signaling pathway could be developed for the treatment of specific disease states. In this regard, GPR120 has received increasing interest in recent years as a therapeutic target for the treatment of both metabolic and inflammatory diseases. Our results clearly demonstrated a distinct anti-inflammatory role of GPR120 in intestinal epithelial cells via inhibition of NF-κB. The transcription factor NF-κB is a key regulator of inflammation, and activation of NF-κB plays a key role in the initiation and perpetuation of the inflammatory diseases, including Crohn's disease and ulcerative colitis, the two major forms of IBD (1, 27). Omega-3 polyunsaturated FAs, as well as conjugated linoleic acids (2, 6), are known to have potent anti-inflammatory properties and have been used in multiple clinical trials and experimental models to assess the therapeutic benefits in treatment and/or prevention of various inflammatory diseases, including IBD. However, the mechanistic details of the role of GPR120 in mediating the beneficial effects of these diet and/or gut microbiota-derived compounds is pivotal for designing targeted therapeutic approaches for IBD. Our future studies, therefore, will attempt to delineate the mechanisms of the anti-inflammatory role of GPR120 in intestinal epithelial cells in normal physiology as well as under inflammatory conditions.
GRANTS
These studies were supported by Bill & Melinda Gates Foundation Grant OPP1058288 (A. Borthakur); National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-54016, DK-81858, DK-92441 (P. K. Dudeja), and DK-71596 (W. A. Alrefai); and Department of Veteran Affairs Grants BX 002011 (P. K. Dudeja) and BX 000152 (W. A. Alrefai).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.N.A., S.P., T.G., and S.B. performed experiments; A.N.A., S.P., and A.B. analyzed data; A.N.A., W.A.A., P.K.D., and A.B. interpreted results of experiments; A.N.A., S.P., and T.G. prepared figures; A.N.A. and A.B. drafted manuscript; W.A.A., P.K.D., and A.B. edited and revised manuscript; P.K.D. and A.B. approved final version of manuscript; A.B. conception and design of research.
REFERENCES
- 1.Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med 263: 591–596, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Bassaganya-Riera J, Hontecillas R. Dietary conjugated linoleic acid and n-3 polyunsaturated fatty acids in inflammatory bowel disease. Curr Opin Clin Nutr Metab Care 13: 569–573, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borthakur A, Bhattacharyya S, Kumar A, Anbazhagan AN, Tobacman JK, Dudeja PK. Lactobacillus acidophilus alleviates platelet-activating factor-induced inflammatory responses in human intestinal epithelial cells. PLoS One 8: e75664, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borthakur A, Gill RK, Tyagi S, Koutsouris A, Alrefai WA, Hecht GA, Ramaswamy K, Dudeja PK. The probiotic Lactobacillus acidophilus stimulates chloride/hydroxyl exchange activity in human intestinal epithelial cells. J Nutr 138: 1355–1359, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borthakur A, Saksena S, Gill RK, Alrefai WA, Ramaswamy K, Dudeja PK. Regulation of monocarboxylate transporter 1 (MCT1) promoter by butyrate in human intestinal epithelial cells: Involvement of NF-kappaB pathway. J Cell Biochem 103: 1452–1463, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calder PC. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim Biophys Acta 1851: 469–484, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep 9: 1202–1208, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diakogiannaki E, Gribble FM, Reimann F. Nutrient detection by incretin hormone secreting cells. Physiol Behav 106: 387–393, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Druart C, Neyrinck AM, Vlaeminck B, Fievez V, Cani PD, Delzenne NM. Role of the lower and upper intestine in the production and absorption of gut microbiota-derived PUFA metabolites. PLoS One 9: e87560, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11: 90–94, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Hudson BD, Shimpukade B, Mackenzie AE, Butcher AJ, Pediani JD, Christiansen E, Heathcote H, Tobin AB, Ulven T, Milligan G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol 84: 710–725, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudson BD, Shimpukade B, Milligan G, Ulven T. The molecular basis of ligand interaction at free fatty acid receptor 4 (FFA4/GPR120). J Biol Chem 289: 20345–20358, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichimura A, Hara T, Hirasawa A. Regulation of energy homeostasis via GPR120. Front Endocrinol (Lausanne) 5: 111, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol 5: 236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasaki K, Harada N, Sasaki K, Yamane S, Iida K, Suzuki K, Hamasaki A, Nasteska D, Shibue K, Joo E, Harada T, Hashimoto T, Asakawa Y, Hirasawa A, Inagaki N. Free fatty acid receptor GPR120 is highly expressed in enteroendocrine K cells of the upper small intestine and has a critical role in GIP secretion after fat ingestion. Endocrinology 156: 837–846, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Jeon MK, Klaus C, Kaemmerer E, Gassler N. Intestinal barrier: molecular pathways and modifiers. World J Gastrointest Pathophysiol 4: 94–99, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katsuma S, Hatae N, Yano T, Ruike Y, Kimura M, Hirasawa A, Tsujimoto G. Free fatty acids inhibit serum deprivation-induced apoptosis through GPR120 in a murine enteroendocrine cell line STC-1. J Biol Chem 280: 19507–19515, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Mo XL, Wei HK, Peng J, Tao YX. Free fatty acid receptor GPR120 and pathogenesis of obesity and type 2 diabetes mellitus. Prog Mol Biol Transl Sci 114: 251–276, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Mobraten K, Haug TM, Kleiveland CR, Lea T. Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis 12: 101, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, Sasik R, Hah N, Chi TJ, Cox JM, Powels MA, Di Salvo J, Sinz C, Watkins SM, Armando AM, Chung H, Evans RM, Quehenberger O, McNelis J, Bogner-Strauss JG, Olefsky JM. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med 20: 942–947, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–698, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulsen SJ, Larsen LK, Hansen G, Chelur S, Larsen PJ, Vrang N. Expression of the fatty acid receptor GPR120 in the gut of diet-induced-obese rats and its role in GLP-1 secretion. PLoS One 9: e88227, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talukdar S, Olefsky JM, Osborn O. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci 32: 543–550, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71: 241–260, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Wellhauser L, Belsham DD. Activation of the omega-3 fatty acid receptor GPR120 mediates anti-inflammatory actions in immortalized hypothalamic neurons. J Neuroinflammation 11: 60, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams-Bey Y, Boularan C, Vural A, Huang NN, Hwang IY, Shan-Shi C, Kehrl JH. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-kappaB activation and enhancing autophagy. PLoS One 9: e97957, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wullaert A. Role of NF-kappaB activation in intestinal immune homeostasis. Int J Med Microbiol 300: 49–56, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Zhang D, Leung PS. Potential roles of GPR120 and its agonists in the management of diabetes. Drug Des Devel Ther 8: 1013–1027, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]