

Abstract
Genetic studies of Drosophila melanogaster have provided a paradigm for RNA interference (RNAi) in arthropods, in which the microRNA and antiviral pathways are each mediated by a single Argonaute (Ago1 and Ago2) and germline suppression of transposable elements is mediated by a trio of Piwi-subfamily Argonaute proteins (Ago3, Aub, and Piwi). Without a suitable evolutionary context, deviations from this can be interpreted as derived or idiosyncratic. Here we analyze the evolution of Argonaute genes across the genomes and transcriptomes of 86 Dipteran species, showing that variation in copy number can occur rapidly, and that there is constant flux in some RNAi mechanisms. The lability of the RNAi pathways is illustrated by the divergence of Aub and Piwi (182–156 Ma), independent origins of multiple Piwi-family genes in Aedes mosquitoes (less than 25Ma), and the recent duplications of Ago2 and Ago3 in the tsetse fly Glossina morsitans. In each case the tissue specificity of these genes has altered, suggesting functional divergence or innovation, and consistent with the action of dynamic selection pressures across the Argonaute gene family. We find there are large differences in evolutionary rates and gene turnover between pathways, and that paralogs of Ago2, Ago3, and Piwi/Aub show contrasting rates of evolution after duplication. This suggests that Argonautes undergo frequent evolutionary expansions that facilitate functional divergence.
Keywords: RNAi, Argonaute, Piwi, gene duplication, Diptera
Introduction
Argonaute genes of the Ago and Piwi subfamilies mediate a broad range of processes from development to antiviral immunity, and are found in almost all eukaryotes (Cerutti and Casas-Mollano 2006). They constitute an ancient gene family that was present in the common ancestor of extant prokaryotes and eukaryotes (reviewed in Swarts et al. 2014), and which diverged into Ago and Piwi subfamilies early in eukaryotic evolution (Cerutti and Casas-Mollano 2006; Mukherjee et al. 2013). The Argonautes are effectors in the RNA interference (RNAi)-related pathways, which can be broadly defined as a system of nucleic acid manipulation through complementary base pairing between small RNA (sRNA) guides and long nucleic acid targets. Each sRNA is loaded into an Argonaute protein, which it guides to a target nucleic acid, resulting in cleavage or translational inhibition of the target (reviewed in Sarkies and Miska 2014). Three broad classes of sRNA can be defined based on their sizes and interactors (reviewed in Kim et al. 2009): short interfering RNAs (siRNAs) are ∼21–24 nt long and are produced from viruses, transposable elements (TEs), and some long double-stranded RNA (dsRNA) products in the soma; microRNAs (miRNAs) are generally ∼22–23 nt long and are derived from host-encoded hairpin loops; and Piwi-interacting RNAs (piRNAs) are 24–29 nt long, derived largely from intergenic repetitive elements (e.g., TEs) in the germline, and exclusively bind Piwi-subfamily Argonaute proteins.
RNAi is well studied in Arabidopsis thaliana, where the Argonaute gene was first identified (Bohmert et al. 1998), and in the nematode Caenorhabditis elegans, where the RNAi mechanism was first characterized (Fire et al. 1998). Subsequent studies have reported Argonautes with diverse functions and differences in copy number across different eukaryotic clades (Mukherjee et al. 2013), illustrating that RNAi pathways have a dynamic evolutionary history. For example, in plants RNAi-mediated suppression of TEs is directed by shorter sRNAs than in animals, and is mediated by Agos not Piwis (which they lack completely; Cerutti and Casas-Mollano 2006; reviewed in Parent et al. 2012). Differences in Argonaute copy number and function are also found in the animals. In the protostomes, the planarian Schmidtea mediterranea has nine Piwi homologs (Palakodeti et al. 2008), two of which (smedwi-2 and smedwi-3) play vital roles in regeneration by facilitating the differentiation of pluripotent neoblasts (Reddien et al. 2005; Palakodeti et al. 2008). In contrast, Piwi and their associated piRNAs have been lost independently in several lineages of nematodes, with TE suppression carried out instead by DNA methylation mediated by RNA-dependent RNA polymerase and Dicer (Sarkies et al. 2015). Interestingly, this loss of Piwi has been accompanied by a massive expansion of other Argonaute genes in nematodes, with Ca. elegans encoding 25 Argonautes, 18 of which fall into the divergent worm-specific Ago (WAGO) clade: These associate with a novel class of sRNA (22G-RNAs) and carry out derived functions such as epigenetic memory formation (reviewed in Buck and Blaxter 2013).
Recent genome sequences and experimental data from isolated taxa have also revealed numerous arthropods with duplicates of Argonautes, some of which have novel and divergent functions. For example, the tick Ixodes scapularis has three Ago2 paralogs, only two of which appear to function in antiviral defense (Schnettler et al. 2014). Larger expansions are seen in the aphid Acyrthosiphon pisum, which has two paralogs of Ago3 and eight paralogs of Piwi, some of which are expressed in the soma (in contrast to Drosophila melanogaster, where they are predominantly germline specific; Lu et al. 2011). Additionally, these Piwi paralogs are differentially expressed in aphid reproductive morphs, suggesting that they may have specialized to function in different reproductive strategies (Lu et al. 2011).
Despite this diversity, much of our functional understanding of arthropod Argonautes comes from studies of D. melanogaster (Kataoka et al. 2001; Li et al. 2002; Pal-Bhadra et al. 2004; Vagin et al. 2004; Kalmykova et al. 2005; van Rij et al. 2006; Chung et al. 2008; Czech et al. 2008), which has two Ago-subfamily genes. Ago1 binds miRNAs and regulates gene expression by inhibiting translation of host transcripts (reviewed in Eulalio et al. 2008). Ago2 binds siRNAs from two sources: first, virus-derived small interfering RNAs, which guide Ago2 to cleave viruses or their transcripts, forming an integral part of the antiviral defense mechanism (Li et al. 2002; van Rij et al. 2006); second, endogenous siRNAs, which are derived from TEs, overlapping untranslated regions (UTRs) and other repetitive sequences in the soma (Chung et al. 2008; Czech et al. 2008). Drosophila melanogaster also encodes three Piwi-subfamily proteins, which bind piRNAs in the germline and surrounding tissues: Ago3, Aubergine (Aub), and Piwi (reviewed in Iwasaki et al. 2015). The piRNAs are differentiated from miRNAs and siRNAs in D. melanogaster by their Dicer-independent production and their amplification through the “Ping-Pong” pathway, a positive feedback loop involving Ago3 and Aub (Li et al. 2009). In D. melanogaster, piRNAs guide Piwi to TEs in euchromatin, where it inhibits transposition (Kalmykova et al. 2005) by directing the formation of heterochromatin (Sienski et al. 2012).
However, comprehensive analysis of Argonaute evolution at a eukaryotic, or even metazoan, scale is hindered by limited taxon sampling, wide variation in evolutionary rate, and the presence of ancient and recent duplications and losses (discussed by Philippe et al. 2011). The Diptera provide an opportunity to study Argonaute evolution in an order that is densely sampled and less divergent, but still shows variation in Argonaute copy number and function. Previous reports of Argonaute duplication in the Diptera have been limited to isolated taxa, such as the house fly Musca domestica (Scott et al. 2014), Drosophila pseudoobscura (Hain et al. 2010), and three mosquito species (Campbell et al. 2008). These mosquito duplicates appear to have evolved derived functions: Several Piwi paralogs in Aedes aegypti (Vodovar et al. 2012; Schnettler et al. 2013) and Aedes albopictus (Morazzani et al. 2012) are expressed in the soma, and at least one of the somatically expressed Piwi duplicates in Ae. aegypti appears to have functionally diverged to a novel antiviral function (Schnettler et al. 2013).
Gene duplications, such as those that gave rise to the diversity of eukaryotic RNAi pathways, are often associated with changes in evolutionary rate (reviewed in Hahn 2009), and Drosophila duplicates that evolve a new function often evolve more rapidly (Assis and Bachtrog 2013). However, the subsequent duration of this rate change can vary considerably, either changing only briefly following duplication (Nielsen et al. 2010), or persisting in all branches subtending the duplication event (Morandin et al. 2014). Additionally, rate change after duplication can be symmetrical or asymmetrical between the resulting paralogs: If both paralogs specialize to different pre-existing functions (subfunctionalization) they are expected to have roughly symmetrical evolutionary rates, whereas if one paralog undergoes neofunctionalization it is expected to evolve more rapidly than the other paralog, resulting in asymmetrical rates (Hittinger and Carroll 2007). Such a difference is seen after duplication of desaturase genes in Drosophila, which play key roles in evolutionary divergence and speciation through their contribution to the formation of cuticular hydrocarbons (Keays et al. 2011). These characteristic patterns of selection following duplication therefore enable us to use analyses of evolutionary rate to gain an insight into functional evolution.
Here we take advantage of the diversity available in the sequenced genomes and transcriptomes of Diptera to analyze patterns of Argonaute duplication and sequence evolution across 86 species. Contrasting rates of duplication and evolution are commonly associated with differences in function and selection pressure. We find a higher rate of protein evolution in Ago2 and Ago3, a higher rate of gene turnover in Ago2 and Piwi/Aub, and we estimate the date of the duplication that led to the separate Piwi and Aub subclades. We also find that paralogs of Ago2, Ago3, and Piwi/Aub evolve more rapidly after duplication, indicating potential divergence into novel and strongly selected functions.
Materials and Methods
Identification of Argonaute Homologs
We used TBLASTX and TBLASTN (Altschul et al. 1997) to identify Argonaute homologs in the genomes and transcriptomes of 86 Dipteran species found in GenBank, Flybase, Vectorbase, Diptex, the NCBI Transcriptome Shotgun Assembly, or other unpublished transcriptomes (see Supplementary Materials for a detailed list of sources; novel sequences have been submitted to GenBank as KR012647–KR012696). For each species, we used Argonautes from the closest well-annotated relative as queries, or D. melanogaster if no homolog from a close relative was available. Where BLAST returned multiple partial hits, we assigned hits to the correct query sequence by aligning all hits from the target species to all Argonautes from the query species, and inferring a neighbor-joining tree. For each query sequence, partial BLAST hits were then manually curated into complete genes using Geneious v5.6.2 (http://www.geneious.com/, last accessed April 15, 2012; Kearse et al. 2012). For some species of Drosophila, polymerase chain reaction (PCR) and Sanger sequencing was used as no transcriptomic or genomic data were available (see Supplementary Materials for details of genes).
Phylogenetic Analysis of Dipteran Argonautes
We initially assigned homologs into subclades (Ago1, Ago2, Ago3, and Piwi/Aub) based on a Bayesian gene tree rooted between the Ago and Piwi subfamilies, with ambiguous alignment positions removed using Gblocks (Castresana 2000) and with the wasp Nasonia vitripennis as the outgroup for each subclade. We repeated this analysis with three other arthropod species as outgroup (Bemisia tabaci, Bombyx mori, and Tribolium castaneum), and found that in each case the same Dipteran genes were classified into the Ago1, Ago2, Ago3, and Piwi/Aub subclades. To minimize the loss of information when removing ambiguous positions, we reinferred separate Bayesian gene trees for each subclade with no outgroup, using new alignments with ambiguous positions identified by eye and removed (see Supplementary Materials for alignments). Sequences were aligned using translational MAFFT (Katoh et al. 2002) with default parameters. All phylogenies were inferred using the Bayesian approach implemented in MrBayes v3.2.1 (Ronquist and Huelsenbeck 2003) under a nucleotide model, assuming a general time reversible (GTR) substitution model with three unlinked codon-position classes, gamma-distributed rate variation between sites with no invariant sites, and inferred base frequencies. We ran each analysis for a minimum of 50 million steps, or as long as necessary for the tree topologies to reach stationarity (standard deviation of split frequencies between duplicate independent runs <0.01; potential scale reduction factor (PSRF)∼1 and effective sample size (ESS) > 1,000 for all parameters). Samples from the posterior were recorded every 10,000 steps, and a maximum clade credibility tree was inferred from 2 duplicate runs using TreeAnnotator (Drummond et al. 2012).
Gene Turnover Rates
To quantify the rate of gene duplication and loss during Argonaute evolution, we estimated the rate of gene turnover (λ, the number of gains or losses per million years) for each Argonaute subclade using CAFE v3.1 (Han et al. 2013). We also tested whether subclades differed significantly in their rates of gene turnover by using 1,000 replicates of CAFE’s Monte Carlo resampling procedure. This generates an expected distribution of gene family sizes under a birth–death model, conditioned on the species topology and a set λ value (which we fixed at the value estimated for each subclade), thus providing an estimate of the P value for each of the other subclades. To mitigate potential bias introduced by incomplete genome assemblies, turnover analyses only included species that had at least one gene in each subclade (66 of total 86 species). To assess the potential impact of searching transcriptomes, which will only detect expressed genes (and may therefore lead to erroneous inference of gene loss and falsely inflate the rate of gene turnover), we repeated these analyses with rates of gene gain and loss estimated separately. We find similar results when comparing rates of gene gain and gene turnover, suggesting that missing data have a negligible effect on our estimates of gene turnover rate.
To provide the independent species-level tree topology for all 66 species that is required for this analysis, we manually combined the high-confidence multigene phylogenies presented in Wiegmann et al. (2011) and Misof et al. (2014). Where these reference trees lacked the relevant taxa (e.g., relationships below the level of family), we either referred to other published multigene phylogenies—van der Linde et al. (2010), Zhang et al. (2010), and Dyer et al. (2008) for Drosophilidae, Bactrocera, and Glossina, respectively—or inferred a Bayesian phylogeny using the arginine kinase gene (Culicidae, MrBayes parameters as above). Conditional on this species topology, we estimated relative branch lengths using BEAST v1.7 (Drummond et al. 2012) and a translational MAFFT alignment of the 1:1:1 ortholog Ago1, constraining the dates of key nodes to previously inferred dates derived from fossil evidence (as used by Wiegmann et al. 2011: Root = 245 Ma, Brachycera = 200 Ma, Cyclorrhapha = 150 Ma, Schizophora = 70 Ma). As our primary concern is the difference in relative rates of gene gain and loss for the different subclades, inaccuracies of the absolute timescale will have minimal impact on our conclusions.
Evolutionary Rate and Positively Selected Residues
To infer the relative rates of synonymous and nonsynonymous substitution (dN/dS = ω) averaged across all sites, we used codeml (PAML; Yang 1997) to fit model M0 (single ω) separately for each subclade (Ago1, Ago2, Ago3, and Piwi/Aub), conditional on the alignment and tree topology. To test for significant differences between these subclade-specific rates, we fixed ω for each subclade at the value estimated for each of the other subclades, and used Akaike weights to compare the likelihood of these fixed ω values with the likelihood of the ω value estimated from the data for that subclade.
To estimate the change in evolutionary rate after duplication, and to test whether duplicates experienced a transient or sustained change in evolutionary rate, we fitted two variants of the M0 model, each with two separate ω parameters estimated for different branches of the gene tree (fig. 1). To test for a transient change in evolutionary rate directly after duplication, we fitted a model (which we term “Immediate”) that specified one ω for branches immediately after a duplication event, and another ω for all other branches. To test for a sustained change in evolutionary rate following duplication, we fitted a second model (which we term “All descendants”) that specified one ω for all branches arising from a duplication event, and another ω for all other branches. For each subclade, Akaike weights were used to estimate the relative support for the M0, Immediate, and All descendants models.
Fig. 1.—
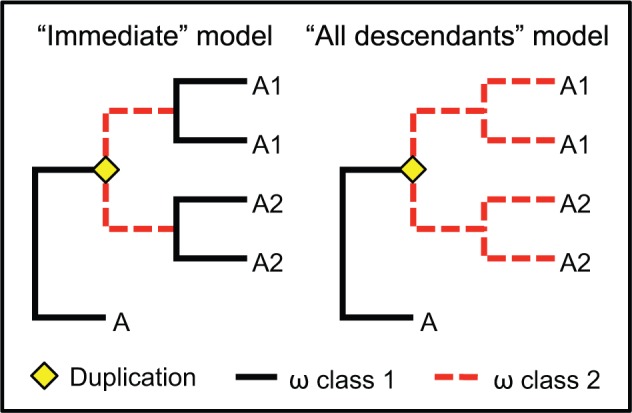
The two models fitted to branches after duplication events. Immediate models the expectation if selection pressures change only briefly after duplication, whereas All descendants models the expectation if paralogs evolve at a consistently different rate.
To test for asymmetrical evolutionary rates after a particular duplication event, we fitted two variants of the M0 model. The first model (which we term “Asymmetrical rates”) estimated three separate ω parameters for different branches of the gene tree: One ω for the branches in one lineage produced by the duplication event; a second ω for the branches in the other lineage; and a third ω for the rest of the tree. The second model (which we term “Symmetrical rates”) estimated two ω ratios: One ω for both lineages arising from the duplication event, and a second ω for the rest of the tree. The large number of nested duplication events means that an exhaustive test of all cases in which some duplication events result in asymmetrical rates is intractable; we therefore focused on ten key duplication events in the Ago2 and Piwi/Aub subclades (supplementary fig. S1, Supplementary Material online). For each duplication event, we used a likelihood ratio test (LRT) to compare the fit of the Asymmetrical and Symmetrical rates models. Following the method outlined above, we fitted Immediate and All descendants versions of each of these models.
To test for positively selected residues in each subclade, we used LRTs to compare the fit of two models, each with two site classes. In both models, ω of the first “constrained” site class was a discretized beta-distribution with eight classes. The models differ in that in the first model (the null model “M8a” in codeml) ω of the second “positively selected” site class is fixed at 1 (neutrality), while in the second model (the “M8” model) ω of the second site class is constrained to exceed 1. If the LRT indicated a significantly better fit for M8 than M8a given the parameters in the model, individual residues were classed as positively selected if they had a Bayes Empirical Bayes (BEB) posterior probability of >95% that ω > 1.
To assess the potential impact of false positives introduced by misalignments (Jordan and Goldman 2012), we ran M0 and M8 codeml analyses on two alignments for each subclade, the first with no trimming of ambiguous alignment positions (which may represent genuinely rapidly evolving sites), and the second with ambiguous alignment positions identified by eye and removed. All estimates and statistical comparisons of evolutionary rates outlined above were very similar with and without alignment screening: We therefore report results estimated from the untrimmed alignments. Although we could not rule out gene conversion between paralogs (which can lead to erroneous support for positive selection; Casola and Hahn 2009), we found very few positively selected sites, so this effect is likely to have little or no effect on our analyses.
Domain Mapping and Structural Modeling
To investigate the distribution of rapidly evolving sites across the domain architecture of each Argonaute gene, we inferred the location of each domain in each Argonaute gene by searching the Pfam database (Finn et al. 2009), and then mapped the mean estimate of ω for each residue across the gene (derived from the BEB posterior distribution under the M8 model in PAML; Yang 1997). To describe evolutionary rate heterogeneity in the protein structures of each gene, we built structural models based on published X-ray crystallography structures: The D. melanogaster Ago1 structure was based on human Ago1 (Faehnle et al. 2013), and the structures of D. melanogaster Ago2, Ago3, and Piwi were based on human Ago2 (Schirle and Macrae 2012). We used the MODELER software in the Discovery Studio 4.0 Modeling Environment (Accelrys Software, Inc., San Diego) to calculate ten models, and selected the most energetically favorable for each protein. The model optimization level was set to High, and loop refinement was included. The model quality was assessed with the three-dimensional (3D) profile option in the software, which compares the compatibility of the 3D structure and the sequence. For D. melanogaster Ago2, we replaced the inferred PAZ domain structure with the D. melanogaster Ago2 PAZ domain structure that has previously been resolved using X-ray crystallography (Song et al. 2003). We then mapped ω onto each residue of the structure using PyMol v.1.7.4.1 (Schrödinger, LLC). For both analyses, we used estimates of ω from trimmed alignments to provide a conservative estimate of residue-specific evolutionary rate. Sites that were trimmed out of the alignment were excluded when mapping ω across domains, and were set as ω = 0 when mapping ω across structures.
Results
Duplications of Ago2, Ago3, and Piwi/Aub Occur in Different Dipteran Lineages
To explore the evolutionary dynamics of Argonautes in the Diptera, we quantified the rate of duplication and evolution of Argonautes from 86 Dipteran species. We find numerous expansions of Ago2 and Piwi/Aub (including the origin of canonical Piwi and Aub themselves from their Piwi-subfamily ancestor; figs. 2 and 3). This is in sharp contrast to Ago1, which is present as a single copy ortholog in all Diptera (fig. 2 and supplementary fig. S2, Supplementary Material online), and Ago3, which has duplicated only rarely (fig. 2 and supplementary fig. S3, Supplementary Material online).
Fig. 2.—

Counts of each Argonaute subclade. Shown are counts for a subsample of 66 Dipteran species with at least one gene in each subclade (out of a total of 86 species). Gene duplication events were inferred by parsimony, and are illustrative only (gene loss is not depicted due to space constraints, thus for some taxa gene counts do not correspond to the number of gene duplications). The rate of gene turnover differs between different Argonautes and lineages, and the divergence of Piwi and Aub occurred 182–156 Ma. Silhuoettes by Warren Photographic and Ramiro Morales-Hojas.
Fig. 3.—

Bayesian gene trees of Ago2 and Piwi/Aub. Ago2 has expanded rapidly in Glossina and the obscura group of Drosophila, whereas Piwi/Aub has undergone numerous duplications in Aedes, Anopheles, and many other Nematoceran taxa.
We also find that the expansions of Ago2 and Piwi/Aub have occurred in different taxa and at different times (figs. 2 and 3). Most duplications of Ago2 have occurred in the Brachycera, with numerous duplications within the Glossina (<84 Ma), and the Drosophila obscura group (<50 Ma) (Hain et al. 2010). Perhaps surprisingly, even in the melanogaster group there appear to have been at least three duplications: A duplicate shared between Drosophila rhopaloa and Drosophila takahashii (DRHO009538 and DTAK011769, respectively) implying multiple losses, and a duplication within the lineage leading to Drosophila kikkawai. Although incomplete genomes and some uncertainty in the gene tree topology mean that the losses are uncertain, implications for our study are minimal, as our analysis of gene turnover uses only gene counts, and losses do not factor into our comparison of evolutionary rates before and after duplication. Single duplications of Ago2 have occurred in the Brachycerans Drosophila willistoni, Scaptodrosophila deflexa, M. domestica, and Megaselia abdita, and in the Nematocerans Belgica antarctica, Culex pipiens, and Sitodiplosis mosellana (fig. 3).
In contrast, most duplications of Piwi/Aub have occurred in the Nematocera. Numerous duplications have occurred in the mosquitoes (Aedes spp., Anopheles spp., and Culex quinquefasciatus) <65 Ma, and multiple copies are seen in Lutzomyia longipalpis, Si. mosellana, Chironomus riparius, B. antarctica, and Corethrella appendiculata (fig. 3). A duplication at the base of the Brachycera between 182 and 156 Ma gave rise to the separate Aub and Piwi subclades (as they occur in D. melanogaster, labeled in figs. 2 and 3). Within these subclades duplications have occurred rarely, only being observed in Piwi of the drosophilids Phortica variegata and Sc. deflexa, and in Aub of Teleopsis species.
Ago2 and Piwi/Aub Have Significantly Higher Duplication Rates than Ago1 and Ago3
To quantify these contrasting patterns of duplication, we used CAFE (Han et al. 2013) to estimate the rate of gene turnover (λ, the number of gains or losses per million years) in each Argonaute subclade. We find that gene turnover rate varies considerably among the subclades, with Ago2 (λ = 0.0022), Ago3 (λ = 0.0003), and Piwi/Aub (λ = 0.0012) having significantly higher gene turnover rates than Ago1 (λ = 1.1516 × 10−10) (P < 0.001 based on the expected distribution of gene family sizes under a birth–death model, with λ fixed at the value estimated for Ago1). We also find that Ago2 and Piwi/Aub have significantly (P < 0.001) higher gene turnover rates than Ago3, but do not differ significantly from each other (P = 0.198).
Argonautes Show Contrasting Rates of Protein Evolution Before and After Duplication
To quantify the rate of protein evolution in each Argonaute subclade, and to identify any sites evolving under positive selection, we fitted models using codeml (PAML; Yang 1997). These analyses revealed that Ago2 has the highest nonsynonymous to synonymous substitution ratio (ω = 0.14 ± 0.0015), followed by Ago3 (ω = 0.12 ± 0.0015), Piwi/Aub (ω = 0.09 ± 0.0009), and finally Ago1 (ω = 0.01 ± 0.0002). All rates were significantly different from each other (Akaike weight = 1.000 to 3 decimal places (d.p.) in all cases). Scans for positively selected sites identified five candidate sites in Ago3 and one in Piwi/Aub; however, in both cases the M8 model was not significantly more likely than the null M8a model (for ω estimates and likelihoods under all models, see supplementary tables 1–3, Supplementary Material online).
To test whether the relative rate of protein evolution changes following duplication, we calculated the likelihood of the data for Ago2, Ago3, and Piwi/Aub under two models: The first with a separate evolutionary rate for branches immediately after a duplication event (the Immediate model); and the second with a separate rate for all branches subtending a duplication event (the All descendants model) (fig. 1). For Ago2 and Ago3, the All descendants model had all support (Akaike weight = 1.000 to 3 d.p. for each). For Piwi/Aub, however, the Immediate model had all support (Akaike weight = 1.000 to 3 d.p.). In each case the evolutionary rate increased after duplication, with Ago2 having the highest rate and Piwi the lowest (fig. 4).
Fig. 4.—
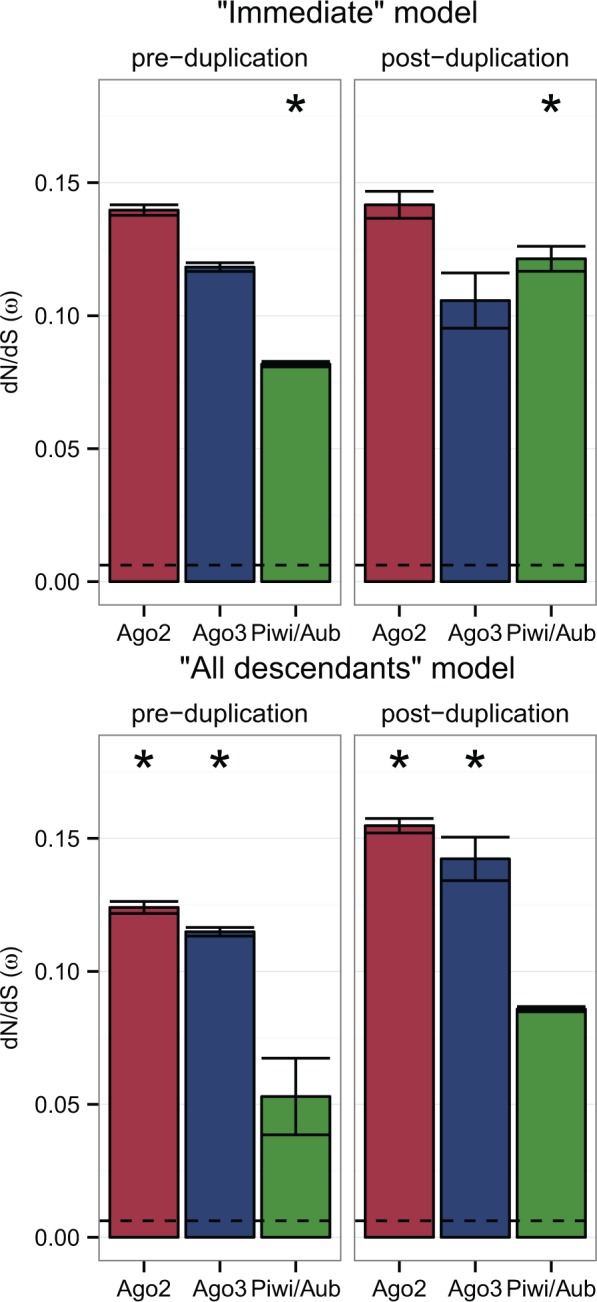
Evolutionary rate estimates before and after duplication, under the Immediate and All descendants models. Asterisks indicate the most highly supported model, and the dashed line indicates the ω value for Ago1 under the M0 model. Duplicates of Piwi/Aub evolve more quickly immediately after duplication, whereas Ago2 and Ago3 paralogs experience a sustained increase in evolutionary rate.
To test for asymmetry between the evolutionary rates of paralogs after duplication, we calculated the likelihood of the data for ten key duplication events in the Ago2 and Piwi/Aub subclades under two models: The Asymmetrical rates model specified one evolutionary rate for one lineage produced by the duplication event, a second rate for the other lineage, and a third for the rest of the tree; and the Asymmetrical rates model specified one evolutionary rate for both lineages produced by the duplication event, and a second rate for the rest of the tree. For Ago2, we find that the Asymmetrical rates model does not provide a significantly better fit for the Glossina sp., B. antarctica, or Cu. pipiens duplication events (LRT, P > 0.1 in all cases). However, the Asymmetrical rates model fits significantly better for the two branches immediately after the obscura group Ago2e-Ago2a/f event (LRT, P < 0.005), and for all branches subtending this event (LRT, P < 0.005). Under the Asymmetrical rates (All descendants) model, the Ago2e clade (ω = 0.17 ± 0.011) and Ago2a/Ago2f clade (ω = 0.22 ± 0.009) are evolving considerably faster than the rest of the tree (ω = 0.13 ± 0.001). For the obscura group Ago2a-Ago2f event, the Asymmetrical rates (All descendants) model does not provide a significantly better fit (LRT, P > 0.1), but the Immediate version of this model does give a significantly better fit (LRT, P < 0.005). Under this model, the branch at the base of the Ago2a clade has a much lower evolutionary rate (ω = 0.06 ± 0.029) than the rest of the tree (ω = 0.14 ± 0.001), and the branch at the base of the Ago2f clade has a much higher evolutionary rate (ω = 0.42 ± 0.142).
For Piwi/Aub, we find that the Asymmetrical rates (All descendants) model provides a significantly better fit for the Piwi–Aub divergence (LRT, P < 0.01) and the duplication event early in mosquito evolution (LRT, P < 0.005). The Aub lineage (ω = 0.09 + 0.002) has a higher evolutionary rate than the Piwi lineage (ω = 0.08 ± 0.002); however, the similarity between the rate of Aub and the rest of the tree (ω = 0.09 ± 0.002) suggests that this difference may be caused by constraint on Piwi rather than positive selection on Aub. After the mosquito Piwi/Aub duplication event, we see a large difference in evolutionary rates, with one clade evolving much more rapidly (ω = 0.14 ± 0.004) than the other (ω = 0.04 ± 0.003), and the rest of the tree (ω = 0.08 ± 0.001).
Ago2 Displays Hotspots of Evolution at the RNA Binding Pocket Entrance
To investigate the distribution of rapidly evolving residues, we mapped ω estimates onto the domains and structures of each Argonaute. We found that rapidly evolving residues are spread across all domains of Ago2, Ago3, and Piwi/Aub (supplementary fig. S4, Supplementary Material online). We also found that Ago2 appears to have clusters of more rapidly evolving residues at the entrance to the RNA binding pocket (fig. 5), which are not found in Ago3 (supplementary fig. S5, Supplementary Material online) or Piwi/Aub (fig. 5). In contrast, the residues that directly contact the sRNA guide are conserved in all Argonautes (fig. 5 and supplementary fig. S5, Supplementary Material online).
Fig. 5.—
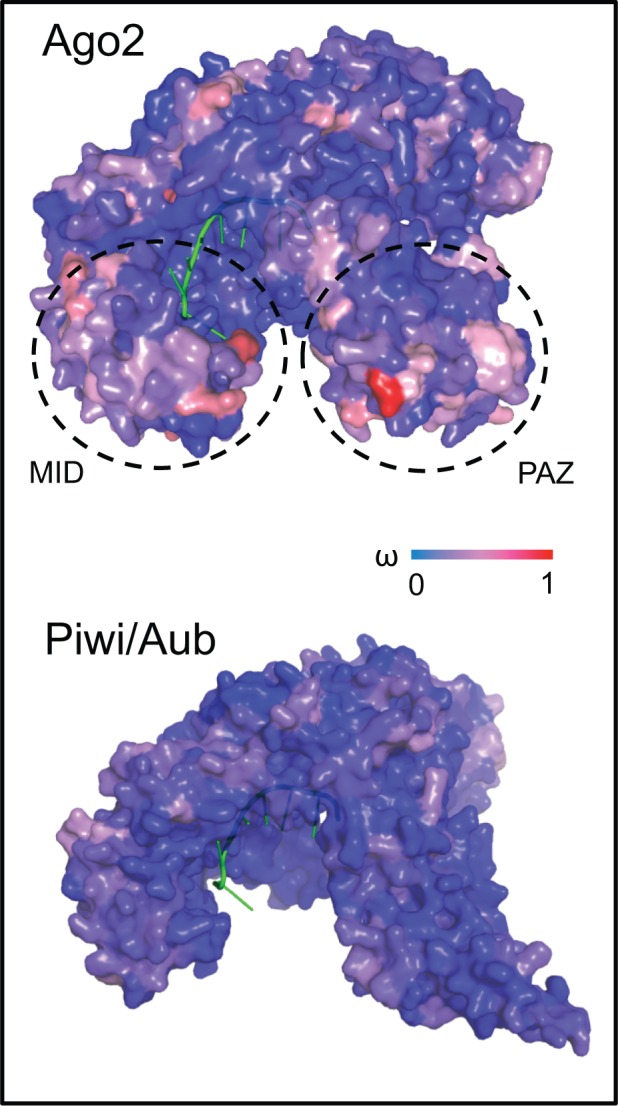
Evolutionary rates mapped onto 3D structures of Ago2 and Piwi/Aub, each binding a sRNA guide. In Ago2, hotspots of evolution are seen at the entrance of the RNA binding pocket; in contrast, evolutionary rate (ω) across the structure of Piwi/Aub is uniformly low. The MID and PAZ protein domains are indicated for Ago2.
Discussion
Our results reveal contrasting patterns of selection and duplication during Dipteran Argonaute evolution. The low evolutionary rate and lack of gene turnover in Ago1 are in agreement with previous studies in D. melanogaster (Obbard et al. 2006, 2009), and are consistent with the idea that Ago1 is carrying out a conserved gene regulatory role in the Diptera as a whole. In contrast, the better fit of the All descendants model to duplications in Ago2 and Ago3 (fig. 4) indicates that paralogs in these subclades have experienced a sustained increase in evolutionary rate, possibly driven by the acquisition of new functions.
This result is particularly noteworthy in Ago2, which is already among the top 3% of the fastest evolving proteins in D. melanogaster (Obbard et al. 2006). Our structural modeling suggests that one possible hotspot of adaptive evolution for these paralogs may be the entrance of the RNA binding pocket (fig. 5). Relaxation of selection pressures on these residues is unexpected as they form alpha-helices, rigid secondary structures that are needed for the stability of the tertiary structure of the protein (Panchenko et al. 2005; Liu et al. 2008); instead, their rapid evolution may be caused by undetected positive selection. The pocket is formed by the PAZ and MID domains, which bind the sRNA guide and form the channel in which the target RNA sits during cleavage (Schirle and Macrae 2012). Although the molecular interactions between the sRNA guide and the inside of the binding pocket have been characterized (reviewed in Swarts et al. 2014), less is known about the function of the residues at the entrance to the pocket. However, given the location of these rapidly evolving residues at the mouth of the binding pocket away from the sRNA guide (fig. 5), such positive selection could be driving differences in target RNA binding and cleavage. Alternatively, selection could be imposed by viral suppressors of RNAi, which are encoded by numerous viruses to inhibit the antiviral RNAi response, and several of which prevent target cleavage by Ago2 (Wang et al. 2006; van Mierlo et al. 2012, 2014). Although we do not find evidence of positive selection in our site analysis across the Diptera as a whole, signatures of selection are evident when we apply branch-sites analyses to Ago2 in the Drosophilidae, as has been reported previously (Kolaczkowski et al. 2011). Such selection may be acting on Ago2 in the Diptera as a whole, but its signature may be masked by saturation of synonymous sites (Anisimova et al. 2001; Clark et al. 2007).
Functional differences between most Dipteran Argonaute paralogs have not been characterized experimentally. However, transcriptome data are available for some G. morsitans tissues, including “lactating” and nonlactating females (Benoit et al. 2014) and salivary glands from parasitized and unparasitized individuals (Telleria et al. 2014) (SRA accessions SRX287393, SRX287395, SRX342351, and SRX342350, respectively). Using these data we explored the possibility of functional divergence in G. morsitans Ago2 and Ago3 paralogs, and found differential expression between both sets of paralogs, as well as high expression of Ago3b in the salivary glands, which increased upon infection with Trypanosoma brucei (supplementary fig. S6, Supplementary Material online). Although this observation awaits replication, the canonical germline-specific role of Ago3 in D. melanogaster (Li et al. 2009) makes any expression of G. morsitans Ago3b in the salivary glands unexpected, and suggests that this paralog has undergone rapid functional divergence to a role beyond TE suppression. Strikingly, this reflects the general patterns noted for somatically expressed Piwis across the eukaryotes, which have evolved diverse roles in epigenetic regulation, genome rearrangement, and somatic development (reviewed in Ross et al. 2014).
The better fit of the Immediate model to duplications of Piwi/Aub (fig. 4) suggests that the evolutionary rate of paralogs in these subclades has been constrained soon after duplication, which may indicate a burst of adaptation to specialize to existing (but distinct) roles. For many duplicates, the branches immediately after duplication are also terminal branches, which clouds the difference between the Immediate and All descendants models. In contrast, the divergence of separate Aub and Piwi (sensu stricto) lineages resulted from a much older duplication in the Piwi-subfamily Piwi/Aub subclade. Our asymmetry analysis suggests that this divergence was accompanied by a reduction in evolutionary rate, particularly in the Piwi lineage, indicating that these lineages are evolving under tight constraint. We estimate that this divergence, which happened at the base of the Brachycera, occurred between 182 and 156 Ma (fig. 2). However, the ambiguous identities of the two Piwi/Aub paralogs in Hermetia illucens and Tabanus bromius (fig. 3) mean that this duplication could have occurred slightly earlier (∼200 Ma).
Under either scenarios, Piwi/Aub paralogs in the vast majority of Nematoceran taxa (including all mosquitoes) are equally homologous to Aub and Piwi, which in D. melanogaster have specialized to distinct roles in the Ping-Pong piRNA amplification cycle and TE silencing, respectively, suggesting that the ancestral Piwi/Aub gene may have had multiple conflicting functions (reviewed in Luteijn and Ketting 2013). It may be that the increased duplication rate of Piwi/Aub in the Nematocera is a result of multiple independent resolutions of this conflict, causing piRNA biogenesis and TE silencing to rely on different suites of Argonaute genes in the Nematocera. This is supported by our asymmetry analysis, which finds that the Piwi/Aub expansion in mosquitoes resulted in asymmetrical evolutionary rates in the resulting lineages, with the rapid evolution of one lineage consistent with the evolution of a novel function. Notably, this rapidly evolving lineage includes a Piwi/Aub paralog in Ae. aegypti (Piwi5) that has recently been shown to have a highly derived function in the production of virus-derived piRNAs (Miesen et al. 2015). Aedes aegypti is a major vector of several arboviruses including yellow fever virus and chikungunya virus, and also has an exceptionally high TE load (Arensburger et al. 2011); although little is known about the total viral load of Ae. aegypti, it is possible that the combined viral and TE loads impose contrasting selection pressures, thereby driving the expansion of Piwi/Aub. Moreover, the numerous instances of expansion followed by functional divergence demonstrate that the Piwi/Aub subclade is not constrained to a germline-specific anti-TE role, but can evolve novel and highly derived functions.
Conclusion
We show that Dipteran Argonautes differ widely in their rates of gene turnover and protein evolution, with duplication driving an increase in evolutionary rate that suggests frequent functional divergence. Our results provide an insight into the selection pressures driving the evolution of RNAi mechanisms across the eukaryotes, which are integral to a range of cellular and genomic processes. Our finding that Argonautes undergo frequent expansions and contractions indicates that expansions in other taxa, such as the WAGO clade of nematodes (Buck and Blaxter 2013) and the Piwi clade of the aphid Ac. pisum (Lu et al. 2011), are not isolated cases; instead, these are further examples of a general pattern of rapid gene turnover in some Argonaute clades. Additionally, our finding that duplication drives rapid evolution suggests that Argonautes evolve new functions frequently and rapidly, as exemplified by Ago4 in the shrimp Penaeus monodon (Leebonoi et al. 2015) and the smedwi clade of planarians (Reddien et al. 2005). This combination of rapid gene turnover and frequent functional divergence illustrates a high degree of evolutionary lability in Argonaute function across a wide range of taxa, and may drive the functional overlap frequently observed between different Argonaute subclades across the eukaryotes. Our work also highlights the selection pressures exerted by parasites, shown by the higher rate of gene turnover for Piwi/Aub and higher evolutionary rate of Ago2, which play roles in defense against TEs and viruses, respectively. This provides further evidence of the importance of parasites in evolution (Dawkins and Krebs 1979), and demonstrates how host–parasite interactions can drive genome evolution and generate phenotypic novelty.
Supplementary Material
Supplementary tables S1–S3 and figures S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by a Natural Environment Research Council Doctoral Training Grant (NERC DG NE/J500021/1 to S.H.L.), the Academy of Finland (265971 to H.S.), a University of Edinburgh Chancellor’s Fellowship (to D.J.O.), and a Wellcome Trust strategic award to the Centre for Immunity, Infection and Evolution (WT095831 to the CIIE). We thank all authors who gave us access to unreleased genomes and transcriptomes. We also thank Esther Schnettler for reading an earlier version of the manuscript, Mark Kunitomi for alerting us to an error in the introduction, and two anonymous reviewers for valuable feedback.
Literature Cited
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z. 2001. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 18:1585–1592. [DOI] [PubMed] [Google Scholar]
- Arensburger P, Hice RH, Wright JA, Craig NL, Atkinson PW. 2011. The mosquito Aedes aegypti has a large genome size and high transposable element load but contains a low proportion of transposon-specific piRNAs. BMC Genomics 12:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Bachtrog D. 2013. Neofunctionalization of young duplicate genes in Drosophila. Proc Natl Acad Sci U S A. 110:17409–17414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit JB, et al. 2014. A novel highly divergent protein family identified from a viviparous insect by RNA-seq analysis: a potential target for tsetse fly-specific abortifacients. PLoS Genet. 10:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmert K, et al. 1998. AGO1 defines a novel locus of Arabidopis controlling leaf development. EMBO J. 1:170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck AH, Blaxter M. 2013. Functional diversification of Argonautes in nematodes: an expanding universe. Biochem Soc Trans. 41:881–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CL, Black WC, Hess AM, Foy BD. 2008. Comparative genomics of small RNA regulatory pathway components in vector mosquitoes. BMC Genomics 9:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola C, Hahn MW. 2009. Gene conversion among paralogs results in moderate false detection of positive selection using likelihood methods. J Mol Evol. 68:679–687. [DOI] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552. [DOI] [PubMed] [Google Scholar]
- Cerutti H, Casas-Mollano JA. 2006. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 50:81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Okamura K, Martin R, Lai EC. 2008. Endogenous RNA interference provides a somatic defense against Drosophila transposons. Curr Biol. 18:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. [DOI] [PubMed] [Google Scholar]
- Czech B, et al. 2008. An endogenous small interfering RNA pathway in Drosophila. Nature 453:798–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawkins R, Krebs JR. 1979. Arms races between and within species. Proc R Soc Lond B. 205:489–511. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer NA, et al. 2008. Molecular phylogenetics of tsetse flies (Diptera: Glossinidae) based on mitochondrial (COI, 16S, ND2) and nuclear ribosomal DNA sequences, with an emphasis on the palpalis group. Mol Phylogenet Evol. 49:227–239. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Izaurralde E. 2008. Getting to the root of miRNA-mediated gene silencing. Cell 132:9–14. [DOI] [PubMed] [Google Scholar]
- Faehnle CR, Elkayam E, Haase AD, Hannon GJ, Joshua-Tor L. 2013. The making of a slicer: activation of human argonaute-1. Cell Rep. 3:1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. 2009. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, et al. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811. [DOI] [PubMed] [Google Scholar]
- Hahn MW. 2009. Distinguishing among evolutionary models for the maintenance of gene duplicates. J Hered. 100:605–617. [DOI] [PubMed] [Google Scholar]
- Hain D, et al. 2010. Natural variation of the amino-terminal glutamine-rich domain in Drosophila argonaute2 is not associated with developmental defects. PLoS One 5:e15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MV, Thomas GWC, Lugo-Martinez J, Hahn MW. 2013. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 30:1987–1997. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Carroll SB. 2007. Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449:677–681. [DOI] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H. 2015. PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem. 84:405–433. [DOI] [PubMed] [Google Scholar]
- Jordan G, Goldman N. 2012. The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol Biol Evol. 29:1125–1139. [DOI] [PubMed] [Google Scholar]
- Kalmykova AI, Klenov MS, Gvozdev VA. 2005. Argonaute protein PIWI controls mobilization of retrotransposons in the Drosophila male germline. Nucleic Acids Res. 33:2052–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka Y, Takeichi M, Uemura T. 2001. Developmental roles and molecular characterization of a Drosophila homologue of Arabidopsis Argonaute1, the founder of a novel gene superfamily. Genes Cells. 6:313–325. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keays MC, Barker D, Wicker-Thomas C, Ritchie MG. 2011. Signatures of selection and sex-specific expression variation of a novel duplicate during the evolution of the Drosophila desaturase gene family. Mol Ecol. 20:3617–3630. [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 10:126–139. [DOI] [PubMed] [Google Scholar]
- Kolaczkowski B, Hupalo DN, Kern AD. 2011. Recurrent adaptation in RNA interference genes across the Drosophila phylogeny. Mol Biol Evol. 28:1033–1042. [DOI] [PubMed] [Google Scholar]
- Leebonoi W, Sukthaworn S, Panyim S, Udomkit A. 2015. A novel gonad-specific Argonaute 4 serves as a defense against transposons in the black tiger shrimp Penaeus monodon. Fish Shellfish Immunol. 42:280–288. [DOI] [PubMed] [Google Scholar]
- Li C, et al. 2009. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137:509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li WX, Ding SW. 2002. Induction and suppression of RNA silencing by an animal virus. Science 296:1319–1321. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang Y, Lei X, Zhang Z. 2008. Natural selection of protein structural and functional properties: a single nucleotide polymorphism perspective. Genome Biol. 9:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HL, et al. 2011. Expansion of genes encoding piRNA-associated argonaute proteins in the pea aphid: diversification of expression profiles in different plastic morphs. PLoS One 6:e28051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn MJ, Ketting RF. 2013. PIWI-interacting RNAs: from generation to transgenerational epigenetics. Nat Rev Genet. 14:523–534. [DOI] [PubMed] [Google Scholar]
- Miesen P, Girardi E, van Rij RP. 2015. Distinct sets of PIWI proteins produce arbovirus and transposon-derived piRNAs in Aedes aegypti mosquito cells. Nucleic Acids Res. 43:6545–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B, et al. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science 346:763–767. [DOI] [PubMed] [Google Scholar]
- Morandin C, et al. 2014. Not only for egg yolk—functional and evolutionary insights from expression, selection, and structural analyses of Formica ant vitellogenins. Mol Biol Evol. 31:2181–2193. [DOI] [PubMed] [Google Scholar]
- Morazzani EM, Wiley MR, Murreddu MG, Adelman ZN, Myles KM. 2012. Production of virus-derived ping-pong-dependent piRNA-like small RNAs in the mosquito soma. PLoS Pathog. 8:e1002470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee K, Campos H, Kolaczkowski B. 2013. Evolution of animal and plant dicers: early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Mol Biol Evol. 30:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MG, Gadagkar SR, Gutzwiller L. 2010. Tubulin evolution in insects: gene duplication and subfunctionalization provide specialized isoforms in a functionally constrained gene family. BMC Evol Biol. 10:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. 2009. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond B Biol Sci. 364:99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Jiggins FM, Halligan DL, Little TJ. 2006. Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr Biol. 16:580–585. [DOI] [PubMed] [Google Scholar]
- Pal-Bhadra M, et al. 2004. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303:669–672. [DOI] [PubMed] [Google Scholar]
- Palakodeti D, Smielewska M, Lu YC, Yeo GW, Graveley BR. 2008. The PIWI proteins SMEDWI-2 and SMEDWI-3 are required for stem cell function and piRNA expression in planarians. RNA 14:1174–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko AR, Wolf YI, Panchenko LA, Madej T. 2005. Evolutionary plasticity of protein families: coupling between sequence and structure variation. Proteins Struct Funct Genet. 61:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JS, Martínez de Alba AE, Vaucheret H. 2012. The origin and effect of small RNA signaling in plants. Front Plant Sci. 3:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, et al. 2011. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 9:e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Oviedo NJ, Jennings JR, Jenkin JC, Sánchez Alvarado A. 2005. SMEDWI-2 is a PIWI-like protein that regulates planarian stem cells. Science 310:1327–1330. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. [DOI] [PubMed] [Google Scholar]
- Ross RJ, Weiner MM, Lin H. 2014. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Miska EA. 2014. Small RNAs break out: the molecular cell biology of mobile small RNAs. Nat Rev Mol Cell Biol. 15:525–535. [DOI] [PubMed] [Google Scholar]
- Sarkies P, et al. 2015. Ancient and novel small RNA pathways compensate for the loss of piRNAs in multiple independent nematode lineages. PLoS Biol. 13:e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, Macrae IJ. 2012. The crystal structure of human Argonaute2. Science 336:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnettler E, et al. 2013. Knockdown of piRNA pathway proteins results in enhanced Semliki Forest virus production in mosquito cells. J Gen Virol. 94:1680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnettler E, et al. 2014. Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Res. 42:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JG, et al. 2014. Genome of the house fly, Musca domestica L., a global vector of diseases with adaptations to a septic environment. Genome Biol. 15:466–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Dönertas D, Brennecke J. 2012. Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151:964–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, et al. 2003. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat Struct Biol. 10:1026–1032. [DOI] [PubMed] [Google Scholar]
- Swarts DC, et al. 2014. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol. 21:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telleria EL, et al. 2014. Insights into the trypanosome-host interactions revealed through transcriptomic analysis of parasitized tsetse fly salivary glands. PLoS Negl Trop Dis. 8:e2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin VV, Klenov MS, Stolyarenko AD, Kotelnikov RN, Gvozdev VA. 2004. The RNA interference proteins and Vasa locus are involved in the silencing of retrotransposons in the female germline of Drosophila melanogaster. RNA Biol. 1:54–58. [PubMed] [Google Scholar]
- van der Linde K, Houle D, Spicer GS, Steppan SJ. 2010. A supermatrix-based molecular phylogeny of the family Drosophilidae. Genet Res (Camb). 92:25–38. [DOI] [PubMed] [Google Scholar]
- van Mierlo JT, et al. 2012. Convergent evolution of argonaute-2 slicer antagonism in two distinct insect RNA viruses. PLoS Pathog. 8:e1002872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo JT, et al. 2014. Novel Drosophila viruses encode host-specific suppressors of RNAi. PLoS Pathog. 10:e1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rij RP, et al. 2006. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev. 20:2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, et al. 2012. Arbovirus-derived piRNAs exhibit a ping-pong signature in mosquito cells. PLoS One 7:e30861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, et al. 2006. RNA interference directs innate immunity against viruses in adult Drosophila. Science 312:452–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmann BM, et al. 2011. Episodic radiations in the fly tree of life. Proc Natl Acad Sci U S A. 108:5690–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 13:555–556. [DOI] [PubMed] [Google Scholar]
- Zhang B, Liu YH, Wu WX, Wang ZL. 2010. Molecular phylogeny of Bactrocera species (Diptera: Tephritidae: Dacini) inferred from mitochondrial sequences of 16S rDNA and COI sequences. Fla Entomol. 93:369–377 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.