

Abstract
Lung epithelial cell death is a prominent feature involved in the development of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Hyperoxia-induced ALI is an established animal model mimicking human ARDS. Small noncoding RNAs such as microRNAs (miRNAs) have potent physiological and pathological functions involving multiple disease processes. Emerging interests focus on the potential of miRNAs to serve as novel therapeutic targets and diagnostic biomarkers. We found that hyperoxia highly induces miR-185 and its precursor in human lung epithelial cells in a time-dependent manner, and this observation is confirmed using mouse primary lung epithelial cells. The hyperoxia-induced miR-185 is mediated by reactive oxygen species. Furthermore, histone deacetylase 4 (HDAC4) locates in the promoter region of miR-185. We found that hyperoxia suppresses HDAC4 specifically in a time-dependent manner and subsequently affects histone deacetylation, resulting in an elevated miR-185 transcription. Using MC1586, an inhibitor of class IIa HDACs, we showed that inhibition of class IIa HDACs upregulates the expression of miR-185, mimicking the effects of hyperoxia. Functionally, miR-185 promotes hyperoxia-induced lung epithelial cell death through inducing DNA damage. We confirmed functional roles of miR-185 using both the loss- and gain-of-function approaches. Moreover, multiple 14-3-3δ pathway proteins are highly attenuated by miR-185 in the presence of hyperoxia. Taken together, hyperoxia-induced miR-185 in lung epithelial cells contributes to oxidative stress-associated epithelial cell death through enhanced DNA damage and modulation of 14-3-3δ pathways.
Keywords: miR-185, epithelial cell, cell death, oxidative stress, hyperoxia
acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) have high mortality and morbidity, reflecting the inefficacy of specific therapies despite decades of research. Features of ALI/ARDS include an intense inflammatory response, diffuse alveolar cell damage, severe injury to the epithelial and endothelial barriers, and alveolar edema. Hyperoxia-induced ALI in mice is an established animal model that mimics human ARDS (20, 21, 42). Hyperoxia induces lung epithelial cell death via multiple mechanisms, including but not limited to apoptosis, necrosis, and autophagic cell death (7, 35, 38). Hyperoxia-associated oxidative stress has been reported to mediate lung epithelial cell damage (35). However, the detailed underlying mechanism involving this process remains incompletely understood, impeding the development of specific and effective therapy.
miRNA is a small noncoding RNA molecule (∼22 nucleotides) that is highly conserved from plants to mammals (19, 51). They function in RNA silencing and posttranscriptional regulation for tight control of target gene expression (18). Emerging evidence suggests that miRNAs are involved in numerous disease processes and are interesting targets for the development of diagnostic and therapeutic reagents (26, 50). Despite a small number of reports (22, 29), the function and regulation of miRNAs in lung epithelial cell death remain largely unexplored.
Previous reports have demonstrated that miRNAs bind their target genes via sequence complementary within the 3′-untranslated region or open reading frame of coding miRNAs (17), which subsequently trigger mRNA degradation and result in inhibiting transcription and translation of the target genes (4). Multiple miRNAs have been implicated in regulating cell death via multiple pathways (43). After hyperoxia, miRNA microarray and real-time PCR analyses have demonstrated upregulation of miR-21 and miR-34a in rat neonatal lungs (8). Furthermore, a previous study shows that miR-30a decreases in lung fibrosis (31). Upregulation of miR-30a potentially decreases apoptosis of type II alveolar epithelial cells (31). Many more miRNAs have been reported involving lung cancer cell apoptosis, metastasis, and proliferation (3, 15, 34). In fact, >2,000 unique mature human miRNAs have been reported on miRBase v. 20 in 2013 (25). Among the majority of these miRNAs, we do not have clear understanding on their expression, regulation, and/or functions, particularly in the pathogenesis of human diseases.
Presently, the entire miRNA profiles involving the development of ALI/ARDS are unknown. Here we report a novel finding on an miRNA that has not been studied in this field but has potential important function(s) in lung diseases. We found that miR-185 expression is highly induced in lung epithelial cells after hyperoxia. In this report, we further explored the regulation and function of miR-185 in lung epithelial cells. miR-185 is located in the 22q11.2 region of the chromosome and is encoded within an intron of the transport and Golgi organization 2 homolog (TANGO2) gene that currently has an unknown function (53). We also investigated the effects of hyperoxia on TANGO2. Insights from our studies on miR-185 described herein may shed light on future understandings of the role of TANGO2.
MATERIALS AND METHODS
Animal, cell culture, and isolation of primary lung epithelial cells.
Wild-type C57BL/6 mice (male, 6–8 wk of age) were obtained from Jackson Laboratory (Bar Harbor, ME). All the protocols involving animals in this study were approved by the institutional animal care and use committee (IACUC) of Boston University.
Beas2B cells were obtained from ATCC (Manassas, VA) and cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (GIBCO, Grand Island, NY). All cells were cultured at 37°C in a humidified atmosphere with 5% CO2-95% air. For hyperoxia treatment, cells were exposed to hyperoxia (95% oxygen-5% CO2) in modular exposure chambers. For N-acetyl-l-cysteine (NAC), Mito-TEMPO, or H2O2 treatment, 5 mM NAC, 100 μM Mito-TEMPO, or 90 μM H2O2, respectively, was added to the culture medium. MC1568, an inhibitor of class IIa histone deacetylases (HDACs), was purchased from Selleck Chemicals (Houston, TX).
Primary alveolar epithelial cells were isolated from the wild-type mice as described previously (27). Briefly, mouse lung tissue was washed with PBS, followed by 2 ml of dispase, and 0.5 ml of 1% agarose. Lung tissue was then dissociated in DMEM with 25 mM HEPES and 200 U/ml DNase. Isolated cells were plated on CD45- and CD16/32-precoated dishes. After centrifuge, the pellets were resuspended in DMEM containing 10% FBS.
Western blot analysis.
Western Blot analysis was performed as described before (56). Briefly, cells were homogenized in RIPA lysis buffer supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail (Sigma, St. Louis, MO). Protein lysates were resolved on SDS-PAGE gels before being transferred to the PVDF membrane. Anti-HDAC1, anti-HDAC2, anti-HDAC3, and anti-HDAC4 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-HDAC5, anti-HDAC6, anti-histone H3, anti-acetyl-histone H3, anti-histone H4, and anti-acetyl-histone H4 antibodies were obtained from Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-GAPDH antibody (Thermo Fisher Scientific, Waltham, MA) was used as a loading control. The densities of bands were quantitated using ImageJ software.
Caspase activity assay.
Caspase-Glo-3/7 assay kit was purchased from Promega (Madison, WI). The activity of caspase-3/7 was detected according to the manufacturer's protocol. In brief, cells were cultured in 96-well plates at a density of 8,000 cells/well. Caspase-Glo-3/7 reagent (100 μl) was then added to each well of the plate. The plate was mixed gently and incubated at room temperature for 45 min, followed by obtaining the luminescence of each sample as relative light units using a plate-reading luminometer.
Chromatin immunoprecipitation assay.
ChIP-IT Express Chromatin Immunoprecipitation Kit (cat no. 53009) was purchased from Active Motif. Chromatin immunoprecipitation (ChIP) assay was performed according to the manufacturer's instructions. In brief, Beas2B cells were grown in 10-cm cell culture dishes and were exposed to normoxia (room air) or hyperoxia for 2 days. After being cross linked with formaldehyde, chromatin was sheared using enzymatic digestion. HDAC4 antibody (H-92, Santa Cruz Biotechnology) was used to capture HDAC4/DNA complexes. DNA was recovered using Proteinase K digestion and analyzed using PCR. The primers used for human TANGO2 promoter were as follows: forward primer (5′-AGGTGGCAGCCTCCGAGCGA-3′); reverse primer (5′-AAGCCGGCGCGTTCACCATT-3′).
RNA preparation, reverse transcription, and quantitative real-time PCR.
MiRNeasy Mini Kits (cat. no. 217004; Qiagen, Valencia, CA) were used for purification of total RNA from tissue and cells. Single-stranded cDNA was generated according to the manuals of the High-Capacity cDNA Reverse Transcription Kit (cat. no. 4374966, Thermo Fisher Scientific). For miR-185 detection, real-time PCR was performed using TaqMan PCR kit (cat. no. 4427975-002271, Thermo Fisher Scientific) and Applied Biosystems StepOnePlus Real-Time PCR Systems (Foster City, CA). Relative miR-185 expression level was normalized to human HPRT1 (cat. no. 4331182-Hs99999909_m1) or mouse Hprt (cat. no. 4331182-Mm03024075_m1), respectively. For the detection of mouse HDAC1 to HDAC6, human TANGO2, and premiR-185, SYBR green-based real-time PCR technique was used as previously described (55). 18S rRNA was used as a reference housekeeping gene. The list of primers is shown in Table 1.
Table 1.
Primers used in qRT-PCR
| Gene Symbol | Sequence (5′-3′) | Length of Product, bp |
|---|---|---|
| 18S-F | ACCGCAGCTAGGAATAATGGA | 157 |
| 18S-R | CAAATGCTTTCGCTCTGGTC | |
| hTANGO2-F | CTGGCAGCACTCACCAACTAC | 90 |
| hTANGO2-R | GTCAGTGGTCAGAAAGTGGGT | |
| mTANGO2-F | AAGTTTGATCCTCGTCCTGTTTC | 107 |
| mTANGO2-R | TTCCCCCAGAAGTCTGCCAA | |
| premiR-185-F | AGGGATTGGAGAGAAAGGCA | 66 |
| premiR-185-R | AAGGACCAGAGGAAAGCCAG | |
| mHdac1-F | TGCTCGCTGCTGGACTTAC | 67 |
| mHdac1-R | GTAGGGCAGCTCATTAGGGATCT | |
| mHdac2-F | AGAAGATTGTCCGGTGTTTGATG | 76 |
| mHdac2-R | CACAGCCCCAGCAACTGAA | |
| mHdac3-F | TCAGCCCCACCAATATGCA | 92 |
| mHdac3-R | GAACTCGAAAAGTCCTGGAAACA | |
| mHdac4-F | GCTCTCCCAGCTCTCCAGCA | 102 |
| mHdac4-R | GTTGTGAGCTGCTGCACCGT | |
| mHdac5-F | TGAGAGGCAGGCCCTTCAGT | 104 |
| mHdac5-R | CCTCCAGTGCCACTCCCAAC | |
| mHdac6-F | ACCGGTATGACCGTGGCACT | 102 |
| mHdac6-R | TCCAGGGCACATTGACAGTGA | |
| hCCNB3-F | ATGAAGGCAGTATGCAAGAAGG | 100 |
| hCCNB3-R | CATCCACACGAGGTGAGTTGT | |
| hSFN-F | ACTTTTCCGTCTTCCACTACGA | 165 |
| hSFN-R | ACAGTGTCAGGTTGTCTCGC |
F, forward; R, reverse.
Apoptosis antibody array.
The kit of human apoptosis antibody array (cat. no. AAH-APO-1-4) was purchased from RayBiotech (Norcross, Georgia). Assay was performed according to the manufacturer's protocol.
Cell proliferation and viability assay, M30 immunofluorescence, and annexin V propidium iodide assay.
Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto, Japan) was used to determine the proliferation of Beas2B cells and to detect the cell viability after hyperoxia.
For M30 immunofluorescence, fixed Beas2B cells were incubated with M30 CytoDEATH and fluorescein antibody (cat. no. 12156857001; Roche, Indianapolis, Indiana) according to the standard protocol specified by the manufacturer. Images were captured using fluorescence microscope (Eclipse TS100, Nikon) at ×10 magnification and analyzed using ImageJ software.
Annexin V-FITC Apoptosis Detection Kit was used (BioVision, Mountain View, CA) according to the manufacturer's protocol. Data were obtained and analyzed using FACSCanto flow cytometer and Flow-Jo software (BD Biosciences Immunocytometry Systems, San Jose, CA).
ELISA.
Histone H3 total acetylation detection fast kit (ab115124) and histone H4 total acetylation detection fast kit (ab115125) were purchased from Abcam (Cambridge, MA) and used according to the manufacturer's protocol. To detect and quantify the protein amount of HDAC4 in Beas2B cells, EpiQuik HDAC4 assay kit (Epigentek Group, Brooklyn, NY) was used, and assays were performed according to the manufacturer's protocol.
TaqMan array.
TaqMan Array Human DNA Damage Induced 14-3-3 Sigma Signaling 96-well plates (cat. no. 4418771, Thermo Fisher Scientific) were obtained to determine the effect of miR-185 on 14-3-3 sigma signaling pathway after hyperoxia. HPRT1 was used as an endogenous control gene in array data analysis.
Statistical analysis.
All data were presented as means ± SD. Comparisons between two groups were performed using a two-tailed unpaired Student's t-test for statistical significance. Multiple groups were compared using a one-way ANOVA with Tukey's method. P < 0.05 was considered statistically significant; *P < 0.05; **P < 0.01.
RESULTS
Hyperoxia-associated oxidative stress induces miR-185 expression in lung epithelial cells.
First, we found that hyperoxia (95% oxygen) induced the expressions of miR-185 and miR-185 precursor in human lung epithelial Beas2B cells in a time-dependent manner (Fig. 1, A and B). Given that miR-185 is encoded within the intron of TANGO2 gene, we next evaluated the effects of hyperoxia on the expression of TANGO2. As shown in Fig. 1C, hyperoxia also induced TANGO2 expression in Beas2B cells in a time-dependent manner. These observations were confirmed in mouse lung primary epithelial cells (Fig. 1, D and E). To determine whether hyperoxia upregulated miR-185 and TANGO2 via reactive oxygen species (ROS), we added a general ROS inhibitor NAC (5 mM) or a mitochondrial antioxidant Mito-TEMPO (100 μM) (45) into the cell culture followed by exposure of hyperoxia. Both NAC and Mito-TEMPO significantly inhibited the hyperoxia-induced miR-185 (Fig. 1F). Consistent with this, treating Beas2B cells with H2O2 (90 μM) markedly upregulated the expression level of miR-185, and these responses were also attenuated by NAC or Mito-TEMPO (Fig. 1, G and H). Interestingly, hyperoxia treatment resulted in miR-185 level decrease in cells isolated from bronchoalveolar lavage fluid (BALF) (Fig. 1I). Furthermore, we also determined the relative expression of miR-185 in a variety of mouse tissues, including brain, heart, lung, kidney, liver, intestine, muscle, spleen, and adipose tissue, as shown in Fig. 1J.
Fig. 1.

Oxidative stress or hyperoxia induces miR-185 expression in lung epithelial cells. A and B: time course of hyperoxia-induced expressions of miR-185 (A), miR-185 precursor (B), and TANGO2 (C) in Beas2B cells detected using real-time PCR. D and E: primary lung epithelial cells isolated from mice were exposed to hyperoxia for the indicated time points. The expression of miR-185 (D) and TANGO2 (E) in mouse primary lung epithelial cells was determined using real-time PCR. F: Beas2B cells were exposed to normoxia or hyperoxia for 24 h in the presence or absence of N-acetyl-l-cysteine (NAC) (5 mM) or Mito-TEMPO (100 μM). Real-time PCR was performed to determine the level of miR-185. G: Beas2B cells were treated with H2O2 (90 μM) for 6 h in the presence or absence of NAC (5 mM). The expression level of miR-185 was measured using real-time PCR. H: Beas2B cells were treated with H2O2 (90 μM) for 6 h in the presence or absence of Mito-TEMPO (100 μM). The expression level of miR-185 was measured using real-time PCR. I: real-time PCR analysis of miR-185 in mouse bronchoalveolar lavage fluid (BALF) exposed to normoxia (room air) or hyperoxia (3 days, n = 6 for each group). J: real-time PCR analysis of miR-185 in different mouse tissues (n = 3). All the data are representative of 3 independent experiments. *P < 0.05, **P < 0.01.
Histone acetylation and HDAC4 play a key role in the regulation of miR-185 expression in lung epithelial cells after hyperoxia.
Previous reports have suggested that inhibitors of HDAC rapidly alter miRNA levels (41). To determine the mechanisms by which hyperoxia regulates miR-185 expression in lung epithelial cells, we evaluated the effects of hyperoxia on histone acetylation and HDAC activities. Hyperoxia triggered histone H3 and H4 acetylation in Beas2B cells in a time-dependent manner, as determined by Western blot analysis (Fig. 2, A–D). We further showed that hyperoxia increased the acetylation of histone H3 and H4 by ELISA. The induction of H3/H4 acetylation was attenuated by mitochondrial antioxidant Mito-TEMPO (Fig. 2, E and F). HDACs are a class of enzymes that can remove acetyl groups from histones (10), subsequently allowing a tighter wrap between the histones and the DNAs they bind (13). To determine which specific HDACs regulated the hyperoxia-induced histone H3 and H4 deacetylation, we first tested the levels of HDAC expression (1 to 5) in Beas2B cells after hyperoxia. As shown in Fig. 3A, hyperoxia suppressed the protein level of HDAC4 in a time-dependent manner. No significant changes were observed in other HDACs (Fig. 3, A and B). To confirm this observation, we isolated primary alveolar epithelial cells from C57/BJ6 mice and exposed them to hyperoxia (95% oxygen). We confirmed that HDAC4 was significantly downregulated in primary lung epithelial cells after hyperoxia using real-time PCR (Fig. 3C). The hyperoxia-suppressed HDAC4 was confirmed using ELISA (Fig. 3D). Pretreatment of Mito-TEMPO in lung epithelial cells significantly attenuated the effects of hyperoxia on HDAC4 expression (Fig. 3D).
Fig. 2.

Hyperoxia regulates histone acetylation in lung epithelial cells. A and B: Western immunoblot analysis of acetyl-histone H3 (A) and acetyl-histone H4 (B) protein expressions in human lung epithelial cells (Beas2B) after hyperoxia. C and D: relative acetyl-histone H3 (C) and acetyl-histone H4 (D) protein levels were quantified using densitometry and normalized to histone H3 or H4. E and F: Beas2B cells were exposed to normoxia or hyperoxia in the presence or absence of Mito-TEMPO (100 μM) for 48 h. Extracted histones were used to measure total histone H3 (E) and H4 (F) acetylation using ELISA. All results represent 3 independent experiments. *P < 0.05, **P < 0.01.
Fig. 3.

Hyperoxia regulates histone deacetylase 4 (HDAC4) expression in lung epithelial cells. A: expression of HDAC1 to HDAC5 after hyperoxia was detected using Western blot analysis in Beas2B cells. B: relative protein level of HDAC4 was quantified and normalized to GAPDH. C: primary alveolar epithelial cells isolated from C57/BJ6 mice were exposed to normoxia (room air) or to hyperoxia for the indicated time points. Relative levels of HDAC1 to HDAC6 were measured using real-time PCR. D: nuclear protein was isolated from Beas2B cells exposed to normoxia or hyperoxia in the presence or absence of Mito-TEMPO (100 μM) for 48 h. HDAC4 ELISA assay kit was used to measure total HDAC4. Results represent the average values obtained in 3 independent experiments. *P < 0.05, **P < 0.01.
To investigate whether hyperoxia-suppressed HDAC4 and histone deacetylation regulate the expression of miR-185, we first reviewed the promoter region of TANGO2, the host gene of miR-185 (14). As shown in Fig. 4A, the acetylation of lysine 27 of the histone H3 (H3K27Ac) has been reported in the promoter region of TANGO2 in seven different cell lines (12). We therefore, performed a ChIP assay to examine whether DNA binding to HDAC4 was altered by hyperoxia. Hyperoxia reduced the HDAC4/DNA interaction in Beas2B cells in a time-dependent manner (Fig. 4B). Using real-time PCR, we confirmed the observation that hyperoxia significantly reduced the HDAC4/DNA interaction using the purified DNA obtained from the HDAC4 antibody-precipitated samples (Fig. 4C). We next treated Beas2B cells with MC1568, a class II (IIa) HDAC inhibitor (49). A dose-dependent upregulation of miR-185 expression was demonstrated, suggesting that HDAC4 negatively regulates miR-185 expression (Fig. 4D).
Fig. 4.

The expression of miR-185 is controlled by HDAC4. A: schematic representation of the genomic location of the human TANGO2 gene. miR-185 locates in the intron of the TANGO2 gene. The acetylation of lysine 27 of the H3 histone protein (H3K27Ac) mark is found in the promoter region of TANGO2 on 7 different cell lines by chromatin immunoprecipitation (ChIP)-sequencing assay that is thought to enhance transcription. B: ChIP assay was performed using chromatin isolated from Beas2B cells exposed to normoxia or hyperoxia for 2 days. HDAC4 antibody was used for immunoprecipitation. IgG was used as a negative control, and water was added as no-DNA control for PCR. C: purified DNA was analyzed using real-time PCR. Fold enhancement was calculated based on the ratio of positive signal to IgG signal. D: Beas2B cells were treated with MC1568, a class II (IIa) HDAC inhibitor selective for HDAC4. After 24 h, the dose-dependent expression of miR-185 was analyzed using real-time PCR. All the data represent 3 independent experiments. *P < 0.05, **P < 0.01.
miR-185 promotes hyperoxia-induced lung epithelial cell death via inducing DNA damage.
To determine the functional role of miR-185 in lung epithelial cells, we used gain- and loss-of-function approaches. Successful overexpression or suppression of miR-185 was confirmed after administration of miR-185 mimics or inhibitors (Figs. 5A and 6A). In the presence of hyperoxia, overexpression of miR-185 in Beas2B cells using miR-185 mimics significantly increased the relative apurinic/apyrimidinic (AP) sites that are located in DNA that have neither a purine nor a pyrimidine base as a result of DNA damage (Fig. 5B). Consistent with our findings, overexpression of miR-185 mimics suppressed the survival of Beas2B epithelial cells after hyperoxia (Fig. 5C). Using M30 cytodeath antibody, we detected a significant level of apoptosis in Beas2B cells using fluorescent microscopy and flow cytometry (Fig. 5, D–G). We next performed apoptosis protein array analysis. We showed that several apoptotic signaling proteins were altered after miR-185 mimic overexpression. Among the proapoptotic signaling proteins, Bad and Bax were upregulated. Fas ligand was also enhanced, suggesting that the Fas-Fas ligand pathway is involved in this process (Fig. 5H). To further determine the effects of miR-185 on cell death, we measured the activity of caspase-3/7 in Beas2B cells that were transfected with either miR-185 mimics or control mimics, in the presence or absence of hyperoxia (Fig. 5I). Consistently, we found that overexpression of miR-185 promoted the activation of caspase-3/7 in Beas2B cells.
Fig. 5.

miR-185 overexpression promotes DNA damage and cell death after hyperoxia. A: Beas2B cells were transfected with control or miR-185 mimics. 48 h after transfection, relative level of miR-185 was measured using real-time PCR. B: Beas2B cells were transfected with miR-185 mimics or control. After transfection, cells were exposed to hyperoxia for 2 days. Relative change of apurinic/apyrimidinic (AP) sites in genomic DNA was measured using the aldehyde reactive probe (ARP) assay kit. C: effect of miR-185 overexpression on cell viability was detected after transfection of control or miR-185 mimics into Beas2B cells, followed by 0 h (room air) or 48 h of hyperoxia. D and E: M30 immunofluorescence was used to detect apoptosis in Beas2B cells transfected with control or miR-185 mimics in the presence of hyperoxia (24 h). M30 staining indicated early stage of apoptosis. Representative images were captured using a fluorescent microscope at ×10 magnification (D). M30 fluorescence intensity was quantified using ImageJ software (E). F and G: Beas2B cells were transfected with control or miR-185 mimics and exposed to hyperoxia for 2 days. Cells were then examined for apoptotic cells using annexin V-FITC apoptosis detection kit. Representative result of 3 independent experiments was shown (F). PI, propidium iodide. The levels of apoptosis were presented as means ± SD from 3 independent experiments (G). H: human apoptosis antibody array was used to detect proteins related to apoptosis in Beas2B cells. Beas2B cells transfected with control or miR-185 mimics were exposed to hyperoxia for 2 days. Cell lysates were used for antibody array analysis. I: Beas2B cells were transfected with control or miR-185 mimics and exposed to hyperoxia for 2 days. The activity of caspase-3/7 was determined. Data are shown as means ± SD from 3 independent experiments. **P < 0.01.
Fig. 6.

Inhibition of miR-185 attenuates DNA damage and lung epithelial cell death after hyperoxia. A: Beas2B cells were transfected with control or miR-185 inhibitors. 48 h after transfection, relative level of miR-185 was measured using real-time PCR. B: Beas2B cells were transfected with control or miR-185 inhibitors. After transfection, cells were exposed to hyperoxia for 2 days. Relative change of AP sites in genomic DNA was measured using the ARP assay kit. C: effect of miR-185 suppression on cell viability was detected after transfection of control or miR-185 inhibitors into Beas2B cells, followed by 0 h (room air) or 48 h of hyperoxia. D and E: M30 immunofluorescence was used to detect apoptosis in Beas2B cells transfected with control or miR-185 inhibitors, followed by hyperoxia (24 h). M30 staining indicated early stage of apoptosis. Representative images were captured using a fluorescent microscope at ×10 magnification (D). M30 fluorescence intensity was quantified using ImageJ software (E). F and G: Beas2B cells were transfected with control or miR-185 inhibitor and exposed to hyperoxia for 2 days. After treatment, cells were examined for apoptotic cells using annexin V-FITC apoptosis detection kit. Representative result of 3 independent experiments was shown (F). The levels of apoptosis are presented as means ± SD from 3 independent experiments (G). H: human apoptosis antibody array was used to detect proteins related to apoptosis in Beas2B cells. Beas2B cells transfected with inhibitor control or miR-185 inhibitor were exposed to hyperoxia for 2 days. Cell lysates were used for antibody array analysis. I: Beas2B cells were transfected with control or miR-185 inhibitor and exposed to hyperoxia for 2 days. The activity of caspase-3/7 was determined. Data are shown as means ± SD from 3 independent experiments. *P < 0.05, **P < 0.01.
To confirm our observations above, we next transfected Beas2B cells with miR-185 inhibitors. After hyperoxia, suppression of miR-185 in Beas2B cells using miR-185 inhibitors markedly decreased the number of relative AP sites (Fig. 6B). Inhibition of miR-185 enhanced the survival of Beas2B epithelial cells after hyperoxia (Fig. 6C). Using M30 cytodeath antibody, we detected less apoptosis in Beas2B cells using fluorescent microscopy and flow cytometry (Fig. 6, D–G). Apoptosis protein array was used to confirm changes of Bad, Bax, and Fas ligand expression in Beas2B cells after miR-185 suppression and hyperoxia (Fig. 6H). We found that the activity of caspase-3/7 was decreased in Beas2B cells that were treated with miR-185 inhibitors (Fig. 6I). These results further supported our above observations obtained using the miR-185 mimics.
miR-185 inhibits the DNA damage-induced 14-3-3δ signaling pathway.
We next examined the mechanisms by which miR-185 promoted DNA damage-associated apoptosis in the presence of hyperoxia. 14-3-3 proteins are conserved dimeric regulators in eukaryotic cells and can bind multiple cellular protein ligands (1). 14-3-3 proteins have antiapoptotic functions through their interactions with a variety of proapoptotic molecules (1, 16). To screen for multiple regulators, we performed a TaqMan Array on human DNA damage-associated genes. We found that DNA damage-associated 14-3-3δ signaling mediators were robustly downregulated by miR-185 mimics and upregulated by miR-185 inhibitors (Table 2), suggesting a potential mechanism by which miR-185 promoted DNA damage-associated apoptosis. The effects of miR-185 on CCNB3 and SFN expressions were further confirmed using real-time PCR (Fig. 7).
Table 2.
miR-185 controls DNA damage-induced 14-3-3δ signaling pathway
| DNA Damage-Induced 14-3-3δ Signaling (>1.5 in Both Groups) | ||
|---|---|---|
| Gene Symbol | miR-185 Mimics | miR-185 Inhibitor |
| ATR | −1.92 | 1.69 |
| BRCA1 | −1.78 | 1.66 |
| CCNB3 | −7.35 | 6.29 |
| CDC2 | −1.53 | 1.60 |
| HUS1 | −1.58 | 1.52 |
| SFN | −9.69 | 1.56 |
| YWHAE | −1.58 | 1.58 |
| YWHAG | −2.30 | 1.53 |
Fig. 7.
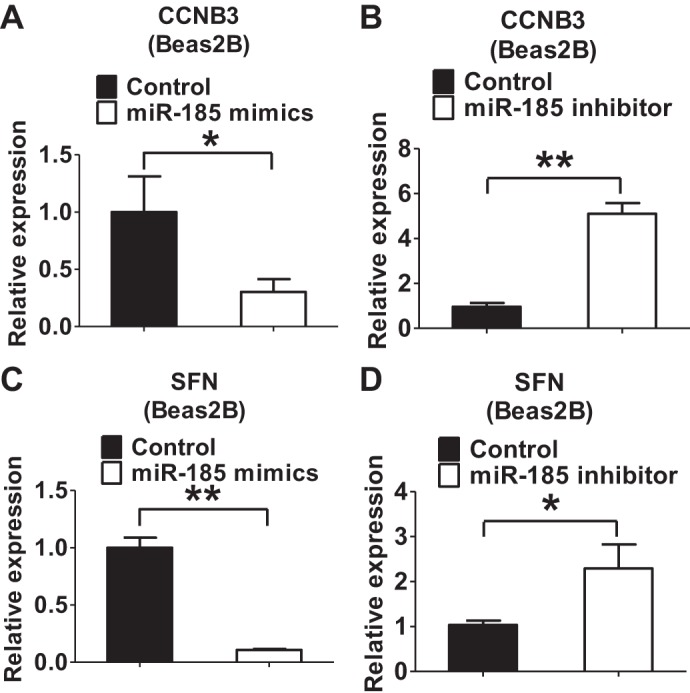
CCNB3 and SFN are regulated by miR-185 under hyperoxic condition. A and B: Beas2B cells were transfected with miR-185 mimics (A) or inhibitor (B) following the hyperoxia exposure (2 days). Total RNA was extracted and reverse transcribed. Relative mRNA level of CCNB3 was detected using real-time PCR. C and D: relative mRNA level of SFN was detected in Beas2B cells transfected with miR-185 mimics (C) or inhibitor (D) following the hyperoxia exposure (2 days).
DISCUSSION
Except for the few reports in cancer research, the expression and function of miR-185 in lung diseases have not been explored, particularly in lung epithelial cells. Deletion of miR-185 is a frequent event in diverse types of cancers (5). miR-185 is reported to induce cell-cycle arrest in human non-small cell lung cancer (44). It shows antitumor effects in gastric cancer via Wnt/β-catenin signaling (37). Similarly, miR-185 functions as a tumor suppressor in prostate cancer cells and clear cell renal cell carcinomas through inhibiting VEGFA (30). Furthermore, miR-185 inhibits hepatocellular carcinoma growth through the DNMT1/PTEN/Akt pathway (36) and inhibits triple-negative breast cancer cell proliferation via DNMT1 and E2F6 (46). Plasma miR-185 also serves as a potential biomarker for glioma and human chondrosarcoma (47). In addition, a low level of miR-185-3p is associated with poor overall survival in patients with nasopharyngeal carcinoma (54).
In noncancer cells, miR-185 effectively suppresses cardiac hypertrophy signaling through multiple targets (24). It directly targets the 3′-untranslated region of Na+/H+ exchanger-1 that is a protein involved in ER stress (23). These studies demonstrated that upregulation of miR-185 results in antihypertrophic effects and that miR-185 deficiency leads to prohypertrophic effects (24).
Our report is the first to explore the expression, regulation, and function of miR-185 in normal lung tissue. Hyperoxia induced miR-185 in lung epithelial cells via mitochondrial ROS (Fig. 1, F and H). Mitochondrial ROS have been shown to mediate hyperoxia-induced lung epithelial cell death via mechanisms involving Bak or Bak-dependent apoptosis (9). Our data provided a novel mechanism by which mitochondrial ROS exert proapoptotic effects via miR-185-mediated pathway.
miR-185 is encoded within an intron of the TANGO2 gene (14). TANGO2 is also referred to as chromosome 22 open-reading frame 25. Presently, the function of TANGO2 remains unclear. However, TANGO is known as a highly conserved gene located in eubacteria to Animalia (40). It has been speculated to be involved in Golgi organization and protein secretion (39). Except in smooth muscle cells, thymus, and lymph nodes, TANGO2 expression has been found in almost all other human tissues (48), suggesting a potential important function for this protein. Our studies showed that hyperoxia induced both miR-185 and TANGO2 expression in lung epithelial cells in a time-dependent manner. However, whether TANGO2 exerts the similar functions as miR-185 requires future exploration.
Fourteen miRNAs have been reported to be upregulated after hyperoxia in lungs; these miRNAs include miR-20b, miR-106a, miR-128, miR-883b-3p, miR-15b, miR-122, miR-30e, miR-365-5p, miR-133a, miR-205, miR-379, miR-449a, and miR-431 (57). We initially discovered highly expressed miR-185 after hyperoxia in lung epithelial cells but not in other BALF cells (Fig. 1I). It has been known that, after hyperoxia, many immunomodulatory cells are recruited to the lungs and can be detected in BALF (33). Therefore, the reduced level of miR-185 in BALF cells suggests that hyperoxia exerts differential effects based on cell types. This prompted us to study the regulation and function of miR-185 in lung epithelial cells, rather than immunomodulatory cells, in the presence of hyperoxia (Fig. 8).
Fig. 8.

Schematic review of the regulation and function of miR-185 in lung epithelial cells after hyperoxia. Hyperoxia induces miR-185 expression in lung epithelial cells via reactive oxygen species. The hyperoxia-upregulated miR-185 probably contributes to oxidative stress-associated epithelial cell death through enhancing DNA damage and modulating 14-3-3δ pathways.
Eighteen different mammalian HDACs can be divided into four classes (10). We found that one of the class II HDACs, HDAC4, is downregulated after hyperoxia. Hyperoxia also significantly suppresses the interaction of HDAC4 and chromatin DNA. The disruption of HDAC4 and chromatin may suppress the deacetylation of histones and subsequently turn on the transcription of miR-185.
Similarly as in the cancer cells, we found that miR-185 exerts proapoptotic effects and limits cell proliferations in normal lung epithelial cells (primary epithelial cells). Overexpression of miR-185 promotes DNA damage, suppresses multiple proteins involved in the 14-3-3δ pathway, and enhances Fas ligand and caspase-mediated apoptosis. 14-3-3 proteins were first found in the brain in 1967 (6). To date, the 14-3-3 family has been shown to be expressed in all eukaryotic cells. There are seven isoforms identified (14-3-3β, γ, ε, η, ζ, σ, and τ/θ). Among these isoforms, 14-3-3σ (also known as SFN) is specifically expressed in epithelial cells. Several studies indicate that 14-3-3 components inhibit the apoptotic pathway (11, 52) through interactions with other proteins, such as BAD, FKHRL-1, and Ask1 (1, 16). Our findings on miR-185-regulated 14-3-3δ pathway provide a novel mechanism by which hyperoxia promotes lung epithelial cell death (Fig. 7). Additionally, Fas ligand and caspase-associated apoptosis have shown crucial roles in the development of epithelial apoptosis in ALI and ARDS (2, 28, 32), consistent with what we have found here.
Our functional studies were conducted using the gain- or loss-of-function approaches. Transfection of miR-185 mimics or inhibitors artificially enhances or suppresses the intracellular level of miR-185. Therefore, these approaches may not necessarily reflect the real physiological status of miR-185. The copy number and concentration required for miR-185 to exert cellular functions at physiological conditions require further investigation. Nevertheless, at the present time, the gain- and loss-of-function approaches remain to be well-accepted methods to determine the cellular functions of miRNAs.
Our future directions will need to focus on building on a reliable method to determine the copy number and concentration of miRNAs in various physiological conditions and in different cell types. We will need to further explore multiple pathways by which miR-185 exerts its proapoptotic functions.
Taken together, we found that the miRNA miR-185 promotes lung epithelial cell apoptosis and limits cell proliferations in the presence of hyperoxia/ROS. This result potentially uncovers a novel mechanism by which hyperoxia-associated ROS stimulates lung epithelial cell death and lung injury, which subsequently provides new targets for the development of therapeutic and diagnostic strategies.
GRANTS
This work was supported by NIH R01HL102076 (Y. Jin), NIH R21AI121644 (Y. Jin), and NIH R01 GM111313 (Y. Jin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.Z., C.S.D.C., and Y.J. conception and design of research; D.Z., H.L., and Y.C. performed experiments; D.Z. and H.L. analyzed data; D.Z. and C.S.D.C. interpreted results of experiments; D.Z. prepared figures; D.Z. and Y.J. drafted manuscript; C.S.D.C. and Y.J. edited and revised manuscript; Y.J. approved final version of manuscript.
REFERENCES
- 1.Aghazadeh Y, Papadopoulos V. The role of the 14-3-3 protein family in health, disease, and drug development. Drug Discov Today. 2015 Oct 9. doi: 10.1016/j.drudis.2015.09.012 [Epub ahead of print] [DOI] [PubMed]
- 2.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and Fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 161: 1783–1796, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barger JF, Nana-Sinkam SP. MicroRNA as tools and therapeutics in lung cancer. Respir Med 109: 803–812, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, function. Cell 116: 281–297, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Bensen JT, Tse CK, Nyante SJ, Barnholtz-Sloan JS, Cole SR, Millikan RC. Association of germline microRNA SNPs in pre-miRNA flanking region and breast cancer risk and survival: the Carolina Breast Cancer Study. Cancer Causes Control 24: 1099–1109, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci 4: 752–762, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Bhandari V. Molecular mechanisms of hyperoxia-induced acute lung injury. Front Biosci 13: 6653–6661, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Bhaskaran M, Xi D, Wang Y, Huang C, Narasaraju T, Shu W, Zhao C, Xiao X, More S, Breshears M, Liu L. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol Genomics 44: 970–980, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budinger GR, Mutlu GM, Urich D, Soberanes S, Buccellato LJ, Hawkins K, Chiarella SE, Radigan KA, Eisenbart J, Agrawal H, Berkelhamer S, Hekimi S, Zhang J, Perlman H, Schumacker PT, Jain M, Chandel NS. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am J Respir Crit Care Med 183: 1043–1054, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen HP, Zhao YT, Zhao TC. Histone deacetylases and mechanisms of regulation of gene expression. Crit Rev Oncog 20: 35–47, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clapp C, Portt L, Khoury C, Sheibani S, Norman G, Ebner P, Eid R, Vali H, Mandato CA, Madeo F, Greenwood MT. 14-3-3 protects against stress-induced apoptosis. Cell Death Dis 3: e348, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics 4: 5, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forstner AJ, Basmanav FB, Mattheisen M, Bohmer AC, Hollegaard MV, Janson E, Strengman E, Priebe L, Degenhardt F, Hoffmann P, Herms S, Maier W, Mossner R, Rujescu D, Ophoff RA, Moebus S, Mortensen PB, Borglum AD, Hougaard DM, Frank J, Witt SH, Rietschel M, Zimmer A, Nothen MM, Miro X, Cichon S. Investigation of the involvement of MIR185 and its target genes in the development of schizophrenia. J Psychiatry Neurosci 39: 386–396, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fortunato O, Boeri M, Verri C, Moro M, Sozzi G. Therapeutic use of microRNAs in lung cancer. Biomed Res Int 2014: 756975, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardino AK, Yaffe MB. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin Cell Dev Biol 22: 688–695, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graves P, Zeng Y. Biogenesis of mammalian microRNAs: a global view. Genomics Proteomics Bioinformatics 10: 239–245, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guarnieri DJ, DiLeone RJ. MicroRNAs: a new class of gene regulators. Ann Med 40: 197–208, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev 87: 3–14, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol 194: 855–860, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallet RH, Matthay MA. Hyperoxic acute lung injury. Respir Care 58: 123–141, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke XF, Fang J, Wu XN, Yu CH. MicroRNA-203 accelerates apoptosis in LPS-stimulated alveolar epithelial cells by targeting PIK3CA. Biochem Biophys Res Commun 450: 1297–1303, 2014. [DOI] [PubMed] [Google Scholar]
- 23.Kim JO, Kwon EJ, Song DW, Lee JS, Kim DH. miR-185 inhibits endoplasmic reticulum stress-induced apoptosis by targeting Na+/H+ exchanger-1 in the heart. BMB Rep 2015. [DOI] [PMC free article] [PubMed]
- 24.Kim JO, Song DW, Kwon EJ, Hong SE, Song HK, Min CK, Kim do H. miR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS One 10: e0122509, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov 13: 622–638, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Liang X, Wei SQ, Lee SJ, Fung JK, Zhang M, Tanaka A, Choi AM, Jin Y. p62 sequestosome 1/light chain 3b complex confers cytoprotection on lung epithelial cells after hyperoxia. Am J Respir Cell Mol Biol 48: 489–496, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin WC, Chen CW, Huang YW, Chao L, Chao J, Lin YS, Lin CF. Kallistatin protects against sepsis-related acute lung injury via inhibiting inflammation and apoptosis. Sci Rep 5: 12463, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Nelson A, Wang X, Kanaji N, Kim M, Sato T, Nakanishi M, Li Y, Sun J, Michalski J, Patil A, Basma H, Rennard SI. MicroRNA-146a modulates human bronchial epithelial cell survival in response to the cytokine-induced apoptosis. Biochem Biophys Res Commun 380: 177–182, 2009. [DOI] [PubMed] [Google Scholar]
- 30.Ma X, Shen D, Li H, Zhang Y, Lv X, Huang Q, Gao Y, Li X, Gu L, Xiu S, Bao X, Duan J, Zhang X. MicroRNA-185 inhibits cell proliferation and induces cell apoptosis by targeting VEGFA directly in von Hippel-Lindau-inactivated clear cell renal cell carcinoma. Urol Oncol 33: 169 e161–e111, 2015. [DOI] [PubMed] [Google Scholar]
- 31.Mao C, Zhang J, Lin S, Jing L, Xiang J, Wang M, Wang B, Xu P, Liu W, Song X, Lv C. MiRNA-30a inhibits AECs-II apoptosis by blocking mitochondrial fission dependent on Drp-1. J Cell Mol Med 18: 2404–2416, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163: 2217–2225, 1999. [PubMed] [Google Scholar]
- 33.Okuma T, Terasaki Y, Sakashita N, Kaikita K, Kobayashi H, Hayasaki T, Kuziel WA, Baba H, Takeya M. MCP-1/CCR2 signalling pathway regulates hyperoxia-induced acute lung injury via nitric oxide production. Int J Exp Pathol 87: 475–483, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Othman N, Nagoor NH. The role of microRNAs in the regulation of apoptosis in lung cancer and its application in cancer treatment. Biomed Res Int 2014: 318030, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pagano A, Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann NY Acad Sci 1010: 405–416, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Qadir XV, Han C, Lu D, Zhang J, Wu T. miR-185 inhibits hepatocellular carcinoma growth by targeting the DNMT1/PTEN/Akt pathway. Am J Pathol 184: 2355–2364, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu F, Xiong JP, Deng J, Xiang XJ. TRIM29 functions as an oncogene in gastric cancer and is regulated by miR-185. Int J Clin Exp Pathol 8: 5053–5061, 2015. [PMC free article] [PubMed] [Google Scholar]
- 38.Ryter SW, Choi AM. Regulation of autophagy in oxygen-dependent cellular stress. Curr Pharm Des 19: 2747–2756, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R, Malhotra V. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 136: 891–902, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Sasahira T, Kirita T, Yamamoto K, Ueda N, Kurihara M, Matsushima S, Bhawal UK, Bosserhoff AK, Kuniyasu H. Transport and Golgi organisation protein 1 is a novel tumour progressive factor in oral squamous cell carcinoma. Eur J Cancer 50: 2142–2151, 2014. [DOI] [PubMed] [Google Scholar]
- 41.Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 66: 1277–1281, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Standiford TJ, Ward PA. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl Res 167: 183–191, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 6: 8474–8490, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi Y, Forrest AR, Maeno E, Hashimoto T, Daub CO, Yasuda J. MiR-107 and MiR-185 can induce cell cycle arrest in human non small cell lung cancer cell lines. PLoS One 4: e6677, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol 46: 507–514, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang H, Liu P, Yang L, Xie X, Ye F, Wu M, Liu X, Chen B, Zhang L, Xie X. miR-185 suppresses tumor proliferation by directly targeting E2F6 and DNMT1 and indirectly upregulating BRCA1 in triple-negative breast cancer. Mol Cancer Ther 13: 3185–3197, 2014. [DOI] [PubMed] [Google Scholar]
- 47.Tang H, Liu Q, Liu X, Ye F, Xie X, Xie X, Wu M. Plasma miR-185 as a predictive biomarker for prognosis of malignant glioma. J Cancer Res Ther 11: 630–634, 2015. [DOI] [PubMed] [Google Scholar]
- 48.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten Proteomics F. Tissue-based map of the human proteome. Science 347: 1260419, 2015. [DOI] [PubMed] [Google Scholar]
- 49.Venza I, Visalli M, Oteri R, Cucinotta M, Teti D, Venza M. Class II-specific histone deacetylase inhibitors MC1568 and MC1575 suppress IL-8 expression in human melanoma cells. Pigment Cell Melanoma Res 26: 193–204, 2013. [DOI] [PubMed] [Google Scholar]
- 50.Vijayarathna S, Oon CE, Jothy SL, Chen Y, Kanwar JR, Sasidharan S. MicroRNA pathways: an emerging role in identification of therapeutic strategies. Curr Gene Ther 14: 112–120, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Xie M, Zhang S, Yu B. microRNA biogenesis, degradation and activity in plants. Cell Mol Life Sci 72: 87–99, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J 19: 349–358, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu B, Hsu PK, Stark KL, Karayiorgou M, Gogos JA. Derepression of a neuronal inhibitor due to miRNA dysregulation in a schizophrenia-related microdeletion. Cell 152: 262–275, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Ai Q, Cao H, Liu Q. MiR-185-3p and miR-324-3p predict radiosensitivity of nasopharyngeal carcinoma and modulate cancer cell growth and apoptosis by targeting SMAD7. Med Sci Monit 21: 2828–2836, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang D, Li X, Chen C, Li Y, Zhao L, Jing Y, Liu W, Wang X, Zhang Y, Xia H, Chang Y, Gao X, Yan J, Ying H. Attenuation of p38-mediated miR-1/133 expression facilitates myoblast proliferation during the early stage of muscle regeneration. PLoS One 7: e41478, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang D, Wang X, Li Y, Zhao L, Lu M, Yao X, Xia H, Wang YC, Liu MF, Jiang J, Li X, Ying H. Thyroid hormone regulates muscle fiber type conversion via miR-133a1. J Cell Biol 207: 753–766, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Peng W, Zhang S, Wang C, He X, Zhang Z, Zhu L, Wang Y, Feng Z. MicroRNA expression profile in hyperoxia-exposed newborn mice during the development of bronchopulmonary dysplasia. Respir Care 56: 1009–1015, 2011. [DOI] [PubMed] [Google Scholar]