

Abstract
Pulmonary endothelial prostacyclin appears to be involved in the pathogenesis of chronic obstructive pulmonary disease (COPD). The effect of treatment with a prostacyclin analog in animal models of previously established COPD is unknown. We evaluated the short- and long-term effect of iloprost on inflammation and airway hyperresponsiveness (AHR) in a murine model of COPD. Nineteen mice were exposed to LPS/elastase, followed by either three doses of intranasal iloprost or saline. In the long-term treatment experiment, 18 mice were exposed to LPS/elastase and then received 6 wk of iloprost or were left untreated as controls. In the short-term experiment, iloprost did not change AHR but significantly reduced serum IL-5 and IFN-γ. Long-term treatment with iloprost for both 2 and 6 wk significantly improved AHR. After 6 wk of iloprost, there was a reduction in bronchoalveolar lavage (BALF) neutrophils, serum IL-1β (30.0 ± 9.2 vs. 64.8 ± 7.4 pg/ml, P = 0.045), IL-2 (36.5 ± 10.6 vs. 83.8 ± 0.4 pg/ml, P = 0.01), IL-10 (75.7 ± 9.3 vs. 96.5 ± 3.5 pg/ml, P = 0.02), and nitrite (15.1 ± 5.4 vs. 30.5 ± 10.7 μmol, P = 0.01). Smooth muscle actin (SMA) in the lung homogenate was also significantly reduced after iloprost treatment (P = 0.02), and SMA thickness was reduced in the small and medium blood vessels after iloprost (P < 0.001). In summary, short- and long-term treatment with intranasal iloprost significantly reduced systemic inflammation in an LPS/elastase COPD model. Long-term iloprost treatment also reduced AHR, serum nitrite, SMA, and BALF neutrophilia. These data encourage future investigations of prostanoid therapy as a novel treatment for COPD patients.
Keywords: chronic obstructive pulmonary disorder, prostacyclin, inflammation, airway hyperresponsiveness
chronic obstructive pulmonary disease (COPD) is characterized by progressive airflow limitation associated with an enhanced chronic inflammatory response to noxious particles (31). Although cigarette smoke is the proximal etiology of COPD, the disease progresses despite smoking cessation due to self-amplifying loops of apoptosis, oxidative stress, and inflammation (29). No currently available pharmacologic therapy conclusively alters the natural history of COPD. Treatments that focus on attenuating these self-amplifying loops of disease progression are desperately needed in COPD.
Although COPD is commonly regarded as a disease confined to the airways and the parenchyma, it is now accepted that the pulmonary vasculature is affected early in the disease as well. Changes in the vessels have been shown in humans with mild COPD (23, 24), as well as animal models of COPD, in which pulmonary vascular changes actually precede emphysema development (26, 33). More provocative studies have suggested that early pulmonary vasculopathy may “contribute” to the pathogenesis of COPD (10, 14).
Prostacyclin (PGI2), a vasodilatory prostanoid released by pulmonary endothelial cells (ECs), is reduced in lung tissue of patients with emphysema. Furthermore, iloprost, a PGI2 analog, protected against cigarette smoke extract-induced EC apoptosis (20). Beraprost, another prostacyclin analog, reduced emphysema formation, inflammation, and apoptosis when given as pretreatment prior to cigarette smoke in a murine model of COPD (6).
On the basis of on these findings, we hypothesized that iloprost, when given as a “treatment” rather than a prevention, would reduce systemic inflammation, oxidative stress, and bronchial hyperresponsiveness in an animal model of COPD. To test this hypothesis, we investigated the effects of intranasal iloprost given either as an acute or chronic treatment in mice previously exposed to intrapulmonary LPS and elastase.
METHODS
Animal Selection and Care
Two separate experiments were performed, the first to evaluate the acute effects of intranasal iloprost and the second to investigate the effects of longer-term (chronic) iloprost administration. Nineteen mice [5-wk-old C57BL/6 males (Jackson Laboratory, Bar Harbor, ME)] were used in the acute iloprost experiments and 18 were used in the chronic experiment. Data obtained from contemporary mice not exposed to LPS/elastase or iloprost were used as a control. Ethical approval was obtained from the Louisiana State University Internal Animal Care and Use Committee (no. 2986).
LPS and Elastase COPD Model
To induce a murine model of COPD (8, 9, 25), all mice were exposed to intratracheal LPS (10 μg/mouse/dose) and elastase (1 U/mouse/dose), as detailed in Fig. 1. For the acute iloprost treatment experiment, elastase was given on days 1 and 15, and LPS on days 6, 13, 19, 27, and 50 (Fig. 1A). Histologic emphysema was induced by this exposure strategy (Fig. 1B). In the chronic iloprost treatment experiment, elastase was given on days 1 and 7, and LPS was given on days 5, 12, 19, and 26 (Fig. 1C).
Fig. 1.

Experimental design. A: in the short-term treatment study, 19 mice were exposed to LPS and elastase, as outlined in the figure. They were then separated into two groups, one receiving three doses of intranasal iloprost over a 24-h period (n = 9), while the other group received a saline placebo (n = 10). B: histologic emphysema was induced by LPS/elastase (right) compared with controls (left). C: in the long-term treatment study, 18 mice were exposed to LPS and elastase. One group (n = 9) received no treatment after model induction, while the other group (n = 9) received intranasal iloprost five times per week for 6 wk, starting at the 2-wk time point. A single dose of elastase (D) resulted in histologic emphysema (right) compared with controls (left).
Treatment Protocol and Sample Processing
After intratracheal exposure to LPS and elastase, mice in the acute treatment experiment were divided into two groups (Fig. 1A): intranasal iloprost (n = 9), 5 μg/mouse/dose, and intranasal saline (n = 10), each given at 0, 6, and 24 h. In the chronic treatment experiment (Fig. 1C), mice were divided into either no treatment (n = 9) or intranasal iloprost (n = 9), 5 μg/mouse/dose, given five times per week for 6 wk, starting 2 wk into the LPS/elastase exposure period. We chose this time point to begin iloprost treatment, as histologic emphysema is clearly present 2 wk after a single dose of elastase (Fig. 1D). At the end of each experiment, all mice were killed by CO2 asphyxiation. All lungs were subjected to bronchoalveolar lavage or lung fixation and processing for histological analysis. Lavage was performed by surgically inserting polyethylene tubing into the trachea. One milliliter of ice-cold sterile PBS was injected into the lung, collected in the tube, and stored at −20°C for cytokine and nitrite assessment. This process was repeated five times for collection of cells in bronchoalveolar fluid (BALF) and subjected to cytospin using a Shandon Cytocentrifuge (Thermo Fisher Scientific, Waltham, MA).
Unrestrained Whole Body Plethysmography
Unrestrained whole body plethysmography (EMKA Systems, Falls Church, VA) in response to escalating doses of methacholine was performed on all mice. In the acute iloprost treatment experiment, enhanced pause (Penh) was measured after the first (0 h) and third dose (24 h) of treatment (iloprost or saline). For the chronic treatment experiment, Penh was measured at 4 and 8 wk. Baseline readings were obtained and averaged for 3 min after animals were placed in the chamber. Increasing concentrations (12.5, 25, 50, 100 mg/ml) of aerosolized methacholine were nebulized, and 3-min average readings were taken, from which Penh was calculated.
Organ Recovery, Staining, Cytokine, Immunohistochemistry, Western Blot, and Oxidative Stress Assessment
BALF inflammatory cells were differentiated by Diff-Quick stain (Siemens Medical Solution, Malvern, PA), and cell counts were calculated. The lungs were fixed by gentle infusion of the fixative (10% formalin) through the cannula by a continuous release pump under pressure and volume-controlled conditions (12 ml/h; 10 min). After the trachea was tied off with a ligature, the lungs were placed in a tube containing 10% formalin. Formalin-fixed lungs were sectioned (5 μm) and stained with hematoxylin and eosin or trichrome staining. Fresh chilled tissues were diced into small pieces and rinsed three times with PBS to remove any external contaminants such as blood. Tissues were then sonicated in RIPA lysis buffer supplemented with protease inhibitors. After removal of debris by centrifugation, the protein concentration of the tissue lysate was determined with the Bio-Rad protein assay reagent. A portion (20 μg of protein) of each lysate was then fractionated by SDS-PAGE on a 4–20% gradient gel, and the separated proteins were transferred to a nitrocellulose filter. The filter was stained with Ponceau S to confirm equal loading and transfer of samples and was then probed with antibodies to VCAM-1 (rabbit polyclonal IgG isotype) and smooth muscle actin (SMA; mouse monoclonal IgG1; both Santa Cruz Biotechnology, Dallas TX).
Immune complexes were detected with appropriate secondary antibodies and chemiluminescence reagents (Pierce). Using ImageJ software, the band intensities for SMA were quantified, averaged, and plotted. Immunoreactivity for smooth muscle actin was assessed in images of stained sections captured with a high-resolution Nikon digital camera (DXM1200F). Briefly, images of representative areas were obtained using 40× objective under the same camera settings (exposure time and light intensity). Thickness of SMA-positive areas was measured using a micrometer ruler. Small and medium vessels were measured in, at least, three representative sites. The hydroxyproline content in lung tissues was calculated using a commercial assay kit (Quickzyme Biosciences, Leiden, Netherlands).
Cytokines were quantified using the Bio-Rad Multiplex System for mouse cytokines. Serum nitrite levels were measured by using a colorimetric assay kit (Cayman Chemical, Ann Arbor, MI). Glutathione peroxidase (GPx) activity in plasma was carried out according to the manufacturer's instructions (Cayman Chemicals). Briefly, plasma was added to a 96-well plate containing assay buffer and GPx cosubstrate mixture (NADPH, glutathione, and glutathione reductase). The reaction was initiated by adding cumene hydroperoxide, and the plate was shaken to mix. The absorbance was then read at 340 nm once every minute for six time points. The analysis was done by plotting absorbance values as a function of time to obtain the slope (rate) of the linear portion of the curve (DA340) and calculating change in absorbance per minute. GPx activity was then calculated using the formula (DA340/min × 0.19 ml × sample dilution)/(0.00373 μM−1 × 0.02 ml), where the extinction coefficient of NAPDH was adjusted to 0.00373 μM−1, as suggested by the manufacturer.
Data Analysis
All data are presented as means ± SD. For comparisons between two groups, a Student's t-test was performed. For comparison of three groups, ANOVA with a Tukey's post hoc test was conducted. Penh analysis was performed using a two-way ANOVA with repeated measures and Bonferroni's post hoc test. Statistical analyses were conducted using GraphPad Prism (version 5, La Jolla, CA) and Stata (version 13, College Station, TX). A P value <0.05 was considered to be statistically significant.
RESULTS
Acute Iloprost Treatment
Airway hyperresponsiveness.
Airway hyperresponsiveness to methacholine, as estimated by Penh, was significantly higher in mice exposed to LPS/elastase compared with unexposed wild-type mice (P < 0.05 for all methacholine doses, Fig. 2A). Additionally, baseline Penh (prior to methacholine dosing) was significantly higher in the LPS/elastase-exposed mice compared with unexposed wild-type mice (0.73 ± 0.30 for exposed vs. 0.24 ± 0.19 for controls, P = 0.005 when measured after one dose of saline; 1.13 ± 0.36 for exposed vs. 0.24 ± 0.19 for controls, P < 0.0001 when measured after three doses of saline). There was no statistically significant differences in Penh after either one (Fig. 2A) or three doses (Fig. 2B) of iloprost vs. saline in the LPS/elastase mice.
Fig. 2.
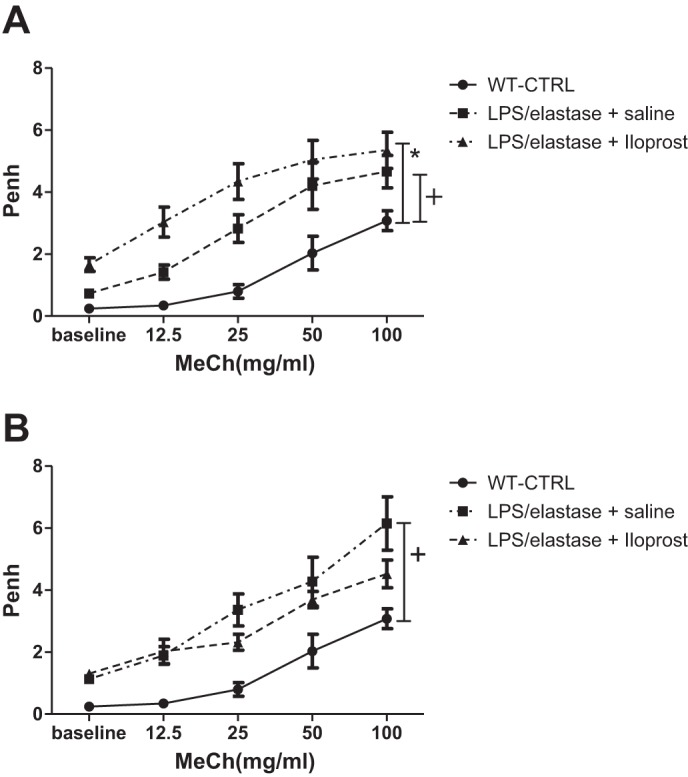
Unrestrained whole body plethysmography for the short-term treatment experiment. After LPS/elastase model induction and treatment with iloprost or saline placebo, Penh was measured in response to increasing doses of methacholine (MeCh). Penh was significantly higher in the LPS/elastase-exposed mice compared with wild-type, unexposed controls (WT-CTRL). There was no difference in Penh between the iloprost and saline groups after either one dose (A) or three doses (B). Comparisons were made using two-way ANOVA with repeated measures and Bonferroni's post hoc test. n = 6 for WT-CTRL, n = 8 for LPS/elastase + saline, and n = 8 for LPS/elastase + iloprost. *P ≤ 0.05 for LPS/elastase + iloprost vs. WT-CTRL; +P ≤ 0.05 for LPS/elastase + saline vs. WT-CTRL.
Cytokine analysis.
There were no significant differences in any of the measured cytokines in BALF obtained from mice treated with three doses of iloprost vs. saline (all P > 0.05; Table 1). However, there were significantly lower levels of serum IL-5 (2.5 ± 0.7 vs. 11.3 ± 0.7 pg/ml, P = 0.0001) and IFN- γ (11.1 ± 8.6 vs. 83.4 ± 14.3 pg/ml, P = 0.0004) in the Iloprost-treated Group compared with the Saline Group. Serum TNF-α was significantly increased in the Iloprost Group compared with saline treatment (24.1 ± 12.5 vs. 7.6 ± 1.8 pg/ml, P = 0.04).
Table 1.
Serum and bronchoalveolar fluid cytokines in response to three doses of iloprost or saline placebo (acute treatment experiment)
| Serum |
BALF |
|||||
|---|---|---|---|---|---|---|
| LPS/Elastase + Placebo | LPS/Elastase + Iloprost | P Value | LPS/Elastase + Placebo | LPS/Elastase + Iloprost | P Value | |
| IL1-β | 49.2 ± 29.5 | 20.1 ± 5.3 | 0.1 | 7.5 ± 0.7 | 6.3 ± 2.7 | 0.51 |
| IL-2 | 14.6 ± 2.2 | 11.5 ± 1.8 | 0.07 | 2.9 ± 1.8 | 4.8 ± 1.1 | 0.14 |
| IL-4 | 22.1 ± 10.1 | 23.6 ± 7.7 | 0.82 | 6.6 ± 1.1 | 12.4 ± 4.5 | 0.09 |
| IL-5 | 11.3 ± 0.7 | 2.5 ± 0.7 | 0.0001 | 2.7 ± 0.6 | 3.0 ± 1.3 | 0.67 |
| IL-10 | 10.2 ± 10.7 | 7.7 ± 7.1 | 0.71 | 4.6 ± 2.0 | 4.8 ± 2.1 | 0.93 |
| GM-CSF | 63.5 ± 34.0 | 18.9 ± 17.4 | 0.07 | 4.7 ± 1.5 | 4.3 ± 2.7 | 0.84 |
| IFN-γ | 83.4 ± 14.3 | 11.1 ± 8.6 | 0.0004 | 2.2 ± 0.3 | 2.6 ± 0.6 | 0.31 |
| TNF-α | 7.6 ± 1.8 | 24.1 ± 12.5 | 0.04 | 8.0 ± 2.9 | 8.9 ± 1.8 | 0.65 |
All data are presented as means ± SD. All units are given in picograms per milliliter. BALF, bronchoalveolar fluid. Comparisons were made using unpaired t-tests. n = 10 for LPS/elastase + placebo, and n = 9 for LPS/elastase + iloprost.
Chronic Iloprost Treatment
Airway hyperresponsiveness.
Baseline Penh (prior to methacholine) tended to be higher after 4 wk of LPS/elastase compared with unexposed controls (1.37 ± 1.21 for exposed vs. 0.24 ± 0.23 for controls, P = 0.07), but there was no difference when measured at 8 wk (P > 0.05). Penh was significantly lower in response to methacholine in the Iloprost-treated Group compared with the untreated mice (P = 0.02; Fig. 3A) when measured at 4 wk (after full exposure to LPS/elastase and 2 total wk of intranasal iloprost, see Fig. 1C). At the 8-wk time point (4 wk after the last LPS exposure and 6 total wk of intranasal iloprost), Penh was significantly lower in the Iloprost-treated Group (P = 0.007, Fig. 3B).
Fig. 3.
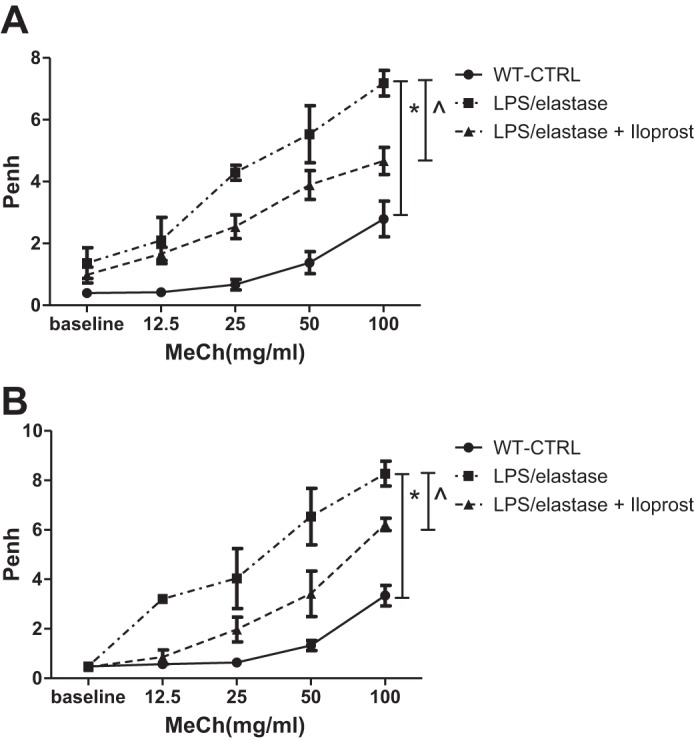
Unrestrained whole body plethysmography for the long-term treatment experiment. Penh was reduced in response to MeCh after 2 wk of iloprost (A; 4 wk of LPS/elastase) compared with LPS/elastase-exposed control values. There also was a reduction in Penh after 6 wk of iloprost (B; 4 wk after last LPS dose) compared with LPS/elastase-exposed control values. See Fig. 2 for abbreviations. Comparisons were made using two-way ANOVA with repeated measures and Bonferroni's post hoc test. At 4 wk (A): n = 3 for WT-CTRL, n = 6 for LPS/elastase, and n = 5 for LPS/elastase + iloprost. At 8 wk (B): n = 5 for WT-CTRL, n = 5 for LPS/elastase, and n = 4 for LPS/elastase + iloprost. *P ≤ 0.05 for LPS/elastase vs. WT-CTRL; ^P ≤0.05 for LPS/elastase + iloprost vs. LPS/elastase.
BALF cell counts.
BALF neutrophil, macrophage, and lymphocyte counts were all significantly higher in the LPS/elastase-exposed mice compared with the unexposed wild-type controls (Fig. 4). There were significantly lower BALF neutrophil counts in LPS/elastase mice treated with iloprost compared with those not treated (6,507 ± 808 vs. 10,410 ± 1,564 cells/mouse, respectively, P = 0.01, Fig. 4). There were no statistically significant differences in macrophage (P = 0.34), lymphocyte (P = 0.11), or total cell counts (P = 0.18) following iloprost.
Fig. 4.

Bronchoalveolar lavage fluid cell counts in the long-term treatment experiment, demonstrating a significant reduction in neutrophil count in response to iloprost treatment: neutrophils (A), macrophages (B), lymphocytes (C), total cells (D), expressed in cells/mouse. WT-CTRL, unexposed, untreated wild-type controls. Comparisons were made using one-way ANOVA with Tukey's post hoc test. n = 3 for each group.
Cytokine and nitrite analysis.
Except for IL-5 and IFN-γ, all cytokines in the serum were increased in the LPS/Elastase Group compared with the unexposed wild-type controls. BALF levels of IL-1β, IL-2, IL-5, and IL-10 were higher in the LPS/Elastase Group compared with unexposed wild-type controls (P < 0.05, data not shown). Six weeks of iloprost caused significant reductions in serum IL-1β (30.0 ± 9.2 vs. 64.8 ± 7.4 pg/ml, P = 0.045), IL-2 (36.5 ± 10.6 vs. 83.8 ± 0.4 pg/ml, P = 0.01), and IL-10 (75.7 ± 9.3 vs. 96.5 ± 3.5 pg/ml, P = 0.02, Fig. 5). Iloprost treatment also reduced BAL IL-2 levels (14.3 ± 0.6 vs. 18.5 ± 0.9 pg/ml, P = 0.002). There were no other significant differences in any of the other measured serum or BALF cytokines (data not shown). LPS and elastase caused a nonsignificant increase in serum NO concentration compared with unexposed wild-type controls (30.5 ± 10.7 vs. 13.8 ± 1.7 μmol, P = 0.08, Fig. 6A). Iloprost treatment reduced serum NO to a level similar to healthy control mice (15.1 ± 5.4 μmol, P = 0.01 compared with untreated LPS-elastase mice, Fig. 6A). Neither LPS/elastase alone nor iloprost treatment had an effect on BAL nitrite levels [0.41 ± 0.58 (unexposed wild-type controls) vs. 0.93 ± 0.83 (untreated LPS/elastase mice) vs. 1.07 ± 0.33 (iloprost treated mice) micromoles, P = 0.48, Fig. 6B]. Another marker of oxidative stress, glutathione peroxidase, was significantly reduced in plasma of mice treated with iloprost (Fig. 6C). GPX was not detected in the BAL fluid.
Fig. 5.

Serum and bronchoalveolar lavage fluid (BALF) cytokines in the long-term treatment experiment. There were significant reductions in serum IL-1β (A), IL-2 (B), and IL-10 (C), as well as BALF IL-2 (D) in the Iloprost-treated Group. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Comparisons were made using one-way ANOVA with Tukey's post hoc test. n = 2 for WT-CTRL, n = 3 for LPS/elastase, and n = 3 for LPS/elastase + iloprost.
Fig. 6.

Serum (A) and bronchoalveolar lavage fluid (B) nitrite in the long-term treatment experiment, demonstrating a significant reduction in serum nitrite in the Iloprost-treated Group. Plasma glutathione peroxidase levels (C) were significantly lower in mice treated with iloprost. *P ≤ 0.05. GPX, glutathione peroxidase. Comparisons were made using one-way ANOVA with Tukey's post hoc test. Serum: n = 2 for WT-CTRL, n = 6 for LPS/elastase, and n = 6 for LPS/elastase + iloprost; BALF: n = 2 for WT-CTRL, n = 5 for LPS/elastase, and n = 5 for LPS/elastase + iloprost.
SMA, trichrome, hydroxyproline, and VCAM-1.
SMA in the lung homogenate was significantly increased in response to LPS/elastase, and reduced toward control levels after 6 wk of iloprost (0.43 ± 0.13 [unexposed wild-type controls] vs. 8.12 ± 3.63 [untreated LPS/elastase mice] vs. 3.07 ± 0.71 [iloprost treated mice] relative band intensity, P = 0.02, Fig. 7A). SMA thickness of the small and medium vessels was increased in the LPS/elastase mice (6.0 ± 1.0 μm) compared with unexposed controls (1.8 ± 0.8 μm, P < 0.001), and there is a significant reduction in SMA thickness after treatment with iloprost (2.8 ± 1.2 μm, P < 0.001 compared with LPS/Elastase Group, Fig. 7, B and C). There did not appear to be any difference in trichrome staining between untreated LPS/elastase mice and iloprost-treated mice (data not shown). There were no significant differences among wild-type mice, LPS/elastase iloprost-treated and untreated LPS/elastase mice in hydroxyproline levels (P = 0.09, Fig. 7D). Iloprost treatment did not reduce the expression of VCAM-1 [0.64 ± 0.003 (unexposed wild-type controls) vs. 0.63 ± 0.12 (untreated LPS/elastase mice) vs. 0.55 ± 0.11 (iloprost-treated mice) VCAM-1/actin ratio, P = 0.45, Fig. 8].
Fig. 7.

Smooth muscle actin (SMA) (A) levels in lung homogenate between unexposed, untreated wild-type controls (WT-CTRL), LPS/elastase mice, and iloprost-treated LPS/elastase mice. Iloprost significantly reduced SMA levels compared with untreated LPS/elastase mice (P < 0.05). Units are relative band intensity. Western blot is also displayed. B: immunohistochemistry stained for SMA. C: staining was increased in the LPS/Elastase Group, and reduced after exposure to iloprost, which was confirmed by quantification of SMA thickness in small and medium vessels. D: There were no significant differences in hydroxyproline levels between the three groups. Comparisons were made using one-way ANOVA with Tukey's post hoc test. n = 3 for WT-CTRL, n = 5 for LPS/elastase, and n = 5 for LPS/elastase + iloprost. *P ≤ 0.05; ***P < 0.0001.
Fig. 8.

VCAM-1 Western blot (A) and densitometric analysis (B), demonstrating no difference between the three groups. Densitometric analysis quantified as VCAM-1/actin ratio. Comparisons were made using one-way ANOVA with Tukey's post hoc test.
DISCUSSION
We have demonstrated, for the first time, that a prostacyclin analog may be beneficial as a treatment (rather than prevention) in a murine model of COPD. Both after acute and chronic iloprost, there were significant and consistent reductions in important markers of systemic inflammation and oxidative stress. Penh, considered a surrogate marker for airway hyperresponsiveness (11), was also reduced in mice treated with 2 wk of iloprost, which persisted after 6 wk of iloprost. Since COPD is a condition characterized by airway obstruction, as well as increased level of systemic inflammation and oxidative stress, these data suggest that iloprost might be beneficial in humans with disease. Although other studies have shown the ability of prostacyclin analogs to prevent the development of COPD manifestations when given prior to model induction (6, 20), ours is the first description of benefit when given in a treatment fashion, which has a much higher degree of clinical relevance.
PGI2 is synthesized from arachadonic acid in the pulmonary ECs under the influence of prostacyclin synthase. Prostacyclin analogs, used clinically to treat pulmonary arterial hypertension, have also been investigated in preclinical models relevant to COPD. Nana-Sinkam et al. (20) demonstrated that there is decreased expression of PGI2 synthase in lung tissue from emphysema patients. Additionally, there was decreased PGI2 synthase expression from in vitro ECs cells after exposure to cigarette smoke extract (CSE). These investigators also found that iloprost prevented CSE-induced EC apoptosis. A different PGI2 analog, beraprost, was shown to reduce CSE-induced emphysema, apoptosis, and inflammation in rats when given in a preventative fashion (6). Another vascular-targeted therapy, a soluble guanylate cyclase stimulator, prevented emphysema formation, pulmonary hypertension, alveolar apoptosis, and neutrophilia in response to cigarette smoke (CS) in animal models of COPD (33). All of the studies discussed above gave the drug prior to CS exposure, which has very limited clinical applicability. To our knowledge, we are the first to demonstrate that a vascular-targeted prostacyclin had beneficial effects on systemic inflammation, oxidative stress, and response to methacholine in a COPD-like animal model when given as a treatment rather than as a prevention.
We found significant reductions in serum IL-5 and IFN-γ after only 3 doses of iloprost in our LPS/elastase murine model. It has been demonstrated that IL-5 is increased in sera of patients with COPD (19) and that treatment with an anti-IL-5 receptor antibody can improve FEV1 in COPD (5). In a murine model of COPD, IFN-γ overexpression increased neutrophilia, alveolar enlargement, and lung compliance (32). It is noteworthy that IFN-γ levels are significantly higher in patients with COPD compared with healthy controls (18, 22). Therefore, serum cytokines that were reduced after three doses of iloprost may have relevance to humans with COPD. Although serum TNF-α was increased in response to acute iloprost treatment, there were trends toward reductions in downstream cytokines IL-1β and GM-CSF. Most importantly, TNF-α was not increased in the sera of mice given long-term iloprost.
After 6 wk of iloprost treatment, we found reductions in serum IL-1β, IL-2, IL-10, and nitrite, as well as BAL fluid neutrophils. IL-1β plays an important role in the pathogenesis of COPD. Transgenic mice overexpressing IL-1β have neutrophilic inflammation, emphysema, and increased thickness of the conducting airways (16). Serum IL-1β is increased in COPD smokers (27), and there is a higher expression of IL-1β in airway epithelium of COPD patients compared with smokers (12). Because this cytokine is important for airway remodeling, reductions in IL-1β may partially explain the improvement in Penh seen with iloprost. Smooth muscle actin was reduced by iloprost, which may also be relevant in airway remodeling. Alternatively, reductions in SMA after iloprost could be related to improvements in pulmonary vascular remodeling, as we have demonstrated by a reduction in smooth muscle thickness of the small and medium vessels. T-regulatory cells (Tregs), controlled in part by IL-2, appear to be involved in COPD (13). In COPD patients, CD4+ T cells producing IL-2 had a significant negative correlation to FEV1 (34). In our experiments, IL-10, an anti-inflammatory cytokine, was reduced in response to iloprost. This paradoxical finding may be explained by the fact that IL-2 was reduced, as Tregs under the control of IL-2 partially regulate IL-10 production (21). Alternatively, since there was an overall decrease in systemic inflammation, there may have been less need for the anti-inflammatory effects of IL-10. Serum nitrite was also reduced by iloprost, which is important since oxidative stress is a critical driver of COPD development and progression (29, 35). Lastly, in humans with COPD, sputum neutrophilia is common and correlates with disease severity (15). In our experiments, neutrophil counts in the BAL fluid were lower after iloprost. In summary, critical mediators of COPD pathogenesis were reduced in response to 6 wk of intranasal iloprost, which may indicate that chronic treatment could be beneficial to humans with COPD.
Our experiments were not set up to comprehensively investigate mechanisms of benefit, but iloprost could improve systemic inflammation by restoring EC barrier function (4), reducing adhesion molecule expression (24), attenuating neutrophil adhesion to ECs (17), and/or by reducing apoptosis (20). We did not see a difference in VCAM-1 expression, although adhesion molecules may play a role early in disease onset, which may have been missed by the timing of our investigations. Iloprost could also have direct effects on inflammatory cells activation, as PGI2 receptors are found on human inflammatory cells (7). It could be hypothesized that, since iloprost was delivered intranasally, there was a direct effect of iloprost on the airways and parenchyma that caused a “downstream” reduction in serum cytokines. However, it is noteworthy that we observed preferential improvement in systemic inflammation and oxidative stress with almost no change in BAL fluid cytokines or nitrite. This observation increases the likelihood that intranasal iloprost is affecting the vasculature rather than directly modifying the airways or parenchyma, although this remains speculative.
There are limitations to our study, which deserve mention. Although no animal model truly replicates human COPD, the most commonly used model is CS exposure (2, 15). We chose to use the LPS/elastase model, as has been done by others in previous published work (8, 9), due to its shorter time course and ability to lead to emphysema, systemic inflammation, and decreased elastic recoil (8). It appears superior to using elastase alone, as the combination causes more lung inflammation, emphysema, and increased lung compliance (25). While invasive measures of lung function are more sensitive than Pehn for detecting changes in airway resistance (30), Penh allowed us to examine the same mice at multiple time points. Moreover, Pehn correlates with invasive measures, and increases in Penh after methacholine correspond with increased intrapleural pressure (11). We started treating mice in the long-term treatment experiment 2 wk into LPS/elastase exposure, to mimic less severe forms of COPD. Therefore, it could be argued that this is just another prevention study. However, it is evident from the literature that a COPD-like phenotype is established 3 wk after a single elastase dose (1, 28), and we have demonstrated that histologic emphysema is clearly present 2 wk after elastase administration (Fig. 1D). While iloprost may be expected to have effects on pulmonary vascular disease in this model, we did not perform invasive measures of hemodynamics.
Conclusions
In conclusion, we demonstrated, for the first time, that treatment with a prostacyclin analog led to reductions in systemic inflammation, oxidative stress, and response to methacholine in a COPD-like murine model. These are novel observations that should be confirmed with more detailed mechanistic investigations that, if validated, could lead to clinical trials of iloprost treatment in emphysema patients.
GRANTS
This study was supported, in part, by HL-072889 from the National Institutes of Health (NIH) (to A. H. Boulares), by the COBRE Pilot Program Grant 1P306M106392-01A1 (M. R. Lammi), and by 1 U54 GM-104940 from the National Institute of General Medical Sciences of the NIH, which funds the Louisiana Clinical and Translational Science Center (M. R. Lammi). A. S. Naura acknowledges grant BT/RLF/Re-entry/36/2012 from DBT, Government of India. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.R.L., A.S.N., K.I.H., B.P.d., J.S., and A.H.B. conception and design of research; M.R.L., M.A.G., K.P., A.S.N., S.V.I., C.J.D., S.C.O., and A.H.B. performed experiments; M.R.L., M.A.G., K.P., S.V.I., C.J.D., and A.H.B. analyzed data; M.R.L., M.A.G., K.P., S.V.I., C.J.D., S.C.O., K.I.H., B.P.d., J.S., and A.H.B. interpreted results of experiments; M.R.L., M.A.G., K.P., and A.H.B. prepared figures; M.R.L. drafted manuscript; M.R.L., A.S.N., B.P.d., J.S., and A.H.B. edited and revised manuscript; M.R.L., M.A.G., K.P., A.S.N., S.V.I., C.J.D., S.C.O., K.I.H., B.P.d., J.S., and A.H.B. approved final version of manuscript.
Matthew Lammi is the guarantor of the paper and takes responsibility for the integrity of the work as a whole, from inception to published article.
ACKNOWLEDGMENTS
Prior abstract publication: Lammi MR, Ghonim M, Pyakurel K, Naura A, Happel K, DeBoisblanc B, Shellito JE, Boulares H. Effect of intranasal iloprost on a murine model of chronic obstructive pulmonary disease. Am J Resp Crit Care Med 189: A6017, 2014.
REFERENCES
- 1.Andersen MP, Parham AR, Waldrep GC, McKenzie WN, Dhand R. Alveolar fractal box dimension inversely correlates with mean linear intercept in mice with elastase-induced emphysema. Int J Chron Obstruct Pulmon Dis 7: 235–243, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballweg K, Mutze K, Konigshoff M, Eickelberg O, Meiners S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 307: L895–L907, 2014. [DOI] [PubMed] [Google Scholar]
- 2a.Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 35: 71–86, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Bella Della S, Molteni M, Mocellin C, Fumagalli S, Bonara P, Scorza R. Novel mode of action of iloprost: in vitro down-regulation of endothelial cell adhesion molecules. Prostaglandins 65: 73–83, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, Leff A, Birukov KG. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur Respir J 41: 165–176, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brightling CE, Bleecker ER, Panettieri RA Jr, Bafadhel M, She D, Ward CK, Xu X, Birrel C, van der Merwe R. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respir Med 2: 891–901, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Hanaoka M, Chen P, Droma Y, Voelkel NF, Kubo K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol 296: L648–L656, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Dorris SL, Peebles RS. PGI 2 as a regulator of inflammatory diseases. Mediators Inflamm 2012: 926968, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganesan S, Faris AN, Comstock AT, Chattoraj SS, Chattoraj A, Burgess JR, Curtis JL, Martinez FJ, Zick S, Hershenson MB, Sajjan US. Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir Res 11: 131, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganesan S, Faris AN, Comstock AT, Sonstein J, Curtis JL, Sajjan US. Elastase/LPS-exposed mice exhibit impaired innate immune responses to bacterial challenge. Am J Pathol 180: 61–72, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giordano RJ, Lahdenranta J, Zhen L, Chukwueke U, Petrache I, Langley RR, Fidler IJ, Pasqualini R, Tuder RM, Arap W. Targeted induction of lung endothelial cell apoptosis causes emphysema-like changes in the mouse. J Biol Chem 283: 29,447–29,460, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 156: 766–775, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Herfs M, Hubert P, Poirrier A, Vandevenne P, Renoux V, Habraken Y, Cataldo D, Boniver J, Delvenne P. Proinflammatory cytokines induce bronchial hyperplasia and squamous metaplasia in smokers: implications for chronic obstructive pulmonary disease therapy. Am J Respir Cell Mol Biol 47: 67–79, 2012. [DOI] [PubMed] [Google Scholar]
- 13.Hou J, Sun Y, Hao Y, Zhuo J, Liu X, Bai P, Han J, Zheng X, Zeng H. Imbalance between subpopulations of regulatory T cells in COPD. Thorax 68: 1131–1139, 2013. [DOI] [PubMed] [Google Scholar]
- 15.John-Schuster G, Hager K, Conlon TM, Irmler M, Beckers J, Eickelberg O, Yildirim AO. Cigarette smoke-induced iBALT mediates macrophage activation in a B cell-dependent manner in COPD. Am J Physiol Lung Cell Mol Physiol 307: L692–L706, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106: 1311–1319, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15a.Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 153: 530–534, 1996. [DOI] [PubMed] [Google Scholar]
- 16.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1β causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol 32: 311–318, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Lindemann S, Gierer C, Darius H. Prostacyclin inhibits adhesion of polymorphonuclear leukocytes to human vascular endothelial cells due to adhesion molecule independent regulatory mechanisms. Basic Res Cardiol 98: 8–15, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Majori M, Corradi M, Caminati A, Cacciani G, Bertacco S, Pesci A. Predominant TH1 cytokine pattern in peripheral blood from subjects with chronic obstructive pulmonary disease. J Allergy Clin Immunol 103: 458–462, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura M, Nakamura H, Minematsu N, Chubachi S, Miyazaki M, Yoshida S, Tsuduki K, Shirahata T, Mashimo S, Takahashi S, Nakajima T, Tateno H, Fujishima S, Betsuyaku T. Plasma cytokine profiles related to smoking-sensitivity and phenotypes of chronic obstructive pulmonary disease. Biomarkers 19: 368–377, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, Cool C, Parr J, Moore MD, Bull TM, Voelkel NF, Geraci MW. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med 175: 676–685, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogawa Y, Duru EA, Ameredes BT. Role of IL-10 in the resolution of airway inflammation. Curr Mol Med 8: 437–445, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paats MS, Bergen IM, Hoogsteden HC, van der Eerden MM, Hendriks RW. Systemic CD4+ and CD8+ T-cell cytokine profiles correlate with GOLD stage in stable COPD. Eur Respir J 40: 330–337, 2011. [DOI] [PubMed] [Google Scholar]
- 23.Peinado VI, Barberà JA, Abate P, Ramirez J, Roca J, Santos S, Rodriguez-Roisin R. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med 159: 1605–1611, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Peinado VI, Barberà JA, Ramírez J, Gomez FP, Roca J, Jover L, Gimferrer JM, Rodriguez-Roisin R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol Lung Cell Mol Physiol 274: L908–L913, 1998. [DOI] [PubMed] [Google Scholar]
- 25.Sajjan U, Ganesan S, Comstock AT, Shim J, Wang Q, Nagarkar DR, Zhao Y, Goldsmith AM, Sonstein J, Linn MJ, Curtis JL, Hershenson MB. Elastase- and LPS-exposed mice display altered responses to rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 297: L931–L944, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, Egemnazarov B, Turowska A, Fuchs B, Nikam S, Roth M, Sydykov A, Medebach T, Klepetko W, Jaksch P, Dumitrascu R, Garn H, Voswinckel R, Kostin S, Seeger W, Schermuly RT, Grimminger F, Ghofrani HA, Weissmann N. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 147: 293–305, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Singh B, Arora S, Khanna V. Association of severity of COPD with IgE and interleukin-1β. Monaldi Arch Chest Dis 73: 86–87, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Tibboel J, Keijzer R, Reiss I, de Jongste JC, Post M. Intravenous and intratracheal mesenchymal stromal cell injection in a mouse model of pulmonary emphysema. COPD 11: 310–318, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest 122: 2749–2755, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verheijden K, Henricks P, Redegeld F, Nijkamp F, Folkerts G. A comparative study between Penh and airway resistance in a mild and severe mouse model for allergic asthma. Am J Respir Crit Care Med 185: A5638, 2012. [Google Scholar]
- 31.Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, Stockley RA, Sin DD, Rodriguez-Roisin R. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 187: 347–365, 2013. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr, Shapiro SD, Elias JA. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med 192: 1587–1600, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weissmann N, Lobo B, Pichl A, Parajuli N, Seimetz M, Puig-Pey R, Ferrer E, Peinado VI, Dominguez-Fandos D, Fysikopolous A, Stasch JP, Ghofrani HA, Coll-Bonfill N, Frey R, Schermuly RT, Garcia-Lucio J, Blanco I, Bednorz M, Tura-Ceide O, Tadele E, Brandes RP, Grimminger J, Klepetko W, Jaksch P, Rodriguez-Roisin R, Seeger W, Grimminger F, Barbera JA. Stimulation of soluble guanylate cyclase prevents cigarette smoke-induced pulmonary hypertension and emphysema. Am J Respir Crit Care Med 189: 1359–1373, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Zhu X, Gadgil AS, Givelber R, George MP, Stoner MW, Sciurba FC, Duncan SR. Peripheral T cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol 182: 3270–3277, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Zuo L, He F, Sergakis GG, Koozehchian MS, Stimpfl JN, Rong Y, Diaz PT, Best TM. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am J Physiol Lung Cell Mol Physiol 307: L205–L218, 2014. [DOI] [PubMed] [Google Scholar]