

Abstract
We describe here the development, verification and validation of the SLE-key® rule-out test for a definitive rule-out of a diagnosis of systemic lupus erythematosus (SLE). The test uses the proprietary iCHIP® micro-array technology platform (Fattal et al., 2010) to identify discriminating patterns of circulating autoantibodies among SLE patients compared to self-declared healthy individuals. Given the challenges associated with the diagnosis of SLE and the healthcare costs of delayed diagnosis and misdiagnosis, a definitive rule-out test can provide significant clinical benefits to patients and potentially major cost savings to healthcare systems.
Keywords: autoantibodies, diagnosis, iCHIP, microarray, systemic lupus erythematosus, multivariate classifier
1. Introduction
`SLE is a chronic inflammatory connective tissue disease of unknown etiology that affects between 1 and 25 of every 100,000 individuals worldwide, primarily women (Petri et al., 1999; Danchenko et al., 2006; Pons-Estel et al., 2013). The disease is mediated by an autoimmune process marked by a large spectrum of autoantibodies within the patient population, targeting more than 100 different autoantigens; individual patients, however, are diverse in the particular autoantibodies they express (Sherer et al., 2004). SLE, moreover, is associated with diverse and variable clinical manifestations that can change often and suddenly (Arbuckle et al., 2003; Li et al., 2007; Lateef and Petri, 2012; Olsen et al., 2012). In addition to gender differences, the prevalence of SLE is associated with racial and geographic clustering, which suggests a role for genetic, hormonal and environmental factors (Danchenko et al., 2006; Hiraki et al., 2009; Borchers et al., 2010; Ramos-Casals et al., 2015).
Because of its complexity, it is not surprising that no single serologic test is diagnostic for SLE; indeed, the definitive diagnosis of SLE is based on a summation of multiple clinical and serologic criteria (Lateef and Petri, 2012), and can take years of concerted monitoring. Difficulties in diagnosis may lead to a protracted process of medical consultations and uncertainty involving long waiting times to see specialist physicians (including second and third opinions), extensive diagnostic testing, undue medical management, and poorer outcomes (Slater et al., 1996). Many of the currently available laboratory tests are inconclusive and may lead to the loss of precious time, during which a patient’s organ systems including kidneys, heart, lungs, brain, blood and skin can undergo irreversible damage (Petri et al., 1999). Furthermore, misdiagnosis in symptomatic patients who do not have SLE impacts both therapeutic management and subsequent insurance coverage based on the lack of conclusive evidence for exclusion of the disease. To further complicate matters, even healthy persons, as well as SLE patients, may manifest positive assays for antinuclear antibodies (ANA positivity) (Slater et al., 1996). Thus definitive, objective diagnostic tools for SLE are an unmet need. A test to rule-out the diagnosis of lupus in patients without disease would be a valuable starting objective.
Here we report the development of a robust and reliable SLE rule-out test based on an antigen microarray and informatics analysis. Since SLE is a heterogeneous and complex disease, no single biomarker is likely to be sufficient. Therefore, our strategy was based on a combination of meaningful SLE antigens derived from multiple molecular classes, using an antigen microarray and advanced informatics analysis. The aim of this study was to overcome the complexity and find a common autoantibody profile among the heterogeneous SLE population by using multivariate analysis. The use of antigen microarrays for the identification of SLE patients has been reported previously (Robinson et al., 2002; Li et al., 2007; Fattal et al., 2010). These approaches have shown significantly greater sensitivity than standard ELISA assays, but none of these systems has been robust enough for dependable clinical application. The iCHIP® (ImmunArray, Rehovot, Israel) described here documents the development and transition of a research microarray platform (Fattal et al., 2010) into a robust and validated clinical test for definitively ruling out a diagnosis of SLE. The SLE-key® rule-out microarray uses a relatively small volume (10–20 ul) of serum and simultaneously detects the presence of a multitude of disease-related autoantibodies to yield a comprehensive profile of the status of the individual patient.
2. Methods
2.1. Patient samples
SLE serum samples and clinical information were obtained from the repositories of four independent, major lupus centers in the US, and were approved by each respective IRB: Albert Einstein College of Medicine, Emory University, Johns Hopkins University and Medical University of South Carolina. Self-declared healthy control samples were obtained from five sites: Baylor College of Medicine, CTI Clinical Research Center, Medical University of South Carolina, Veracis Laboratory, and San Francisco Medical Center and were collected in a manner compliant with the HIPPA and with appropriate informed consent.
The samples were collected from females, not known to be pregnant, between 18–60 years of age at the time of sample collection, and were tested within 10 years from the time of serum collection. All SLE patients fulfilled four or more ACR criteria (mean 5.24±1.2) and samples were collected within three years of diagnosis (mean 1.00±1). Healthy samples were collected from self-declared healthy subjects who had no record of immunologically active disease or steroid use within three months of sample collection, and no first-degree relatives with SLE. For the classifier development phase, sera from 146 SLE patients and 151 healthy individuals were tested. An additional test set of 50 SLE and 50 healthy samples was used in the verification phase, and 50 SLE patients and 51 healthy samples were used in the validation phase. In total, 246 SLE patient and 252 healthy individuals were tested.
2.2. Study design
Classifier development and verification testing was performed on 15 iCHIP® print lots. Validation testing was performed on six new, independently manufactured iCHIP® print lots. To eliminate biases, samples were rationally assigned to each test session based upon subject demographics, sample class, ACR score, time post-diagnosis for SLE samples, and sample source. Samples were then randomized across iCHIP® print lots, test days and operators. The majority of classifier-development stage samples (~85%) were tested in duplicates on two different print lots and in two different test sessions. In order to verify equal distribution of subject sample profiles across test sets, suitable statistical analyses were performed; the Wilcoxon test for numeric variables and the Chi square for categorical variables. An alpha value of 0.2 was used in all tests in order to reduce the risk of a type 2 error. All tests were performed at Immunarray’s CLIA-certified laboratory, Veracis (Richmond, VA). All print lots used for development, verification and validation passed quality control (QC) release testing at Veracis.
2.3. iCHIP® preparation
Glass slides were coated at ImmunArray with an epoxy silane organic layer, using an inhouse developed proprietary coating procedure, using a YES1224p oven (Yield Engineering Systems, CA, USA). After coating, the slides were packed into slide boxes and vacuum-sealed until printing. Antigens were printed on the coated slides using a Scienion S11 non-contact arrayer (Scienion AG, Germany). The slides were blocked with 1% casein (Sigma) (1h, room temperature), and then rinsed with phosphate buffer solution (PBS) for ten minutes, 0.05% PBS- Tween 20 (PBS-T) (Sigma) for ten minutes, and again with PBS for ten minutes. The slides were then dipped in reverse osmosis-grade water and dried in a Nuve centrifuge (10 min, 2180 rpm). All washing solutions were used at room temperature.
2.4. Antigen Array
The proprietary SLE-key® iCHIP® microarray (ImmunArray Ltd., Rehovot, Israel) was used to display approximately 200 antigens, including antigens selected from a wide range of reported SLE-associated biochemical pathways including inflammatory pathways, immune system signaling pathways and lipid and lipoprotein metabolism antigens (Figure 1). Approximately 12% of the antigens were proprietary oligonucleotides of various chain lengths from 7 to 30 nucleotides (Fattal et al., 2015).
Figure 1. Distribution of iCHIP® antigens over multiple biochemical pathways.
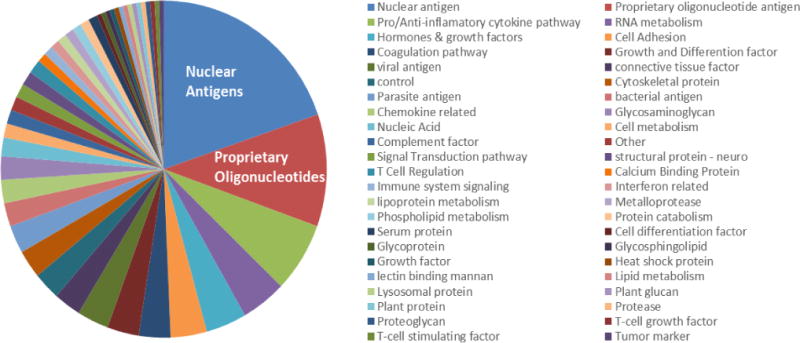
Antigens displayed on the iCHIP® included reported SLE-associated antigens and proprietary antigens developed at ImmunArray.
2.5. Serum testing
The slides were allowed to reach room temperature and serum specimens were fully thawed before testing. Serum samples were diluted 1:20 in freshly filtered 1% casein and then centrifuged (15 min, 13,300 RCF), to remove aggregates. Diluted serum (135 μl) was dispensed using pipette at the center of the slide, covered with a cover slip (24×60 mm, Marienfeld) and then incubated in a humidity chamber (1h, 37 °C). Each slide was then dipped in PBS, to remove the cover slips, and then sequentially washed in PBS, PBS-T, and PBS for ten minutes each, before being dipped in reverse osmosis-grade water. The slides were dried in a Nuve centrifuge NF800 (10 min, 2180 rpm) before being incubated with goat anti-human IgG-Cy3 (Jackson ImmunoResearch Laboratory) and goat anti-human IgM-Dy5 (Jackson ImmunoResearch Laboratory) diluted 1:500 in 1% casein in PBS. All washing solutions were at room temperature. The slides after testing were stored at 4°C, under vacuum, until scanning.
2.6. Slide scanning and data processing
The slides were scanned at two wavelengths (532 nm and 633 nm) using an Agilent C scanner (Agilent Technologies, Santa Clara, CA) and images were extracted using the GenePix 7.0 feature extraction (FE) software (Molecular Devices, Sunnyvale, CA), using default settings. All scanned images were preprocessed, without regard to sample class.
Data preprocessing involves these major steps: subtraction of background, combination of replicates (six spots per antigen per slide), adjustment of overall intensity per slide and correction of print lot effect. Preprocessing was performed as follows:
Signals were represented by the mean intensity of the spot minus the median of the local background, followed by log (base 2) transformation for non-negative spots.
Negative spots (un-printed area) were imputed by artificial low intensity values, a process performed separately for each channel (IgG and IgM).
The median intensities of all slides were adjusted to be equal to 9 intensity units, for each channel separately.
The spot intensity per slide was represented by the median across all spots, excluding flagged spots and outlier spots defined by the Genepix software. The intensity-dependent effect of print lot was globally corrected through a general intensity-based approach (not antigen-specific) based on the in-process QC slides (SLE-SP1) which were included in every test session and on each print lot. The LOWESS-correction (smoothing factor of 0.7) was found to perform best in comparison to other data correction methods that were applied, as it proved more resistant to print lot effects.
3. Results
The samples obtained from the SLE patients and the healthy control subjects were tested for their intensity of binding to the arrayed antigens as described in the Methods section and the data were analyzed in several ways with the aim of devising a robust and reliable rule-out assay for SLE.
The 246 SLE samples were collected from African-American patients (53.0%), White non-Hispanic patients (21.5%) White Hispanic patients (20.0%), Indian/Asian/Middle Eastern patients (2.0%) and others (3.6%). The average age was 34.8±11.4. The 252 healthy control samples were collected from subjects with similar demographic and age distributions as those of the SLE cohort (Table 1).
Table 1.
Classifier development, verification and validation sample demographics.
| Sample Demographics | SLE Patients (n=246) | Healthy Controls (n=252) |
|---|---|---|
| Age in years Mean (±SD) |
34.8 (11.4) | 36.8 (12.0) |
| Ethnic category | ||
| Afro-American: Number (%) | 130 (53.0) | 113 (44.8) |
| White non-Hispanic: Number (%) | 53 (21.5) | 74 (29.4) |
| Indian/Asian/Middle Eastern: Number (%) | 5 (2.0) | 20 (7.9) |
| White Hispanic: Number (%) | 49 (20.0) | 42 (16.7) |
| Other: Number (%) | 9 (3.6) | 3 (1.2) |
3.1 Univariate antigen performance
As a preliminary and necessary step for feature selection as the basis for multi-variate analysis, we initiated the analysis with univariate antigen performance. The intensities of both IgM and IgG antibody binding to each arrayed antigen were analyzed, and 131 antigens binding autoantibodies and viral antibodies were found to successfully separate between the SLE patients and the healthy controls (p <0.05). The informative antigens included known SLE associated antigens such as dsDNA (IgG) which generated a p value of 1.2E-26; Sm (IgG) which generated a p value of 3.5E-24; Ro52 (IgG) which generated a p value of 2.3E-20; Ro60 (IgG) which generated a p value of 1.1E-09; and La (IgG) which generated a p value of 1.3E-06; these antibodies are characteristically tested by physicians in patients suspected of having SLE. In addition, informative antigens also included those not typically included in the clinical evaluation of patients with suspected rheumatic disease, such as Epstein Barr Virus (EBV) (IgG), which generated a p value of 2.4E-22; vimentin (IgM) which generated a p value of 7.8E-11; and others (data not shown). Nevertheless, despite highly significant results on the univariate analysis for specific antigens, it is unlikely that a single biomarker would be able to capture the collective of individual variations among patients with this disease. Therefore, we hypothesized that a comprehensive multivariate classifier development would be the appropriate solution to arrive at a robust autoantibody profile that would characterize the SLE patient population, regardless of disease stage or activity.
3.2 Multivariate Classifier Development
3.2.1. Classifier development, Stage 1
We tested six different multivariate models: Classification and Regression Tree (CART); Random Forest (RF); Logistic Regression (LR); Support Vector machine (SVM); Quadratic Discriminant Analysis (QDA); and Linear Discriminant Analysis (LDA). All six models performed well. The optimal performance, defined as the best balance between sensitivity and specificity, was obtained with the LDA method, as detailed below.
3.2.2. Feature selection
LOWESS-corrected data were used as the input for all six classifier construction methods, all of which demonstrated some success in defining an SLE rule-out classifier. The highest-ranking features obtained with each of the methods were identified through cross validation; 70% of the samples were randomly selected, with class stratification, to serve as the training set, and the remaining 30% served as the test set. Two hundred reiterations of classifier construction using each of the methods were performed in order to select the most informative features. To further filter the data, correlations between all features within a feature group were calculated without regard to sample class. To avoid over-fit, which negatively impacts the generalized ability of the classifier to differentiate between sample groups, one feature was removed from each highly correlated feature pair (Pearson correlation coefficient ≥ 0.9). Cross validation was then repeated using only the selected list of antigens from the previous stage for each classification method. In order to validate each antigen list and verify that each antigen on the list is significant, further filtering was performed by running cross validations on the training data with a decreasing number of features used in each run and following the performance response to this decrease. Eventually and as a result of this examination, an optimal set of features was defined for each of classification methods. Due to repeated use of features in multiple tree nodes, the CART and RF models demonstrated extreme over-fit, with sensitivity and specificity close to 100%, and were therefore abandoned. The four remaining models were used for the next development stages.
3.2.3. Final model fit
After the final feature selection for each model (Table 2), the four final models were fit based on the selected features of the full training data set; this concluded the training phase. At this stage, each of the models could provide a probability score for a blinded sample. We then established a threshold for class prediction in the verification stage.
Table 2.
Selected antigens and univariate performance (p value) by each classification method.
| Antigen Sets | ||||
|---|---|---|---|---|
| SVM | Logistic Regression | QDA | LDA | FDR corrected p value |
| ssDNA (IgG) | ssDNA (IgG) | ssDNA (IgG) | ssDNA (IgG) | 3.1E-45 |
| U1snRNP (IgG) | U1snRNP (IgG) | U1snRNP (IgG) | U1snRNP (IgG) | 9.5E-35 |
| Histone3S (IgM)a | Histone3S (IgM)a | H2a (IgM)b | Histone3S (IgM)a |
a1.6E-22; b2E-18 |
| Sm (IgG) | Sm (IgG) | Sm (IgG) | 3.5E-24 | |
| Ro52 (IgG) | Ro52 (IgG) | Ro52 (IgG) | 2.3E-20 | |
| Apo SAA (IgG) | Apo SAA (IgG) | 2.2E-03 | ||
| DNAse1 (IgG)c | Collagen III (IgG)d | Proprietary Oligonucleotide (IgM)e |
c2.6E-3; d0.16; e1.7E-12 |
|
| U1snRNP (IgM) | 7.8E-09 | |||
3.2.4. Classifier Verification – Stage 2
The goal of the verification stage was to verify the classifier capabilities on a new set of samples by analyzing receiver operating characteristic (ROC) plots using blinded samples.
Since only 17% of the verification samples were tested on duplicate arrays, we expected a certain degree of reduced performance compared to the training data area under the curve (AUC) determinations, in which 85% of the samples were tested in duplicate. To take this into account, a single set of duplicates of 100 of the training set samples was randomly selected to undergo cross-validation analysis; the final classifier models were then applied and a score was obtained per a sample.
For each method, an estimated range spanning 99% of the AUC distributions was calculated by subtracting three standard deviations from the mean value, yielding the acceptance criteria for each method individually.
Following classifier application to the blinded verification data, we scored samples for their likelihood of being from either the SLE or healthy control class. The samples were then unblinded and the predicted diagnoses were compared to the actual subject designations. ROC curves were then generated by combining the predicted and actual scores, and AUCs were determined. The minimal area under the ROC curve defined the verification performance. The AUC values (range: 0.896–0.956) met the preassigned verification test acceptance criteria.
At this point, the continuous prediction score was transformed into a binary classifier by introducing a classification threshold. For a predicted score below this threshold, a patient is ruled out as having SLE, and a score equal to or above this threshold indicates that SLE cannot be ruled out. The establishment of this threshold was performed manually using unblinded verification data sets.
3.2.5. Classifier Validation – Stage 3
The aim of this stage was to validate the SLE iCHIP® Version 1.0 classifiers based on independent testing with a new set of samples on newly manufactured iCHIP® lots.
Fifty SLE patients and 51 healthy control validation samples were run on separate print lots that had not been used for classifier development and verification; this approach introduced maximal variability and thereby tested classifier robustness and reliability. Moreover, the validation sample class and the clinical data were blinded to the analyst. The data were preprocessed, sample duplicates were combined, and then analyzed.
Table 3 shows the validation results; True and False Positive and Negative rates, AUC, Sensitivity, Specificity, Accuracy, Positive Predictive Value (PPV), and Negative Predictive Value (NPV) for each classifier method. Validation ROC curves for each model and prediction distribution results are shown in figure 2A and figure 2B respectively. It can be seen that all four methods differentiated well between the SLE subjects and the healthy controls; however, the LDA method generated the best performance determined by a balance between sensitivity and specificity (sensitivity: 94%, specificity: 75%, NPV: 93%.) (Figure 2A and table 3). The LDA method of analysis was thus chosen to be the informatic model for analyzing the iCHIP® results.
Table 3. Summary of Classifier Validation Results.
AUC, area under the curve; PPV, positive predictive value; NPV, negative predictive value.
| Validation | SVM | Logistic Regression | QDA | LDA |
|---|---|---|---|---|
| Threshold | −0.95 | 0.17 | 0.10 | 0.18 |
| Validation Result | 1.6 | 1.6 | 1.6 | 1.7 |
| AUC | 0.95 | 0.95 | 0.95 | 0.94 |
| Sensitivity | 98% | 98% | 88% | 94% |
| Specificity | 59% | 59% | 76% | 75% |
| Accuracy | 78% | 78% | 82% | 84% |
| PPV | 70% | 70% | 79% | 78% |
| NPV | 97% | 97% | 87% | 93% |
Figure 2. A) ROC curves for the four classification models on validation data set, indicating test performance at selected threshold. B) Validation Prediction Distribution.
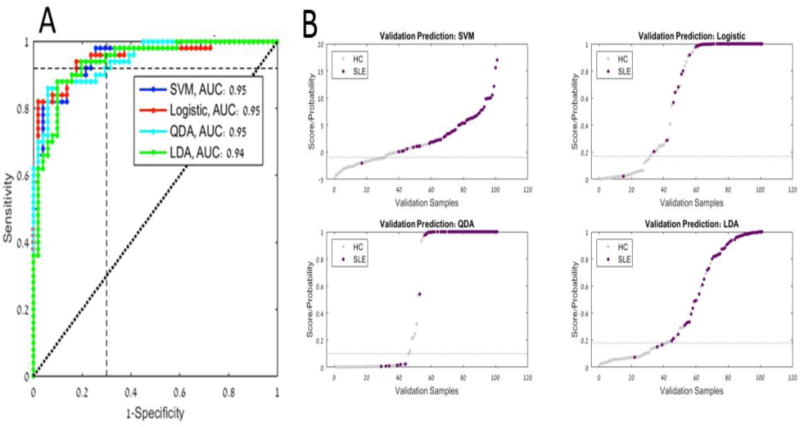
Grey points indicate samples classified as healthy control and purple points indicate samples classified as SLE.
4. Discussion
At present, the diagnosis of SLE relies heavily upon clinical signs and symptoms in combination with the results of laboratory tests. Fulfillment of 4 of 11 American College of Rheumatology (ACR) classification criteria yields a diagnostic sensitivity of 85% and specificity of 95% (Bertsias et al., 2012). Many criteria for classifying SLE have been devised over the years by different international groups such as the Systemic Lupus International Collaborating Clinics (SLICC) criteria (Petri et al., 2012); SLICC criteria are not weighted and do not include subjective symptoms. The SLICC criteria have now been proven to be more sensitive than the classic ACR criteria. However, these criteria are primarily meant for classification of patient cohorts for research; they must be used with caution to diagnose individual patients (Lateef and Petri, 2012). At the early stages of the disease, when serological findings and clinical symptoms might overlap with those of other autoimmune diseases (Wallace, 2015), criteria have not been tested.
This paper documents the development of a novel multivariate test based on an antigen-microarray approach. Multivariate approaches have been successful in the diagnosis of other complex diseases such as breast cancer (van ’t Veer et al., 2002; van de Vijver et al., 2002), cardiovascular disease (Ridker et al., 2007) and diabetes (Kolberg et al., 2009). The strategy of developing a multivariate tool that detects an informative autoantibody signature across broad and diverse SLE patient populations proved to work.
The iCHIP® microarray device developed by ImmunArray combined with the LDA classifier algorithm achieves a sensitivity of 94%, a specificity of 75%, and NPV of 93% across a diverse group of patients manifesting a wide spectrum of SLE disease activity index (SLEDAI scores ranging from 0–25) and representing diverse ethnic backgrounds. A sensitivity of 94% means that it is unlikely that the SLE-key® rule-out serologic test would falsely misdiagnose an SLE patient as not having SLE; a specificity of 75% for SLE means that not all patients with SLE would be designated positive for SLE by the test. This is acceptable because the aim of a rule-out test is to diagnose non-disease subjects. Obviously there is a trade-off between sensitivity and specificity; a test to positively rule-in SLE would be calibrated differently and would probably employ a different set of antigens on the chip.
The SLE-Key® was developed on females, not known to be pregnant, between 18–60 years of age subjects. A question that can be raised is whether test performance would be restricted to this population. However, preliminary results show that the test is not limited, and can be used also in a diverse SLE patient population including males, pregnant females, and without age limitation (data not shown).
The classifier antigens used in our rule-out test (Table 2) include both nucleic acids (complex ssDNA and an oligonucleotide) along with protein biomarkers. Additionally, both IgM and IgG autoantibody subtypes are critical to the test. Note that the different classification methods perform best with somewhat different sets of antigens. Five of the six antigen markers included in the SLE-Key® classifier are known to be associated with SLE. The sixth marker is a proprietary oligonucleotide derived from the nucleotide sequence of telomeres (Fattal et al., 2015). Anti-telomere antibodies were found in 60% of patients with SLE (Salonen et al., 2004). The prevalence of anti-telomere antibodies was much lower (5–30%), or rare (1/15%) in other autoimmune conditions; this finding invites further research on the telomere sequence in SLE. The iCHIP® microarray can thus alert us to factors in the pathophysiology of disease.
The dsDNA antibodies of SLE patients recognize antigenic epitopes involving the sugar-phosphate backbone of DNA, and react with different sources of DNA (Stollar et al., 1994; Salonen et al., 2004). However, a percentage of SLE patients (ranging from 2 to 30%) appear to lack these antibodies (Conti et al., 2015).
The performance of an assay panel comprised of cell-bound complement activation products alongside established markers, such as anti-dsDNA, ANA and anti-MCV, demonstrated 86% specificity and 80% sensitivity in distinguishing SLE from other autoimmune rheumatic disease (Kalunian et al., 2012; Putterman et al., 2014). The test proved 33% more specific than ANA-based tests and 48% more sensitive than the anti-dsDNA test. However, the iCHIP®, which uses a relatively small volume of serum and requires only a simple blood draw at the testing site followed by overnight shipping at ambient temperature, does not require undue processing either at the clinical site of the serum draw or lengthy, cell-based preparation at the testing site.
Beyond the rule-out test described in this paper, we intend to exploit our iCHIP® platform to obtain autoantibody profiles useful in monitoring SLE subtype, activity and progression, organ involvement and responsiveness to therapy (Gunther et al., 2003); indeed, the development of specific therapies for SLE would be aided by iCHIP® autoantibody profiling that could predict responsiveness.
5. Conclusion
The iCHIP® autoantibody detection platform combined with a multi-analyte-based algorithm is able to overcome the complexity and variability inherent in SLE to enable clear discrimination between affected and unaffected individuals. The SLE-key® rule-out test device achieves a sensitivity of 94% and a specificity of 75% and NPV of 93%.
Highlights.
We review the development, verification, and validation of the SLE-key® rule-out test for a definitive exclusion of a diagnosis of systemic lupus erythematosus;
The SLE-key® rule-out test was developed on the iCHIP® microarray platform, and is based on a multi-analyte autoantibody signature;
Analysis of the SLE-key® rule-out test is based on the LDA classifier algorithm, and achieves a sensitivity of 94%, a specificity of 75%, and negative predictive value of 93%.
Acknowledgments
The authors wish to acknowledge the invaluable contributions of Ornit Cohen-Gindi, Miriam Lerner, Naama Shefer, Ilana Gilkaite, Angela Turner, Tim Van Meter, Justin Pitts, Joseph Green and Nazanin Mirshahi.
The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n°115308 BIOVACSAFE, resources of which are composed of financial contributions from the European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies in kind contribution. The Hopkins Lupus Cohort is supported by NIH RO-1 AR43727.
Abbreviations
- SLE
systemic lupus erythematosus
- ANA
antinuclear antibodies
- QC
quality control
- FE
feature extraction
- EBV
Epstein Barr Virus
- CART
Classification and Regression Tree
- RF
Random Forest
- LR
Logistic Regression
- SVM
Support Vector machine
- QDA
Quadratic Discriminant Analysis
- LDA
Linear Discriminant Analysis
- ROC
receiver operating characteristic
- AUC
area under the curve
- PPV
Positive Predictive Value
- NPV
Negative Predictive Value
- ACR
American College of Rheumatology
- SLICC
Systemic Lupus International Collaborating Clinics
- SLEDAI
SLE disease activity index
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
These studies were funded by ImmunArray, to which the disclosures below are related.
Chaim Putterman – Scientific Advisory Board (SAB) member
Alan Wu – Consultant
Anat Reiner-Benaim - Consultant
D. Scott Batty – Employee, equity
Irun Cohen –Consultant, SAB member, Senior advisor and cofounder, equity
Ignacio Sanz- SAB member
Robert Gerwien – Consultant
Drs. Putterman, Sanz, Petri and Oats provided clinical samples for the study.
Keren Jakobi, Pennina Safer, Rachel Sorek, and Yakov Blumenstein are employees of ImmunArray.
References
- Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- Bertsias G, Cervera R, Boumpas DT. EULAR Textbook on Rheumatic Diseases 2012 [Google Scholar]
- Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME. The geoepidemiology of systemic lupus erythematosus. Autoimmun Rev. 2010;9:A277–87. doi: 10.1016/j.autrev.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Conti F, Ceccarelli F, Perricone C, Massaro L, Marocchi E, Miranda F, Spinelli FR, Truglia S, Alessandri C, Valesini G. Systemic Lupus Erythematosus with and without Anti-dsDNA Antibodies: Analysis from a Large Monocentric Cohort. Mediators Inflamm. 2015;2015:328078. doi: 10.1155/2015/328078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308–18. doi: 10.1191/0961203306lu2305xx. [DOI] [PubMed] [Google Scholar]
- Fattal I, Shental N, Ben-Dor S, Molad Y, Gabrielli A, Pokroy-Shapira E, Oren S, Livneh A, Langevitz P, Zandman-Goddard G, Sarig O, Margalit R, Gafter U, Domany E, Cohen IR. Guanine polynucleotides are self-antigens for human natural autoantibodies and are significantly reduced in the human genome. Immunology. 2015;146:401–10. doi: 10.1111/imm.12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattal I, Shental N, Mevorach D, Anaya JM, Livneh A, Langevitz P, Zandman-Goddard G, Pauzner R, Lerner M, Blank M, Hincapie ME, Gafter U, Naparstek Y, Shoenfeld Y, Domany E, Cohen IR. An antibody profile of systemic lupus erythematosus detected by antigen microarray. Immunology. 2010;130:337–43. doi: 10.1111/j.1365-2567.2010.03245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther EC, Stone DJ, Gerwien RW, Bento P, Heyes MP. Prediction of clinical drug efficacy by classification of drug-induced genomic expression profiles in vitro. Proc Natl Acad Sci U S A. 2003;100:9608–13. doi: 10.1073/pnas.1632587100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraki LT, Benseler SM, Tyrrell PN, Harvey E, Hebert D, Silverman ED. Ethnic differences in pediatric systemic lupus erythematosus. J Rheumatol. 2009;36:2539–46. doi: 10.3899/jrheum.081141. [DOI] [PubMed] [Google Scholar]
- Kalunian KC, Chatham WW, Massarotti EM, Reyes-Thomas J, Harris C, Furie RA, Chitkara P, Putterman C, Gross RL, Somers EC, Kirou KA, Ramsey-Goldman R, Hsieh C, Buyon JP, Dervieux T, Weinstein A. Measurement of cell-bound complement activation products enhances diagnostic performance in systemic lupus erythematosus. Arthritis Rheum. 2012;64:4040–7. doi: 10.1002/art.34669. [DOI] [PubMed] [Google Scholar]
- Kolberg JA, Jorgensen T, Gerwien RW, Hamren S, McKenna MP, Moler E, Rowe MW, Urdea MS, Xu XM, Hansen T, Pedersen O, Borch-Johnsen K. Development of a type 2 diabetes risk model from a panel of serum biomarkers from the Inter99 cohort. Diabetes Care. 2009;32:1207–12. doi: 10.2337/dc08-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lateef A, Petri M. Unmet medical needs in systemic lupus erythematosus. Arthritis Res Ther. 2012;14(Suppl 4):S4. doi: 10.1186/ar3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QZ, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, Mohan C, Wakeland EK, Olsen NJ. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen NJ, Li QZ, Quan J, Wang L, Mutwally A, Karp DR. Autoantibody profiling to follow evolution of lupus syndromes. Arthritis Res Ther. 2012;14:R174. doi: 10.1186/ar3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri M, Barr SG, Zonana-Nach A, Magder L. Measures of disease activity, damage, and health status: the Hopkins Lupus Cohort experience. J Rheumatol. 1999;26:502–3. [PubMed] [Google Scholar]
- Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O, Sturfelt G, Ramsey-Goldman R, Bae SC, Hanly JG, Sanchez-Guerrero J, Clarke A, Aranow C, Manzi S, Urowitz M, Gladman D, Kalunian K, Costner M, Werth VP, Zoma A, Bernatsky S, Ruiz-Irastorza G, Khamashta MA, Jacobsen S, Buyon JP, Maddison P, Dooley MA, van Vollenhoven RF, Ginzler E, Stoll T, Peschken C, Jorizzo JL, Callen JP, Lim SS, Fessler BJ, Inanc M, Kamen DL, Rahman A, Steinsson K, Franks AG, Jr, Sigler L, Hameed S, Fang H, Pham N, Brey R, Weisman MH, McGwin G, Jr, Magder LS. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–86. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons-Estel GJ, Alarcon GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2013;39:257–68. doi: 10.1016/j.semarthrit.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putterman C, Furie R, Ramsey-Goldman R, Askanase A, Buyon J, Kalunian K, Chatham WW, Massarotti E, Kirou K, Jordan N, Blanco I, Weinstein A, Chitkara P, Manzi S, Ahearn J, O’Malley T, Conklin J, Ibarra C, Barken D, Dervieux T. Cell-bound complement activation products in systemic lupus erythematosus: comparison with anti-double-stranded DNA and standard complement measurements. Lupus Sci Med. 2014;1:e000056. doi: 10.1136/lupus-2014-000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Casals M, Brito-Zeron P, Kostov B, Siso-Almirall A, Bosch X, Buss D, Trilla A, Stone JH, Khamashta MA, Shoenfeld Y. Google-driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun Rev. 2015;14:670–9. doi: 10.1016/j.autrev.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297:611–9. doi: 10.1001/jama.297.6.611. [DOI] [PubMed] [Google Scholar]
- Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, Fong D, Genovese MC, de Vegvar HE, Skriner K, Hirschberg DL, Morris RI, Muller S, Pruijn GJ, van Venrooij WJ, Smolen JS, Brown PO, Steinman L, Utz PJ. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- Salonen EM, Miettinen A, Walle TK, Koskenmies S, Kere J, Julkunen H. Anti-telomere antibodies in systemic lupus erythematosus (SLE): a comparison with five antinuclear antibody assays in 430 patients with SLE and other rheumatic diseases. Ann Rheum Dis. 2004;63:1250–4. doi: 10.1136/ard.2003.011890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. 2004;34:501–37. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Slater CA, Davis RB, Shmerling RH. Antinuclear antibody testing. A study of clinical utility. Arch Intern Med. 1996;156:1421–5. [PubMed] [Google Scholar]
- Stollar NE, Kim JK, Hollingsworth MJ. Ribosomes pause during the expression of the large ATP synthase gene cluster in spinach chloroplasts. Plant Physiol. 1994;105:1167–77. doi: 10.1104/pp.105.4.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Wallace DJ. Diagnosis and differential diagnosis of systemic lupus erythematosus in adults. 2015 UpToDate http://www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-systemic-lupus-erythematosus-in-adults.