

Abstract
Modern industrial agriculture depends on high-density cultivation of genetically similar crop plants, creating favorable conditions for the emergence of novel pathogens with increased fitness in managed compared with ecologically intact settings. Here, we present the genome sequence of six strains of the cucurbit bacterial wilt pathogen Erwinia tracheiphila (Enterobacteriaceae) isolated from infected squash plants in New York, Pennsylvania, Kentucky, and Michigan. These genomes exhibit a high proportion of recent horizontal gene acquisitions, invasion and remarkable amplification of mobile genetic elements, and pseudogenization of approximately 20% of the coding sequences. These genome attributes indicate that E. tracheiphila recently emerged as a host-restricted pathogen. Furthermore, chromosomal rearrangements associated with phage and transposable element proliferation contribute to substantial differences in gene content and genetic architecture between the six E. tracheiphila strains and other Erwinia species. Together, these data lead us to hypothesize that E. tracheiphila has undergone recent evolution through both genome decay (pseudogenization) and genome expansion (horizontal gene transfer and mobile element amplification). Despite evidence of dramatic genomic changes, the six strains are genetically monomorphic, suggesting a recent population bottleneck and emergence into E. tracheiphila’s current ecological niche.
Keywords: Cucurbita, Cucumis; Erwinia, mobile DNA; transposase; insertion sequence; pseudogene; host specialization; vector; monomorphic; phage; pumpkin; squash; cucumber
Introduction
Human populations have increased exponentially within the past 10,000 years, in part due to the domestication of few species of plants and animals whose numbers have similarly increased (Diamond 2002). This dramatic increase in population size and density of genetically similar plants, animals, and humans in turn create new ecological niches susceptible to invasion by novel microbial variants. Most of the pathogens that affect humans, and their domesticated plants and animals, have large amounts of mobile DNA in their genomes. This feature of recently emerged pathogens is hypothesized to permit large-scale changes in genetic architecture compared with progenitor strains. Despite the dramatic changes in gene content and function compared with close relatives, recently host-restricted pathogen populations generally show negligible levels of genetic diversity, suggesting a recent genetic bottleneck associated with emergence into a new host population ((McCann et al. 2013) and reviewed in Mira et al. (2006) and Achtman (2008, 2012).
Modern agriculture is characterized by extensive plantings of genetically similar crops in simplified agro-ecosystems, inevitably leading to high density plant populations that are susceptible to invasion by pathogens (Mira et al. 2006; Stukenbrock and McDonald 2008; Raffaele et al. 2010). As more intact ecological habitats are converted to simplified agro-ecosystems, crop losses caused by plant disease is an accelerating problem contributing to food insecurity and economic hardship. At least 10% of world crop production is estimated to be lost to pathogen infections (Strange and Scott 2005), and the geographic spread of newly emerged and highly virulent pathogens threatens cash crop and staple food production worldwide (reviewed in Anderson et al. 2004).
Several Erwinia spp. (Enterobacteriaceae) rank among the most economically important plant pathogens (Malnoy et al. 2012), and also represent potentially outstanding models for understanding the ecology of agricultural disease emergence. Erwinia tracheiphila is the causative agent of bacterial wilt disease of cucurbits (squashes, pumpkins, melons, and cucumbers). It is highly virulent, often causing death of the host plant within several weeks after the first onset of wilt symptoms. Although susceptible cucurbits are cultivated worldwide, E. tracheiphila is geographically restricted to Eastern North America. Despite causing millions of dollars in agricultural losses in this area, E. tracheiphila has received little research attention.
To gain insight toward how E. tracheiphila evolved to infect distinct plant hosts compared with other characterized Erwinia spp., we generated a reference genome sequence with PacBio long-read sequencing of one E. tracheiphila strain and Illumina short-read draft genomes of five additional strains isolated from cultivated squash varieties (Cucurbita pepo) in New York, Pennsylvania, Kentucky, and Michigan. Comparative genome analyses of these six E. tracheiphila strains reveal that E. tracheiphila is divergent from other sequenced Erwinia spp. and shows genomic characteristics indicative of a recent restriction to a novel host. Although the estimated genome size of E. tracheiphila (5.05 MB) is within the range of many free-living Enterobacteria (including other Erwinia spp.), extensive pseudogenization, low protein-coding gene density, proliferation of mobile genetic elements, and evidence of large-scale horizontal gene transfer events suggest that E. tracheiphila is experiencing the first stages of host specialization and genome reduction (Mira et al. 2001; Moran and Plague 2004; Gil et al. 2010). Taken together, these findings suggest that E. tracheiphila is undergoing rapid evolution coincident with its niche specialization as a host-restricted, obligately vector-transmitted phytopathogen.
Materials and Methods
The Study System
Cucurbita is a genus of plants native to the Americas that is characterized by the production of toxic tetracyclic triterpenes (cucurbitacins). These bitter compounds are effective herbivory deterrents to the vast majority of insect and mammalian herbivores, but wild and domesticated Cucurbita (squashes, pumpkins, and gourds) are susceptible to herbivory damage and disease exposure when fed upon by several closely related genera of diabroticite leaf beetles (Coleoptera: Chrysomelidae: Luperini) that have coevolved with Cucurbita host plants and are able to sequester and detoxify cucurbitacins (Ferguson and Metcalf 1985). Herbivory by leaf beetles can include exposure to E. tracheiphila, a relationship first recognized by Erwin F. Smith and his students (Rand 1920; Rand and Cash 1920; Rand and Enlows 1920; Smith 1920). Unlike other plant-associated Erwinia spp. that are well adapted for multiplication and survival on leaf surfaces, E. tracheiphila is an obligately vector-transmitted pathogen that replicates only in the xylem of susceptible host plants or the digestive tract of insect vectors (Shapiro et al. 2014). Beetles are attracted to the odors of infected plants, which are easier for them to eat compared with noninfected plants (Shapiro et al. 2012). Transmission to healthy plants can occur when frass from infected beetles falls onto sites of recent feeding damage (Rand and Enlows 1920) or onto the nectaries of flowers where the beetles aggregate (Sasu et al. 2010).
Cucurbita plants have nutritional and cultural significance for humans throughout their native range, and a wild gourd is thought to have been the first plant domesticated in the New World, more than 10,000 years ago (Smith 1997). Although susceptible cucurbit plants grow in wild and agricultural settings worldwide, the distribution of E. tracheiphila is limited to the Northeastern and Midwestern United States (Saalau Rojas et al. 2015). Most of the E. tracheiphila affected area is north of where undomesticated Cucurbita plants are found in wild populations, and far removed from the earliest evidence of Cucurbita domestication in Southern Mexico (Smith 1997). Despite the economic and cultural importance of Cucurbita plants, and the economic losses caused by herbivory and disease exposure, progress toward understanding this pathosystem has languished until recently (reviewed in Saalau Rojas et al. 2015).
Strains
All six strains were isolated from Cucurbita host plants (table 1). Strain BuffGH, previously called PSU-1, was isolated from a wilt-infected wild gourd (C. pepo ssp. texana) grown at the Larson Agricultural Research Station at Rock Springs, PA. Strains GZ4 and NYZuch1 were isolated from cultivated squash (C. pepo L.) in New York. Strain ppHow2 was isolated from C. pepo L. in Pennsylvania, strain BHKY was isolated from C. pepo in Kentucky, and strain MISpSq was isolated from Cucurbita moschata in Michigan (Saalau Rojas et al. 2013).
Table 1.
Overview of Assembly Metrics and Genome Characteristics of the Six Erwinia tracheiphila Strains Examined in This Study
| Strain | BuffGH | GZ4 | ppHow2 | NYZuch1 | BHKY | MISpSq |
|---|---|---|---|---|---|---|
| Plant of origin | Cucurbita pepo ssp. texana | Cucurbita pepo L. | Cucurbita pepo L. | Cucurbita pepo L. | Cucurbita pepo | Cucurbita moschata |
| State of origin | Pennsylvania | New York | Pennsylvania | New York | Kentucky | Michigan |
| Sequencing platform | PacBio RSII | Illumina HiSeq | Illumina HiSeq | Illumina HiSeq | Illumina HiSeq | Illumina HiSeq |
| N50 contig size, bp | 4,281,223 | 4,281,360 | 4,281,255 | 4,281,334 | 4,281,247 | 4,281,228 |
| N90 contig size, bp | 312,225 | 257,693 | 312,224 | 257,695 | 312,230 | 312,225 |
| Hybrid assembly contigs | NA | 99 | 90 | 104 | 7 | 139 |
| De novo assembly contigs | 7 | 1,407 | 1,140 | 1,330 | 3,604 | 3,334 |
| Max mapped contig size, bp | 4,281,223 | 4,281,360 | 4,281,255 | 4,281,334 | 4,281,247 | 4,281,228 |
| Combined size of all mapped contigs, bp | 5,015,962 | 5,108,917 | 5,075,853 | 5,131,136 | 5,016,029 | 5,053,035 |
| Coverage (×) | 94 | 60 | 63 | 45 | 57 | 25 |
| Pseudogenes | 939 | 936 | 936 | 936 | 936 | 935 |
| Intergenic SNPs compared with BuffGH | N/A | 56 | 27 | 53 | 7 | 10 |
| Total SNPs compared with BuffGH | N/A | 209 | 73 | 227 | 37 | 41 |
Isolate Culture Growth and DNA Extraction
Erwinia tracheiphila BuffGH was grown in liquid Difco nutrient broth for 4 days at 28 °C. Genomic DNA was isolated from the pellet using the CTAB extraction protocol (Wilson 1987), including Proteinase K and RNase treatments, as recommended for PacBio Genomic DNA extractions. Strains GZ4, ppHow2, and NYZuch1, MISpSq, and BHKY from Dr. Mark Gleason’s strain collection at Iowa State University were grown in nutrient agar peptone medium (de Mackiewicz et al. 1998) for 4 days at 25 °C. Genomic DNA was isolated using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI).
Genomic DNA Sequencing
The reference genome sequence of strain BuffGH was generated using SMRTbell Template Prep Kit (Pacific Biosciences, Menlo Park, CA) according to the PacBio standard protocol (“20 kb Template Preparation Using BluePippin Size-selection system” with steps for DNA damage and end repair, and ligation to hairpin adapters). After DNA size selection of fragments greater than 7 kb (BluePippin, Sae Science Inc, Beverly, MA), the average library size was 27 kb, based on analysis in a Fragment Analyzer (Advanced Analytical Technologies, Inc, Ames, IA). Three SMRT cells were run on a PacBio RS II instrument using P4-C2 chemistry combination.
Libraries of the genomic DNA from strains GZ4, ppHow2, and NYZuch1, BHKY, and MISpSq were generated using a Nextera DNA Sample Preparation Kit (Illumina, San Diego, CA). The libraries were amplified for eight cycles using the KAPA HiFi Library Amplification Kit (KAPA Biosystems, Wilmington, MA), and the size selection was performed using AMPure XP beads (Agencourt Bioscience Corp, Beverly, MA). Library concentrations were measured using a QuBit DNA Quantification Kit (Life Technologies, Carlsbad, CA) and the fragment size range detection (100–400 bp) was performed using the TapeStation 2200 (Agilent Technologies, Santa Clara, CA). Libraries were pooled using Nextera Index kits and 250-bp paired-end reads were generated with an Illumina HiSeq 2500 Sequencing System (table 2). Assembly metrics of all strains sequenced for this study were determined with GAEMR v. 1.0.1. (http://www.broadinstitute.org/software/gaemr/, last accessed June 6, 2015).
Table 2.
Genome Characteristics between Erwinia tracheiphila BuffGH, Erwinia amylovora CFBP 1430, and Erwinia billingiae Eb661
| E. tracheiphila Strain BuffGH | E. amylovora CFBP 1430a | E. billingiae Eb661a | |
|---|---|---|---|
| Size (bp) | 5,050,000 | 3,805,573 | 5,100,167 |
| G+C content (%) | 51.5 | 54.69 | 56.43 |
| CDS | 5,048 | 3,706 | 4,596 |
| Coding density (%) | 63 | 85.4 | 87.7 |
| Average CDS size (bp) | 830 | 879 | 966 |
| G+C content (%) | 51.6 | 53.6 | 56.4 |
| CDS with assigned function | 3,339 (66%) | 2,822 (76%) | 3,768 (82%) |
| Conserved uncharacterized CDS | 868 | 884 | 515 |
| Phage-related CDS | 1,316 | 777 | 519 |
| Pseudogenes | 939 | 115 | 82 |
| rRNA operons | 19 | 22 | 21 |
| tRNAs | 68 | 77 | 77 |
| Mobile elements | 792 | 10 | 8 |
Chromosomal features are based on the published genome sequences of E. amylovora CFBP1430 (Smits 2010) and E. billingiae Eb661 (Kube et al. 2010).
Genome Assembly
The Hierarchical Genome Assembly Process pipeline (Chin et al. 2013) was used to process reads produced by the PacBio sequencing for the E. tracheiphila BuffGH reference genome by trimming adaptor sequences, filtering for quality, correcting for errors, and then assembling the processed reads using a starting seed length of 10 kb. This resulted in 110,720 reads with a mean length of 6,560 bp and an N50 read length of 9,102 bp and a final assembly into seven contigs. We refer to this genome assembly as the reference genome throughout the manuscript.
Strains GZ4, ppHow2, NYZuch1, BHKY, and MISpSq were sequenced through Illumina to 25–63× coverage (table 1). Adaptor trimming and quality filtering of short read Illumina reads were performed using the FastX toolkit 0.0.13.2 (Pearson et al. 1997), SeqTK 1.0 (https://github.com/lh3/seqtk/, last accessed August 13, 2014), and FastQC 0.10.1 (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). Due to the large number of repetitive elements, de novo assembly of Illumina paired-end reads using Mira 4.1 (Chevreux et al. 1999) and Velvet 1.2.10 (Zerbino and Birney 2008) resulted in poor assembly metrics (> 1,000 contigs). To improve assembly, a hybrid approach was undertaken: Illumina paired-end reads were mapped to the E. tracheiphila strain BuffGH PacBio genome assembly using Mira 4.1 (Chevreux et al. 1999). To assemble strain-specific regions that may not be present in the reference genome, all unmapped reads were extracted and de novo assembled in Mira 4.1 and kept as separate contigs. The mapped and de novo assemblies from each strain were combined into a single FASTA file. All parameters for the hybrid mapping/de novo genome assembly are listed in the supplementary file S1, Supplementary Material online.
For variant calling, quality filtered reads were mapped to the reference sequence with Burrows–Wheeler Alignment (BWA) tool 0.7.4 (Li and Durbin 2009). A pileup was created with SAMTools 0.1.18 (Li et al. 2009), and variants were called with VCFtools 0.1.9 if the phred quality score of the variant site was greater than or equal to 60 (Danecek et al. 2011). Single nucleotide polymorphisms (SNPs) were not called if 1) within 9 bp (three codons) of each other, 2) in repeat regions with ten or more paralogous genomic copies, and 3) with less than 10× or more than 150× coverage, as short Illumina reads cannot be accurately placed over repetitive regions.
Data Accessions
Genome of the strain BuffGH is available in the NCBI's RefSeq database under accession NZ_JXNU00000000.1. Assembled scaffolds for the BuffGH strain are available in the NCBI's WGS database under the accession number JXNU00000000, BioProject PRJNA272881, experiment SRX956319/Et-BuffGH. The PacBio raw sequences used to assemble the BuffGH reference strain are available under run SRR1916166 of the NCBI's Sequence Read Archive (SRA) database. The raw Fastq files for the Illumina sequences of the GZ4, ppHow2, and NYZuch1, MISpSq, and BHKY strains are available under Experiment: SRX1471864/Erwinia tracheiphila Illumina shortreads, Run: SRR2982552/Accessions SRX1473634, SRX1473635, SRX1473636, SRX1473638, and SRX1473639. Hybrid assemblies are available via Data Dryad DOI 10.5061/dryad.v7h28.
Genome Annotation
The PacBio reference genome of E. tracheiphila was annotated using the Rapid Annotation using Subsystem Technology (RAST) 2.0 server (Aziz et al. 2008), Prokka 1.11 (Seemann 2014) and GenePRIMP 1.0 (Pati et al. 2010). Specifically, RAST and Prokka were used for ab initio gene predictions, whereas GenePRIMP was used to scan the intergenic regions of E. tracheiphila to identify pseudogenized coding sequences (CDS) that were too degraded to be detected through ab initio gene prediction. We discarded putative gene annotations that would encode proteins that 1) were < 50 amino acids long, 2) did not have detectable BLASTP matches in GenBank, or 3) resulted from open-reading frames (ORFs) that overlapped other coding regions. Protein motifs were identified using Pfam 28.0 (Finn 2014). Transmembrane domains, signal peptides, and rRNA genes were identified with Prokka 1.11 (Seemann 2014). Rapid Annotation Transfer Tool 1.0 (RATT) (Swain et al. 2012) was used to transfer annotations from the PacBio reference E. tracheiphila genome to the five Illumina-generated E. tracheiphila genomes. Prokka ab initio annotations were then used to predict CDS in the unannotated regions of the Illumina generated assemblies. All annotations and predictions were manually curated using Artemis 15.0.0 (Rutherford et al. 2000). The contig gene content was visualized using OmicCircos 1.8.0 (Hu et al. 2014) in R 3.2.2 (R 2015).
Pseudogene Annotation
Pseudogenes were defined as either a CDS that was truncated with ≤60% amino acid content of the closest functional homolog, or a CDS with at least one internal frame shift or premature stop codon. Each putative pseudogene region was manually inspected by aligning adjacent homologous fragments (defined as having the same BLASTP annotations) against the nearest GenBank homolog. All pseudogene fragments that aligned to the full-length functional protein sequence were merged into one pseudogene record.
Phylogenetic Reconstruction of Enterobacteriaceae Core Genome Phylogeny
In total, 436 protein-coding genes shared by all Enterobacteriaceae strains were identified through OrthoMCL 2.0 (Li et al. 2003), based on all-versus-all BLASTP 2.2.28 + searches with an E-value cutoff of 10−5. The genes were aligned with MAFFT 6.853 (Katoh et al. 2002), trimmed with trimAl 1.2 using the “automated1” option (Capella-Gutiérrez et al. 2009), and concatenated using publically available scripts available through https://github.com/tatumdmortimer/core-genome-alignment, last accessed September 8, 2014. The resulting alignment is available through Data Dryad doi:10.5061/dryad.v7h28. GTR (general time reversible) + CAT substitution model was selected in ProtTest 3.4 (Abascal et al. 2005) as the best-fitting substitution model. The maximum-likelihood phylogeny was reconstructed using RAxML 8.2.4 (Stamatakis 2006) as implemented on the CIPRES server (Miller et al. 2010), under the GTR + CAT model and with 100 bootstrap replicates. Bootstrapped pseudosamples were summarized with SumTrees 4.0.0 (Sukumaran and Holder 2010) and the resulting phylogeny was visualized in FigTree 1.4.2 (Rambaut 2008).
Comparative Analysis with Other Erwinia spp. Genomes
The two most thoroughly characterized Erwinia spp. are Erwinia amylovora, a clonal vascular plant pathogen that emerged in the Northeastern United States in the 1,700s and now threatens pome fruit production worldwide, and Erwinia billingiae, an antagonist of E. amylovora with potential for biocontrol (Kube et al. 2010a). Synteny between the closed chromosomes of E. amylovora (NC_013961.1), E. billingiae (NC_014306.1), and the largest E. tracheiphila contig was assessed in MUMmer 3.23 (Kurtz et al. 2004) to detect all maximal exact matches between the query sequences (no minimum match length was specified). OrthoMCL 2.0 (Li et al. 2003) was used to identify shared and unique gene content among these genomes as follows: All bidirectional BLASTP matches with E-value <10−5 were retained and clustered into gene families using the MCL algorithm. The resulting gene content was visualized in a Venn diagram using nml_parse_orthomcl.pl script (https://github.com/apetkau/orthomcl-pipeline/tree/master/scripts, last accessed October 22, 2014).
Detection of Horizontally Transferred Genes
A customized version of HGTector (Zhu et al. 2014; available through https://github.com/ecg-lab/hgtector) was used to identify putatively horizontally transferred genes in the E. tracheiphila genome. Homologs of each ORF were retrieved from a local copy of NCBI’s “nr” database (downloaded on November 21, 2014) using the “BLASTP” program from BLAST 2.2.28+ (Altschul et al. 1990). Only matches with the E-value <10−5 and sequence coverage ≥ 70% were retained. NCBI Taxonomy database (downloaded on November 21, 2014) was used to classify BLAST (Basic Local Alignment Search Tool) matches. Database matches corresponding to RefSeq entries were expanded according to the “MultispeciesAutonomousProtein2taxname” file from RefSeq release 68. This was necessary as many genes are combined into a single entry in RefSeq, which artificially decreased the representation of these genes in Close and Distal groups and confounded downstream analysis. After the expansion, only the 500 top-scoring matches were used as input for HGTector. The “Self” group was defined as TaxID 65700 (E. tracheiphila), the “Close” group was defined as the TaxIDs 551 (genus Erwinia) and 53335 (genus Pantoea), and the “Distal” group comprised the remaining organisms. The conservative cutoffs (the median between the zero peak and the first local minimum) of 7.78 and 69.7 hits were used for the “Close” and “Distal” groups, respectively. A gene was assigned as putatively transferred if its “Close” score was below the cutoff and its “Distal” score was above the cutoff.
For individual gene phylogenies, additional homologs were retrieved by BLASTP search of the nr database, and aligned with MAFFT 7.0 (Katoh et al. 2002). ProtTest 3.4 (Abascal et al. 2005) was used to identify the amino acid substitution model appropriate for each alignment. The phylogenetic trees were reconstructed under the appropriate substitution model using RAxML 7.7.5 with 100 bootstrap replicates and visualized in FigTree 1.4.2 (Rambaut 2008).
Assignment of Functional Categories to the Protein-Coding Genes
Each E. tracheiphila BuffGH gene was compared with the Clusters of Orthologous Groups (COG) database (2014 update; Galperin et al. 2014) using BLASTP 2.2.28+ (Altschul et al. 1990). Only the top-scoring match (per gene) with E-value <10−5 was kept. Each gene was assigned a COG category of the first functional category of the top-scoring match. Genes without the significant matches to any sequence in the COG database were not assigned a functional category. Out of 5,048 protein-coding genes in the reference E. tracheiphila genome, 3,957 were assigned to a functional category.
Identification of Mobile Genetic Elements and Phage Genes
Insertion sequence (IS) and transposable elements were detected and annotated using PfamScan 1.5 (Finn et al. 2014) and ISFinder (January 2015 update; Siguier et al. 2006). To identify putative phage gene homologs in Erwinia spp. genomes, protein-coding genes of E. tracheiphila, E. amylovora CFBP 1430, and E. billingiae Eb661 were used as queries in BLASTP search against RefSeq database (downloaded on October 14, 2014). Protein-coding genes with matches in the Viral RefSeq database downloaded October 14, 2014 (E-value < 10−10) were assigned as putatively viral in origin. Prophage regions were predicted using PHAST (accessed January 2015; Zhou et al. 2011).
Detecting Clustered Regularly Interspaced Short Palindromic Repeat Regions and Mapping Them onto Erwinia/Pantoea Core Genome Phylogeny
Several Pantoea spp. and all complete and draft Erwinia genomes available in GenBank were queried for clustered regularly interspaced short palindromic repeat (CRISPR) regions using CRISPRFinder (Grissa et al. 2007; database update 2014-08-05). Detected CRISPR regions were classified either as “intact” (those that have direct repeats with 100% sequence identity and many distinct spacers) or “questionable” (those that have few direct repeats or less than 100% sequence identity between putative direct repeats). The CRISPR regions and number of spacers per region were mapped onto Erwinia/Pantoea phylogeny reconstructed from 731 core genes. The core gene detection and phylogeny reconstruction were made using the pipeline described in the “Phylogenetic Reconstruction of Enterobacteriaceae Core Genome Phylogeny” section.
Results and Discussion
Genome Sequencing, Assembly, and Variant Calling
We obtained draft genomes for six E. tracheiphila strains (table 1). PacBio sequencing of E. tracheiphila BuffGH allowed us to assemble a high-quality draft genome with only seven contigs (fig. 1). The largest contig (4,281,223 bp, contig 2 in fig. 1) comprises the majority of the chromosome (table 1). Contig 1 (11,793 bp) is an intact enterobacterial phage that also assembles as part of contig 2. Contig 5 (23,682 bp) only contains Mu-like phage genes, and this region also assembles as part of contig 4. IS4 on phage contig 5 is also present in 39 copies on the chromosome. The presence of these two phage contigs that assemble both independently and as part of the chromosome indicate E. tracheiphila may be coinfected by two phages active in culture. Contig 6 (49,313 bp) contains several mobile elements and plasmid conjugation genes, and therefore is likely a plasmid (fig. 1). The Illumina sequencing of the other three strains resulted in 73–87 contigs from the mapping assemblies and >1,000 contigs from de novo assemblies due to proliferation of repetitive mobile DNA throughout the genomes (table 1 and discussion below). We designate the high-quality E. tracheiphila draft genome from strain BuffGH as the reference genome (Shapiro et al. 2015).
Fig. 1.—

The genome architecture of the reference strain E. tracheiphila BuffGH. Features mapped onto the main chromosome as concentric rings (from outer to inner) correspond to locations of 1) the contigs and unclosed gaps; 2) pseudogenes (brown); 3) the putative horizontal gene acquisitions (green); 4) predicted phage regions, further classified as either intact (dark blue) or incomplete/questionable (light blue), with two phages that also assemble as separate contigs shown in orange and yellow; 5) the SNP sites in the ppHow2 strain from PA; 6) GZ4 strain from NY; 7) NYZuch1 strain from NY; 8) BHKY strain from Kentucky; 9) MISpSq strain from Michigan; and 10) the mobile elements, with the homologous pairs connected by lines.
Despite the high number of phage genes and repetitive regions interfering with short-read assembly, the six strains in this study are monomorphic, with less than 300 variable sites between them (fig. 1, table 1, and supplementary table S1, Supplementary Material online). Midwestern strains BHKY and MISpSq have fewer than 50 variable sites compared with BuffGH, and strain ppHow2, which was also isolated from Pennsylvania, has only 73 SNPs compared with BuffGH (supplementary table S1, Supplementary Material online). The two New York isolates only have 43 variable sites between them, and have 209 and 227 variable sites compared with BuffGH (table 1, fig. 1, and supplementary table S1, Supplementary Material online). This level of between-strain variability is consistent with a very recent genetic bottleneck, likely associated with recent emergence into a novel ecological niche (Cai et al. 2011; McCann et al. 2013).
Phylogenetic Position of E. tracheiphila
The enterobacterial phylogenetic tree based on concatenated alignment of 436 orthologous gene families indicates that plant pathogenicity has arisen independently at least twice within Enterobacteriaceae: As a subgroup in the Pantoea/Erwinia clade and the “soft-rot” Dickeya/Pectobacterium clade (fig. 2A). The “soft-rot” clade is defined by the profuse production of pectinolytic enzymes for extensive plant cell wall degradation abilities, a trait that is absent in characterized Erwinia spp., which are commensal epiphytes or vascular pathogens. Erwinia spp. form a monophyletic group, with Erwinia toletana as a basal strain of the clade. Erwinia tracheiphila emerges within the Erwinia clade with strong bootstrap support. Notably, E. tracheiphila does not cluster with E. amylovora, the best characterized of the Erwinia plant pathogens that also first emerged in Eastern North America (fig. 2A).
Fig. 2.—

Phylogenetic and synteny comparison of E. tracheiphila with other Erwinia spp. and enterobacteria. (A) Phylogenetic position of the six E. tracheiphila strains among sequenced Enterobacteria. The maximum-likelihood tree was reconstructed from a concatenated alignment of 436 core enterobacterial genes and rooted with Haemophilus parahaemolyticus. The outgroup lineage was selected as the top-scoring nonenterobacterial match in a BLAST search with E. tracheiphila 16S rRNA genes as a query. Not all draft Erwinia genomes are included, as their incompleteness results in dramatic decrease of available core genes. Additional draft Erwinia genomes, however, are included in figure 4, in which E. tracheiphila is most closely related to the plant pathogen E. mallotivora, an uncharacterized papaya pathogen (Redzuan et al. 2014). Support values at the nodes correspond to results of 100 bootstrap replicates. (B) Chromosomal inversions and rearrangements among Erwinia spp. genomes. Chromosomes of closed Erwinia spp. genomes and contig 2 of E. tracheiphila BuffGH were compared pairwise. Each dot in the plots corresponds to homologous regions in two examined genomes, as identified in MUMmer 3.23 (Kurtz et al. 2004).
Genomic Rearrangements and Gene Content Comparisons
Whole-genome alignments between several plant-associated Erwinia spp. with closed genomes (both pathogenic and non-pathogenic) show few chromosomal rearrangements and highly conserved gene content (Kube et al. 2010b; Smits, Jaenicke, et al. 2010; Smits, Rezzonico, et al. 2010; and fig. 2B). In contrast, the E. tracheiphila chromosome has undergone extensive chromosomal rearrangements compared with the other characterized strains in the genus (fig. 2B).
The shared core genome of E. billingiae, E. amylovora, and E. tracheiphila consists of 2,149 gene clusters (fig. 3). Erwinia billingiae and E. amylovora share more orthologous gene clusters with each other (2,589 genes) than either E. billingiae or E. amylovora share with E. tracheiphila (2,468 and 2,297, respectively). All previously characterized Erwinia strains to date share broadly similar life histories as pathogenic or commensal associates of woody plants that have facultative, nonspecific interactions with insects. Erwinia tracheiphila has the smallest shared gene content in pairwise comparisons with congeneric strains. This is likely associated with E. tracheiphila’s niche of some susceptible Cucurbitaceae. Among the genes unique to E. tracheiphila, several likely affect plant host range and were putatively acquired through horizontal gene transfer (discussed below).
Fig. 3.—
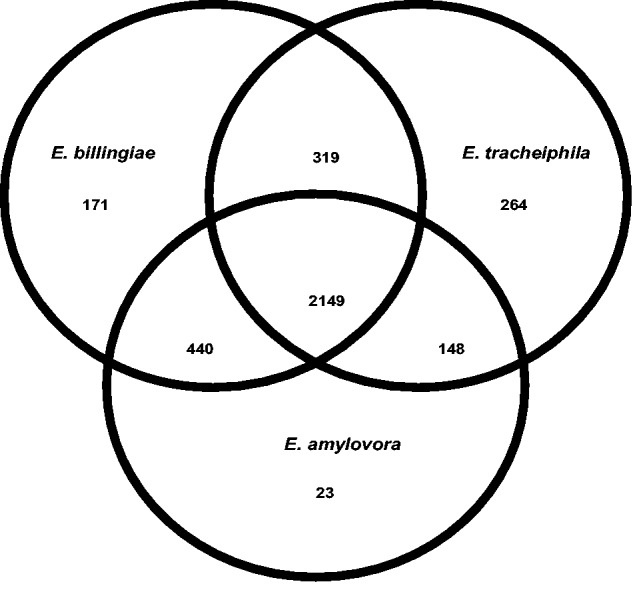
Gene content comparison of the genomes of the plant commensal E. billingiae Eb661, the highly host adapted plant pathogen E. amylovora CFBP 1430, and the cucurbit pathogen E. tracheiphila BuffGH. E. billingiae Eb661 and E. amylovora CFBP 1430 share more gene families with each other than either genome shares with E. tracheiphila BuffGH.
Absence of CRISPR Repeats in E. tracheiphila
To defend against the ubiquitous threat of invasion by bacteriophage and foreign plasmids, most known archaea (∼90%) and many bacteria (∼40%) employ heritable antiviral defenses. CRISPRs are direct repeats between 23 and 47 bp in length separated by short unique spacer regions of putative viral origin, and are used by a cell to detect and neutralize subsequent invasion attempts by the same virus (Horvath and Barrangou 2010). Most Erwinia spp. strains with draft or complete genomes deposited in GenBank have either intact or questionable CRISPR repeats (fig. 4). The exceptions are E. tracheiphila and E. typographi, an uncharacterized strain isolated from specialist bark beetles (Skrodenytė-Arbačiauskienė et al. 2012) (fig. 4). Absence of CRISPR repeats in E. tracheiphila suggests an inability to mitigate invasive DNA infection. This conjecture is consistent with the presence of two distinct complete phages in the current genome assembly, an indication of active phage infection. The E. tracheiphila chromosome contains an additional 37 putative prophage regions, of which 21 are intact, 7 are incomplete, and 9 are questionable (fig. 1). A total of 1,316 protein-coding genes on the four contigs of the E. tracheiphila chromosome (∼25% of the CDS) have viral homologs, compared with 777 genes in E. billingiae and 519 in E. amylovora. IS and transposable elements often gain entry to bacterial cells through phage infections, so the influx of phages containing transposable elements and ISs has likely contributed to the proliferation of mobile DNA genes throughout the E. tracheiphila genome (figs. 1 and 5).
Fig. 4.—

Distribution of CRISPR loci in Erwinia and Pantoea genomes. The distribution is mapped onto a maximum-likelihood tree reconstructed from a concatenated alignment of 731 core genes in all draft or complete Erwinia genomes and several Pantoea genomes. Each colored bar is either “intact” or “questionable” CRISPR region, and the number above each bar corresponds to the number of spacers at each CRISPR locus. Support values at the nodes correspond to results of 100 bootstrap replicates.
Fig. 5.—

Mobile element proliferation and decay in E. tracheiphila strain BuffGH. Mobile elements are grouped into categories by presence of conserved domains, as identified by PfamScan, and further clustered into larger families (brackets), as identified by ISFinder. Each mobile element is additionally classified as either intact (red) or pseudogenized (gray).
Genome Expansion through Transposable Element Proliferation
Erwinia billingiae contains only eight annotated IS elements and E. amylovora contains only ten. The E. tracheiphila genome, on the other hand, harbors at least 792 mobile DNA elements from 43 transposases or IS elements (fig. 5), comprising more than 15% of the CDS in the genome. Five mobile elements from families IS481, IS91, IS200, IS256, and IS1 make up almost half of the mobile element pool in the genome (fig. 5). Because mobile element transposition is often suppressed by the host cell, massive mobile element invasion and proliferation is indicative of an evolutionarily recent transition from a free living to a host-restricted lifestyle (Moran and Plague 2004). Mobile DNA is under strong negative selection in most bacterial genomes (Moran and Plague 2004), so the high copy number together with the relatively low rates of pseudogenization (∼30% for each mobile element; fig. 5) suggests their recent expansion within the E. tracheiphila genome.
Such high number of very similar sequences in mobile element repeats represents potential points for homologous recombination and may explain observed high level of chromosomal rearrangements in E. tracheiphila when compared with E. billingiae or E. amylovora (fig. 2B). At the population level, punctuated transposable element proliferations create mutations and genomic variation and are often directly linked to lineage divergence in both prokaryotic and eukaryotic organisms (Oliver and Greene 2009). Invasion and proliferation of mobile DNA can rapidly generate new genotypes that may be more fit in changing or novel environments, such as colonization of novel hosts (Iranzo et al. 2014).
Genome Decay through Pseudogenization in E. tracheiphila
Of 5,048 predicted CDS in the reference E. tracheiphila genome, 939 are putative pseudogenes, defined here as either 1) putatively encoding a protein that is truncated by at least 40% amino acid length relative to its top-scoring BLASTP match (281 genes) or 2) a CDS containing at least one internal frame shift or nonsense mutation (670 genes) (table 1, supplementary table S4, Supplementary Material online). This results in 68.9% coding density of E. tracheiphila genome. The five other E. tracheiphila genomes in this study also show high rates of pseudogenization (table 1). Erwinia amylovora and E. billingiae both have far fewer pseudogenes, when defined in the same manner, as well as much higher coding densities (table 2).
We cannot attribute such high number of pseudogenes in the E. tracheiphila genome to sequencing errors of PacBio SMRT sequencing technology. Although the error rates of base calls from the PacBio platform is high (∼11%), the errors are randomly distributed and not tied to any homopolymeric regions (Koren et al. 2012; Koren and Phillippy 2015). As the reference E. tracheiphila genome was sequenced to 94× coverage, the consensus sequence is expected to be ∼99.9999% accurate (Korlach 2014). Additionally, genomes of five other E. tracheiphila strains were sequenced using a much less error-prone Illumina technology but nevertheless show similar level of pseudogenization.
Decay in the six E. tracheiphila genomes in this study has occurred predominantly in the accessory genome, comprising genes involved in replication and repair (fig. 6, category L, which includes transposable element and phage genes), and “Mobilome” genes (fig. 6, category X). This pattern of decay is consistent with recent host restriction, where core metabolic genes are expected to be conserved, whereas accessory genes that may no longer be useful in a new niche are pseudogenized, and eventually lost. Only 5 of 939 E. tracheiphila pseudogenes are interrupted by mobile elements (Etr-0776, Etr-0838, Etr-0859, Etr-4570, and Etr-4167), so active mobile element transposition appears as a minor contributor to pseudogenization in E. tracheiphila.
Fig. 6.—

Distribution of E. tracheiphila protein-coding genes across functional categories. The categories are defined by a BLASTP comparison to the COG database and referred by a standard one-letter notation (see supplementary table S3, Supplementary Material online, header for descriptions). The categories are further grouped into functional supercategories. Distributions of specific subsets of all genes are plotted in distinct colors (see inset). Y-axis refers to the percent of genes within each specific gene subset. “Mobilome” supercategory is plotted on a different scale. The functional categories in a specific subset that have significantly different number from expected (i.e., observed for all genes in the genome) are shown with asterisks (Fisher’s exact test, P < 0.05; details are in supplementary table S3, Supplementary Material online). Category L includes genes known to be carried by the IS elements or phages, whereas category X mainly contains the accessory genes unique to E. tracheiphila.
Pseudogenization is observed most dramatically in pathogens that have recently emerged to colonize novel hosts, such as Mycobacterium leprae (Cole et al. 2001) and Bordetella pertussis (Parkhill 2003), or in recently host-restricted vertically transmitted insect symbionts (Andersson and Andersson 1999; Cole et al. 2001; Parkhill 2003; Toh et al. 2006; Holt et al. 2009; Burke 2011). Pseudogenes are assumed to be inactivated and transcriptionally/translationally silent, but transcripts from putatively inactivated genes are often detected in transcriptome data, and some pseudogenes are even translated into proteins with residual functions (Petty et al. 2011; Goodhead and Darby 2015). The metabolic costs of transcription of pseudogenes and/or the interference of transcribed products may have a negative effect on host fitness, resulting in negative selection for most pseudogenes (Kuo and Ochman 2010). The metabolic costs of pseudogenes may be similarly high for E. tracheiphila, particularly given its already slow growth in the low-nutrient environment of the xylem. Thus, the high proportion of pseudogenes in E. tracheiphila suggests relatively recent pseudogenization.
Genome Expansion through Horizontal Gene Acquisitions
Horizontal acquisition of genes provides an immediate source of genetic innovation and a mechanism for rapid niche adaptation and specialization (reviewed by Zhaxybayeva and Doolittle 2011). We predict 1,126 CDS in the E. tracheiphila reference genome as putatively transferred (supplementary fig. S1 and tables S2 and S3, Supplementary Material online).
Functions of Putatively Transferred Genes
Of the 1,126 putatively transferred genes, 613, or just below 50%, were not functionally characterized in COG database, as opposed to 898, or 18%, in the genome as a whole (Pearson’s Chi-squared test, X2 = 463, df = 1, P < 10−15; Fisher’s Exact Test, P < 10−15), suggesting that most transferred genes belong to the accessory genome and not core cellular processes. The distribution of 633 putatively transferred genes with an assigned COG functional category across functional categories was significantly different from that of all genes in the genome (Pearson’s Chi-squared test, X2 = 570, df = 24, P < 10−15; Fisher’s Exact Test, P < 10−7) (supplementary table S3, Supplementary Material online), suggesting either some bias in the function of transferred genes or some bias in the types of transferred genes that confer a fitness advantage and therefore are retained in a recipient genome.
Several functional categories were significantly depleted in the set of horizontally acquired genes (fig. 6 and supplementary table S3, Supplementary Material online). These functions included “translation, ribosomal structure, and biogenesis” (P = 1.4 × 10−9; a one-way Fisher’s Exact Test after multiple hypothesis correction), “amino acid transport and metabolism” (P = 4.9 × 10−9), “cell wall/membrane/envelope biogenesis” (P = 9.0 × 10−9), and “posttranslational modification, protein turnover, chaperones” (P = 6.1 × 10−7). These functions are generally critical to survival and thus are considered less prone to stable disruptions through horizontal gene transfer (Jain et al. 1999).
On the other hand, one functional category, “Mobilome” (X), is significantly enriched in transferred genes (Pearson’s Chi-squared test, X2 = 619.31, df = 1, P < 0.005; Fisher’s Exact Test, P = 0.005). As transposases and phage proteins are included in the “Mobilome” category, the overrepresentation of transferred genes in this category may simply indicate the proliferation of mobile DNA in the E. tracheiphila genome, rather than horizontal gene transfer of truly “cellular” genes.
A Pool of Pseudogenes Is Enriched with Putatively Transferred Genes
In total, 742 pseudogenes with an assigned COG functional category were differently distributed across the categories from genome as a whole (Pearson’s Chi-squared test, X2 = 500, df = 440, P value = 0.025; Fisher’s Exact Test, P < 10−15). In total, 283 (∼30%) of these pseudogenes are classified into functional categories with poorly characterized functions, a significant enrichment in comparison to the genome as a whole (Pearson’s Chi-squared test, x2 = 79, df = 1, P < 10−15). This indicates that accessory genes within the genome may be preferentially undergoing pseudogenization. In addition, genes associated with translation were significantly underrepresented in the set of pseudogenes (Pearson’s Chi-squared test, X2 = 23, df = 1, P = 10−6), reflecting the importance of the translational machinery to cellular functionality. Out of 1,126 putatively transferred genes, 338 were classified as pseudogenes, a 1.5-fold significant enrichment compared with the total of 939 pseudogenes out of 5,048 genes (Pearson’s Chi-squared test, X2 = 26, df = 1, P = 3.3 × 10−7). Significantly higher rates of pseudogenization among HGT candidates may indicate a purging of recently transferred genes that are not compatible with host genetic background or do not provide a fitness benefit to the host (Liu et al. 2004).
Type III Secretion System Pathogenicity Islands
The translocation of protein effectors into host cells through a type III secretion system (T3SS) is a virulence trait that often strongly influences a pathogen’s host specificity and virulence phenotype (Galán and Collmer 1999). Erwinia tracheiphila contains two distinct T3SS loci. One is an hrp (hypersensitive response and pathogenicity) T3SS locus utilized by phytopathogens that colonize intercellular spaces to translocate effectors across the plant cell membrane into the cytoplasm, where they can directly interact with host defense proteins. The hrp T3SS locus is composed of structural pilus genes, and is often flanked by secreted effectors. Erwinia tracheiphila shows strong conservation with E. amylovora and other enterobacterial plant pathogens (Oh et al. 2005) in the pathogenicity island encoding the structural components of the hrp T3SS secretion system, but exhibits differences in the genes for secreted effectors both in this locus and throughout the genome that may affect host specificity (table 3). Relative to E. amylovora, the E. tracheiphila T3SS pathogenicity island is missing the hrp-associated systemic virulence genes hsvABC, which encode toxins required for full virulence by E. amylovora (Oh et al. 2005), although hsvA and hsvC were found as pseudogenes adjacent to transposases elsewhere on the E. tracheiphila chromosome. The gene encoding the chaperone for the T3SS effector protein HrpW is present, but the HrpW effector gene itself is absent relative to the T3SS locus in E. amylovora.
Table 3.
T3SS Effectors and Chaperones Putatively Horizontally Acquired by Erwiniatracheiphila
| Locus Tag | Gene Integrity | Annotated Function | Taxonomic Assignment of Top-Scoring BLASTP Match | Adjacent Mobile Element |
|---|---|---|---|---|
| Etr-0398c | Intact | Type III effector HopV1 | Erwinia mallotivora | IS200/IS605 |
| Etr-0399c | Truncated | Type III chaperone ShcV | Erwinia mallotivora | IS200/IS605 |
| Etr-4629 | Pseudo | Type III chaperone ShcF | Pseudomonas syringae | Intact phage region |
| Etr-4630 | Intact | avrPpiA2 Avr protein | Pseudomonas syringae | Intact phage region |
| Etr-2937c | Intact | Type III chaperone ShcF | Erwinia mallotivora | Integrase catalytic unit |
| Etr-2936c | Two Frameshifts | Type III effector HopF2 | Erwinia mallotivora | IS481 |
| Etr-4372 | Intact | Type III chaperone protein ShcA | Erwinia mallotivora | IS200 |
| Etr-4373 | Intact | Type III effector HopA1 | Erwinia mallotivora | IS200 |
| Etr-1275 | Intact | Type III effector AvrB4-1 | Pseudomonas syringae | IS6, ISL3 |
| Etr-1646 | Truncated | Type III effector HopL | Baeuvaria bessiana | None adjacent |
| Etr-0681c | Intact | Type III effector HopE1 | Pseudomonas syringae | None adjacent |
| Etr-4310 | Intact | Type III effector/insecticidal toxin | Serratia fonticola | IS3 |
| Etr-0975c | Intact | Type III effector NleD | Citrobacter rodentium | ISL3 |
| Etr-2302 | Intact | Type III effector NleD | Citrobacter rodentium | ISL3 |
| Etr-2349 | Intact | Type III effector NleD | Citrobacter rodentium | ISL3 |
| Etr-3560c | Intact | Type III effector NleD | Citrobacter rodentium | ISL3 |
| Etr-4711c | Intact | Type III effector NleD | Citrobacter rodentium | ISL3 |
| Etr-5070c | Intact | Type III effector NleD | Citrobacter rodentium | ISL3 |
| Etr-4520 | Intact | type III effector protein XopAD | Pseudomonas savastonoi | IS200 |
| Etr-2547 | Frameshift | Type III AvrgF1 family effector | Xanthomonas vesicatoria | IS256 |
| Etr-4520 | Intact | Type III effector XopAD | Pseudomonas amygdali | IS605 |
| Etr-4120c | Frameshift | Type III effector XopA1 | Xanthomonas gardneri | IS91 |
Note.—In three cases, both a chaperone and an effector appear to have been horizontally transferred from the same donor strain. All listed genes have highly disjoint phylogenetic distributions (supplementary fig. S2, Supplementary Material online, and other data not shown) and most homologs are absent in other Erwinia spp.
The second T3SS is an Inv/Spa type, best characterized in enterobacterial pathogens such as Salmonella spp. as manipulating host cells in the gut mucosa to uptake enteroinvasive bacteria. The Inv/Spa locus has also been implicated in persistent insect colonization for Pantoea stewartii (Correa et al. 2012) and Sodalis glossinidius (Dale et al. 2001). Erwinia tracheiphila is thought to be confined to plant xylem and the extracellular lumen of the insect gut. The role of both T3SS loci is not clear as the xylem lacks a cell membrane and living cells and E. tracheiphila is thought to localize to the extracellular lumen of the insect digestive tract. Whether the hrp and Inv/Spa T3SS contribute to E. tracheiphila host specialization, colonization of plant xylem, or persistence in the insect vector digestive tract will be a primary focus for a future functional analysis.
Multiple putative T3SS effectors and effector chaperones that are encoded outside of these pathogenicity islands were flagged by HGTector as putatively horizontally acquired. These candidates share sequence similarity with T3SS effectors found in both plant and animal pathogens, including Pseudomonas, Xanthomonas, Citrobacter, Shigella, and Serratia spp. (table 3) (Pieretti et al. 2012). All of these genes are associated with mobile elements, supporting the horizontal acquisition hypothesis, although truncations and putatively deactivating frame shifts suggest that they may not all be functional. Three of these putative secreted effectors, HopA1, HopF2 and HopV1, are each encoded with a chaperone, have homologs in the recently sequenced and uncharacterized papaya pathogen Erwinia mallitovora, lack clear homologs in other enterobacterial plant pathogens, and are highly restricted in their distribution among plant pathogenic bacteria (supplementary fig. S2, Supplementary Material online). Such phylogenetic distribution of these effectors among few bacterial plant pathogens strongly suggests that these effectors are in the “mobilome” and were acquired horizontally by E. tracheiphila. Six copies of the T3SS effector NleD, all of which are adjacent to intact ISL3 transposase, are also putatively transferred. These homologs have 99% sequence identity to the NleD effector found in the rapidly evolving, unstable mouse pathogen Citrobacter rodentium (Petty et al. 2011). In enteric animal pathogens, NleD contributes to blocking bacterial flagella induced innate immune responses (Marchés et al. 2005; Baruch et al. 2011), which is a pathway functionally conserved between plants and animals (Haney et al. 2014).
Conclusions
Here we describe evolutionary insights from the multiple genome sequences of the cucurbit bacterial wilt pathogen, E. tracheiphila. The genome shows signs of incipient reductive evolution and host specialization through extensive pseudogenization, mobile element invasion and proliferation, and horizontal gene acquisitions. Together, these changes are consistent with an evolutionarily recent shift from a putatively free-living or broad host range lifestyle to a host-restricted lifestyle. The high sequence conservation between the strains suggests a recent genetic bottleneck, which may have coincided with emergence of E. tracheiphila as an obligately vector-transmitted plant pathogen with a highly restricted plant host range.
The emergence of human agriculture created dense concentrations of susceptible human, animal, and plant hosts, resulting in the rapid evolution and emergence of pathogens (Mira et al. 2006; Stukenbrock and McDonald 2008; Mennerat et al. 2010), and it seems likely that human cultivation of susceptible host plants contributed to a rapid adaptation of E. tracheiphila to its current ecological niche. Native wild squash varieties (C. pepo ssp. texana) are common from the Southern United States through Central America, and domesticated C. pepo varieties have been grown in Mesoamerica for more than 10,000 years (Smith 1997). However, bacterial wilt epidemics caused by E. tracheiphila are geographically restricted to Eastern North America, where the predominant insect vector (Acalymma vittatum, Coleoptera: Chrysomelidae: Luperini) is an abundant agricultural pest (Saalau Rojas et al. 2015). Agricultural cultivation of high-density cucurbit plantings in this geographic area provided millions of acres of genetically similar native plants (Cucurbita spp.), as well a novel susceptible plant host (Cucumis spp., cucumber and melons of Asian origin), while likely also supporting significantly higher population densities of beetle vectors than would be possible in intact ecological settings.
Changes in host and vector populations in novel agricultural settings, together with invasion of mobile DNA and horizontal acquisition of novel plant virulence genes, may have permitted E. tracheiphila to recently emerge as a host-restricted pathogen. As a result, E. tracheiphila appears to undergoing rapid genomic change and host adaptation, making the E. tracheiphila pathosystem an ideal model system for studying pathogen emergence. Obtaining more detailed knowledge of how agricultural policy and management practices create ecological conditions that may facilitate the emergence of virulent pathogens will be essential to creating more sustainable pathogen control strategies and should be a priority for future work.
Supplementary Material
Acknowledgments
Bioinformatic analyses were performed using computing resources available at the Research Computing Odyssey cluster at Harvard University (Cambridge, MA). The authors thank Miruna Sasu and Erika Salaau Rojas for the bacterial isolations; Bob Freeman and Aaron Kitzmiller for outstanding computational support; Qian Lui for DNA extractions; Andrew Murray for critical reading of the manuscript, and sequencing and library preparation advice; Olga Shechvenko at the Delaware Genotyping and Sequencing Center for PacBio library prep, sequencing, and assembly assistance; and Christian Daly and Jennifer Cougat at Harvard’s Bauer Core for Illumina library preparation advice and for running the Illumina HiSeq samples. Funding for this research was provided by Grants 2008-35302-04577 and 2009-33120-20093 from the United States Department of Agriculture to A.G.S., C.D.M., M.C.M.; National Science Foundation Graduate Research Fellowship to L.R.S.; National Science Foundation Post Doctoral Research Program in Biology (DBI-1202736) to L.R.S.; and in part by a Simons Investigator award from the Simons Foundation to O.Z.
Supplementary Material
Supplementary file S1, figures S1 and S2, and tables S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Literature Cited
- Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. [DOI] [PubMed] [Google Scholar]
- Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 62:53–70. [DOI] [PubMed] [Google Scholar]
- Achtman M. 2012. Insights from genomic comparisons of genetically monomorphic bacterial pathogens. Philos Trans R Soc Lond B Biol Sci. 367:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W., Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of molecular biology 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Anderson PK, et al. 2004. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol. 19:535–544. [DOI] [PubMed] [Google Scholar]
- Andersson JO, Andersson S. 1999. Genome degradation is an ongoing process in Rickettsia. Mol Biol Evol. 16:1178–1191. [DOI] [PubMed] [Google Scholar]
- Aziz R, et al. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, et al. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF‐κB. EMBO J. 30:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke GR, Moran NA. 2011. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol Evol. 3:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai R, et al. 2011. The plant pathogen Pseudomonas syringae pv. tomato is genetically monomorphic and under strong selection to evade tomato immunity. PLoS Pathog. 7:e1002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. 1999. Genome Sequence Assembly using trace signals and additional sequence information. Computer Science and Biology. Proceedings of the German Conference on Bioinformatics (GCB) 99, pp. 45–56, http://mira-assembler.sourceforge.net/docs/DefinitiveGuideToMIRA.html.
- Chin C-S, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 10:563–569. [DOI] [PubMed] [Google Scholar]
- Cole S, et al. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007–1011. [DOI] [PubMed] [Google Scholar]
- Correa VR, et al. 2012. The bacterium Pantoea stewartii uses two different type III secretion systems to colonize its plant host and insect vector. Appl Environ Microbiol. 78:6327–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale C, Young SA, Haydon DT. 2001. The insect endosymbiont Sodalis glossinidius utilizes a type III secretion system for cell invasion. Proc Natl Acad Sci U S A. 98:1883–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCF tools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mackiewicz D, Gildow FE, Blua M, Fleischer SJ, Lukezic FL. 1998. Herbaceous weeds are not ecologically important reservoirs of Erwinia tracheiphila. Plant Dis. 82:521–529. [DOI] [PubMed] [Google Scholar]
- Diamond J. 2002. Evolution, consequences and future of plant and animal domestication. Nature 418:700–707. [DOI] [PubMed] [Google Scholar]
- Ferguson JE, Metcalf RL. 1985. Cucurbitacins. J Chem Ecol. 11(3):311–318. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. 2014. The Pfam protein families database. Nucleic Acids Res. 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán JE, Collmer A. 1999. Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284:1322–1328. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Makarova KS, Wolf YI, Koonin EV. 2014. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43:D261–D269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, et al. 2010. Massive presence of insertion sequences in the genome of SOPE, the primary endosymbiont of the rice weevil Sitophilus oryzae. Int Microbiol. 11:41–48. [PubMed] [Google Scholar]
- Goodhead I, Darby AC. 2015. Taking the pseudo out of pseudogenes. Curr Opin Microbiol. 23:102–109. [DOI] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35(Suppl. 2):W52–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney CH, Urbach J, Ausubel FM. 2014. Innate immunity in plants and animals. Biochemist 36:1–5. [Google Scholar]
- Holt KE, et al. 2009. Pseudogene accumulation in the evolutionary histories of Salmonella enterica serovars Paratyphi A and Typhi. BMC Genomics 10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. [DOI] [PubMed] [Google Scholar]
- Hu Y, et al. 2014. OmicCircos: a simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Inf. 13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranzo J, Gómez MJ, de Saro FJL, Manrubia S. 2014. Large-scale genomic analysis suggests a neutral punctuated dynamics of transposable elements in bacterial genomes. PLoS Comput Biol. 10:e1003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. 1999. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci U S A. 96:3801–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Ki K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 30:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Phillippy AM. 2015. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol. 23:110–120. [DOI] [PubMed] [Google Scholar]
- Korlach J. 2014. Understanding accuracy in SMRT sequencing. In: Biosciences P, editor. Available from: http://www.pacificbiosciences.com/pdf/Perspective_UnderstandingAccuracySMRTSequencing.pdf.
- Kube M, et al. 2010. Genome comparison of the epiphytic bacteria Erwinia billingiae and E. tasmaniensis with the pear pathogen E. pyrifoliae. BMC Genomics 11(1):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C-H, Ochman H. 2010. The extinction dynamics of bacterial pseudogenes. PLoS Genet. 6(8):e1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Harrison PM, Kunin V, Gerstein M. 2004. Comprehensive analysis of pseudogenes in prokaryotes: widespread gene decay and failure of putative horizontally transferred genes. Genome Biol. 5:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnoy M, et al. 2012. Fire blight: applied genomic insights of the pathogen and host. Annu Rev Phytopathol. 50:475–494. [DOI] [PubMed] [Google Scholar]
- Marchés O, et al. 2005. Characterization of two non-locus of enterocyte effacement-encoded type III-translocated effectors, NleC and NleD, in attaching and effacing pathogens. Infect Immun. 73:8411–8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann HC, et al. 2013. Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog. 9:e1003503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerat A, Nilsen F, Ebert D, Skorping A. 2010. Intensive farming: evolutionary implications for parasites and pathogens. Evol Biol. 37:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE); 2010 Nov 14; New Orleans, LA. pp 1–8.
- Mira A, Ochman H, Moran NA. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17:589–596. [DOI] [PubMed] [Google Scholar]
- Mira A, Pushker R, Rodríguez-Valera F. 2006. The Neolithic revolution of bacterial genomes. Trends Microbiol. 14:200–206. [DOI] [PubMed] [Google Scholar]
- Moran NA, Plague GR. 2004. Genomic changes following host restriction in bacteria. Curr Opin Genet Dev. 14:627–633. [DOI] [PubMed] [Google Scholar]
- Oh CS, Kim JF, Beer SV. 2005. The Hrp pathogenicity island of Erwinia amylovora and identification of three novel genes required for systemic infection. Mol Plant Pathol. 6:125–138. [DOI] [PubMed] [Google Scholar]
- Oliver KR, Greene WK. 2009. Transposable elements: powerful facilitators of evolution. BioEssays 31:703–714. [DOI] [PubMed] [Google Scholar]
- Parkhill J. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 35:32. [DOI] [PubMed] [Google Scholar]
- Pati A, et al. 2010. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 7:455–457. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Wood T, Zhang Z, Miller W. 1997. Comparison of DNA sequences with protein sequences. Genomics 46:24–36. [DOI] [PubMed] [Google Scholar]
- Petty NK, et al. 2011. Citrobacter rodentium is an unstable pathogen showing evidence of significant genomic flux. PLoS Pathog. 7:e1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieretti I, et al. 2012. Genomic insights into strategies used by Xanthomonas albilineans with its reduced artillery to spread within sugarcane xylem vessels. BMC Genomics 13:658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. (2015). R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. Retrieved from https://www.R-project.org/.
- Raffaele S, et al. 2010. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330:1540–1543. [DOI] [PubMed] [Google Scholar]
- Rambaut A. 2008. FigTree, A graphical viewer of phylogenetic trees and as a program for producing publication-ready figures. Available from: http://tree.bio.ed.ac.uk/software/figtree/
- Rand FV, Cash LC. 1920. Some insect relations of Bacillus tracheiphilus Erw. Sm. Phytopathology 10:133–140. [Google Scholar]
- Rand FV., Enlows EMA. Bacterial wilt of cucurbits. Vol. 828. US Department of Agriculture, 1920.
- Redzuan RA, et al. 2014. Draft genome sequence of Erwinia mallotivora BT-MARDI, causative agent of papaya dieback disease. Genome Announc. 2:e00375–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944. [DOI] [PubMed] [Google Scholar]
- Saalau Rojas E, et al. 2013. Genetic and virulence variability among Erwinia tracheiphila strains recovered from different cucurbit hosts. Phytopathology 103:900–905. [DOI] [PubMed] [Google Scholar]
- Saalau Rojas E, et al. 2015Bacterial wilt of cucurbits: resurrecting a classic pathosystem. Plant Dis. 99:564–574. [DOI] [PubMed] [Google Scholar]
- Sasu M, Seidl-Adams I, Wall K, Winsor J, Stephenson A. 2010. Floral transmission of Erwinia tracheiphila by cucumber beetles in a wild Cucurbita pepo. Environ Entomol. 39:140–148. [DOI] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- Shapiro LR, et al. 2015. Draft genome sequence of Erwinia tracheiphila, an economically important bacterial pathogen of cucurbits. Genome Announc. 3:e00482–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, Moraes CM, Stephenson AG, Mescher MC. 2012. Pathogen effects on vegetative and floral odours mediate vector attraction and host exposure in a complex pathosystem. Ecol Lett. 15:1430–1438. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Seidl-Adams I, De Moraes C, Stephenson A, Mescher M. 2014. Dynamics of short-and long-term association between a bacterial plant pathogen and its arthropod vector. Sci Rep. 4, doi:10.1038/srep04155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34:D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrodenytė-Arbačiauskienė V, Radžiutė S, Stunžėnas V, Būda V. 2012. Erwinia typographi sp. nov., isolated from bark beetle (Ips typographus) gut. Int J Syst Evol Microbiol. 62:942–948. [DOI] [PubMed] [Google Scholar]
- Smith BD. 1997. The initial domestication of Cucurbita pepo in the Americas 10,000 years ago. Science 276:932–934. [Google Scholar]
- Smith EF. 1920. An introduction to bacterial diseases of plants. Philadelphia (PA: ): W.B. Saunders Company. [Google Scholar]
- Smits THM, Jaenicke S, et al. 2010. Complete genome sequence of the fire blight pathogen Erwinia pyrifoliae DSM 12163T and comparative genomic insights into plant pathogenicity. BMC Genomics 11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits THM, et al. 2010. Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol Plant Microbe Interact. 23:384–393. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Strange RN, Scott PR. 2005. Plant disease: a threat to global food security. Annu Rev Phytopathol. 43:83–116. [DOI] [PubMed] [Google Scholar]
- Stukenbrock EH, McDonald BA. 2008. The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol. 46:75–100. [DOI] [PubMed] [Google Scholar]
- Sukumaran J, Holder MT. 2010. DendroPy: a python library for phylogenetic computing. Bioinformatics 26:1569–1571. [DOI] [PubMed] [Google Scholar]
- Swain MT, et al. 2012. A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nat Protoc. 7:1260–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh H, et al. 2006. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 16:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K. 1987. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol. 2.4.1–2.4.5. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O, Doolittle WF. 2011. Lateral gene transfer. Curr Biol. 21:R242–R246. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39(Web Server issue):W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.