

Abstract
Mutations in the PRSS1 gene encoding human cationic trypsinogen are associated with hereditary and sporadic chronic pancreatitis. High-penetrance PRSS1 mutations found in hereditary pancreatitis alter activation and/or degradation of cationic trypsinogen, thereby promoting intrapancreatic trypsinogen activation. In contrast, a number of rare PRSS1 variants identified in subjects with sporadic chronic pancreatitis cause misfolding and endoplasmic reticulum (ER) stress. Mutation p.L104P is unique among natural PRSS1 variants, since it affects the substrate binding site of trypsin. The aim of the present study was to establish the clinical significance of variant p.L104P through functional analysis. We found that p.L104P trypsin exhibited decreased activity on peptide and protein substrates; however, autoactivation was slightly accelerated. Remarkably, binding of the physiological trypsin inhibitor serine protease inhibitor Kazal type 1 (SPINK1) was decreased by 70-fold. In the presence of the trypsinogen-degrading enzyme chymotrypsin C, mutant p.L104P autoactivated to higher trypsin levels than wild-type trypsinogen. This apparent resistance to degradation was due to slower cleavage at Arg122 rather than Leu81. Finally, secretion of mutant p.L104P from transfected cells was markedly reduced due to intracellular retention and aggregation with concomitant elevation of ER stress markers. We conclude that PRSS1 variant p.L104P exhibits a variety of phenotypic changes that can increase risk for chronic pancreatitis. Mutation-induced misfolding and associated ER stress are the dominant effects that support a direct pathogenic role in chronic pancreatitis.
Keywords: chronic pancreatitis, hereditary pancreatitis, trypsinogen activation, autoactivation, endoplasmic reticulum stress, misfolding, intracellular aggregation
chronic pancreatitis is a relapsing progressive inflammatory disease of the pancreas that often develops on the basis of genetic predisposition (7, 44). Susceptibility genes identified to date include, in the order of discovery, PRSS1 (serine protease 1, human cationic trypsinogen), CFTR (cystic fibrosis transmembrane conductance regulator), SPINK1 (serine protease inhibitor Kazal type 1, pancreatic secretory trypsin inhibitor), CTRC (chymotrypsinogen C), CPA1 (procarboxypeptidase A1), and CEL (carboxyl ester lipase) (8, 12, 26, 31, 42, 45, 46). Mutations in PRSS1, such as p.N29I and p.R122H, are strong risk factors that cause autosomal dominant hereditary pancreatitis with incomplete penetrance and variable expressivity (20). Although less well characterized, CPA1 mutations also confer strong predisposition resulting in early onset disease (45). In contrast, risk variants of other genes typically confer smaller but significant risk and are often found in the trans-heterozygous state in affected carriers (25). More recently, a GWAS study identified common variants in the CLDN2 locus and in the promoter region of PRSS1 that appear to increase disease risk only by a small degree (10, 43).
Mechanistic studies revealed that mutations in most risk genes result in premature intrapancreatic trypsinogen activation. Thus, high-penetrance PRSS1 variants cause higher trypsin levels either by stimulating trypsinogen autoactivation and/or by inhibiting CTRC-dependent trypsinogen degradation (13, 33). Loss-of-function mutations in the protective trypsin inhibitor SPINK1 or the trypsinogen-degrading enzyme CTRC can also result in elevated trypsin activity (1, 4, 5, 15, 18, 26). Consistent with the trypsin paradigm, a rapidly autodegrading variant of PRSS2 encoding human anionic trypsinogen was shown to protect against chronic pancreatitis (47). Finally, variants that diminish CFTR function can stimulate trypsinogen activation through impaired ductal flushing and/or altered intraluminal pH (24).
Investigations into the cellular effects of pancreatitis-associated mutations revealed an alternative disease mechanism unrelated to trypsin activity, i.e., mutation-induced protein misfolding and consequent endoplasmic reticulum (ER) stress (1, 14, 30, 39, 45). CPA1 mutations and some PRSS1 variants are most likely to exert their pathogenic effect via this pathway (14, 30) although misfolding was also observed with a handful of CTRC (1, 39) and SPINK1 (4, 5, 18) variants, and it may also underlie the mechanism of action of the recently described CEL hybrid variant (12). The PRSS1 variants that appeared to cause misfolding (p.K92N, p.D100H, p.R116C, p.S124F, p.C139F, p.C139S, p.G208A) were typically rare and found in sporadic idiopathic cases (14, 30, and references therein).
The c.311T>C (p.L104P) human cationic trypsinogen variant was first reported in three heterozygous carriers of a German pedigree with a history of abdominal pain and diabetes but without clinically proven chronic pancreatitis (40). The same variant was later identified in a subject of Chinese origin with idiopathic chronic pancreatitis (6). The limited clinical and genetic data do not allow conclusive determination whether or not the p.L104P variant is pathogenic. The Leu104 residue forms part of the conserved hydrophobic S2 subsite (Schechter-Berger nomenclature) (29) of the substrate binding site of trypsin. Natural PRSS1 variants affecting the substrate binding site of trypsin have not been described before, and in this regard variant p.L104P is unique. Therefore, in the present study, we set out to investigate the biochemical and cellular effects of variant p.L104P to confirm or rule out a possible pathogenic role in chronic pancreatitis.
MATERIALS AND METHODS
Materials.
Expression and purification of recombinant CTRC, CTRB1, CTRB2, CPA1, SPINK1, and ecotin have been described previously (18, 19, 23, 33, 34, 37, 38, 45).
Nomenclature.
Amino acid residues in human cationic trypsinogen were numbered starting with the initiator methionine of the primary translation product according to the recommendations of the Human Genome Variation Society. The reference sequence used was NM_002769.4.
Plasmid construction and mutagenesis.
The pTrapT7 PRSS1, pTrapT7 intein-PRSS1, and pcDNA3.1(−) PRSS1 expression plasmids were constructed previously (17, 22, 27, 28). Missense mutation p.L104P was generated by overlap extension PCR mutagenesis and cloned into the expression plasmids.
Expression and purification of recombinant trypsinogen.
Wild-type and p.L104P cationic trypsinogens were expressed in Escherichia coli BL21(DE3) as cytoplasmic inclusion bodies (27, 28). For autoactivation experiments in the presence of CTRC, trypsinogens were expressed as fusions with a self-splicing mini-intein in the aminopeptidase P-deficient LG-3 E. coli strain (16, 17). After self-splicing, this construct generates homogeneous intact NH2-termini not processed by cellular aminopeptidases. Refolding and purification of trypsinogen on immobilized ecotin was carried out as reported previously (16). Concentrations of purified trypsinogen preparations were determined from the ultraviolet absorbance at 280 nm using the extinction coefficient 37,525 M−1·cm−1
SPINK1 binding assays.
Binding of wild-type and p.L104P mutant human cationic trypsin to the trypsin inhibitor SPINK1 was characterized by measuring the KD value of the reaction in equilibrium, as we described previously (32, 35). Trypsin concentrations were determined by titration against ecotin, and SPINK1 concentration was determined by titration against wild-type human cationic trypsin. Wild-type (50 pM) and mutant (500 pM) trypsin were incubated overnight (for 16 h) with increasing concentrations of the inhibitor ranging from 0 to 100 pM for wild-type trypsin and 0 to 1,000 pM for the mutant trypsin. Residual trypsin activity was measured with 150 μM N-CBZ-Gly-Pro-Arg-7-amido-4-methylcoumarin in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20. The increase in fluorescence was monitored at 380 nm excitation and 460 nm emission wavelengths continuously for 10 min at 22°C. The free protease concentration was plotted as a function of the total inhibitor concentration, and the experimental points were fitted with the following equation: y = E − {E + x + K-sqrt[(E + x + K)2 − 4Ex]}/2, where the independent variable x represents the total inhibitor concentration, the dependent variable y is the free protease concentration in equilibrium, K is KD, and E denotes the total protease concentration.
Trypsinogen autoactivation.
Wild-type and mutant trypsinogen at the indicated concentration (1 or 2 μM) were incubated in the absence or presence of 5, 10, 25, and 50 nM human CTRC and 10 nM cationic trypsin in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20 (final concentrations) at 37°C. At given times, 2-μl aliquots were withdrawn and mixed with 48 μl assay buffer containing 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20. Trypsin activity was measured by adding 150 μl of 200 μM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate and following the release of the yellow p-nitroaniline at 405 nm in a SpectraMax plus384 microplate reader (Molecular Devices, Sunnyvale, CA) for 1 min. Reaction rates were calculated from fits to the initial linear portions of the curves. The trypsin substrate was dissolved in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20.
Enzyme kinetic analysis.
Michaelis-Menten parameters were measured in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20 at 22°C using 1–2 nM (wild type) or 2–5 nM (p.L104P) trypsin (final concentrations). The concentration of the peptide substrates was varied between 10 and 180 μM (wild type) or 40 and 360 μM (p.L104P mutant). Rates of substrate cleavage were plotted as a function of substrate concentration, and data points were fitted with the Michaelis-Menten hyperbolic equation. To determine the apparent inhibitory constant (Ki) of benzamidine, kinetic measurements were performed in the presence of increasing inhibitor concentrations (0–80 μM). Apparent KM values were plotted as a function of inhibitor concentration, and Ki was derived from the negative intercept of the linear fit on the horizontal axis.
Cell culture and transfection.
Human embryonic kidney (HEK) 293T cells were cultured in six-well tissue culture plates (106 cells/well) in DMEM supplemented with 10% fetal bovine serum, 4 mM glutamine, and 1% penicillin/streptomycin solution at 37°C in a humidified atmosphere containing 5% CO2. Transfections were performed using 4 μg pcDNA3.1(−) PRSS1 plasmid and 10 μl Lipofectamine 2000 (Invitrogen) in 2 ml DMEM. After overnight incubation at 37°C, cells were rinsed and overlaid with 2 ml OptiMEM medium (Invitrogen). Media and cells were collected 48 h after this medium change.
Measurement of trypsin activity in conditioned media.
Aliquots (50 μl) of conditioned media were supplemented with 0.1 M Tris·HCl (pH 8.0) by adding 5 μl from a 1 M stock solution and 10 mM calcium by adding 1 μl of a 0.5 M CaCl2 solution. Trypsinogen was then activated by adding 1 μl human enteropeptidase from a 1.4 μg/ml stock solution (R&D Systems, Minneapolis, MN). After incubation for 1 h at 37°C, 50-μl aliquots were removed and mixed with 150 μl of 200 μM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate. Trypsin activity was measured as described under Trypsinogen autoactivation.
Preparation of cell lysates.
Transfected cells were rinsed two times with phosphate-buffered saline. Reporter lysis buffer (200 μl; Promega) and 4 μl protease inhibitor cocktail (Sigma) were added to each well, and the cells were scraped and vortexed briefly. After 15 min incubation on ice, the lysates were cleared by centrifugation. The protein concentration of the supernatant was measured with a Micro BCA Protein Assay Kit (Thermo Scientific).
SDS-PAGE and Western blotting.
Conditioned media (200 μl) were precipitated with 10% trichloroacetic acid (final concentration), resuspended in 20 μl Laemmli sample buffer containing 100 mM dithiothreitol, heat-denatured at 95°C for 5 min, and run on 15% SDS-polyacrylamide gels. The gels were stained with Coomassie blue R-250. For Western blotting, conditioned media (5 μl) or cell lysates (20 μg total protein) were directly mixed with sample buffer and electrophoresed as described above. Proteins were transferred to Immobilon-P membranes (Millipore, Bedford, MA) at 350 mA for 1 h. Trypsinogen was detected with a sheep polyclonal antibody (no. AF3848; R&D Systems) used at a dilution of 1:5,000 followed by horseradish peroxidase (HRP)-conjugated donkey polyclonal anti-sheep IgG (no. HAF016; R&D Systems) used at 1:2,000 dilution. Incubations with primary and secondary antibodies were performed at room temperature for 1 h each. HRP was detected using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Reverse transcription-PCR analysis.
RNA was isolated from HEK 293T cells transfected with given plasmids using the RNeasy Mini Kit (Qiagen), and 2 μg RNA were reverse-transcribed with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Semiquantitative measurements of X-box-binding protein 1 (XBP1) mRNA splicing were performed by PCR using primers that amplify both the spliced and unspliced forms and generate 441- and 415-bp amplicons, respectively. XBP1 primers were as follows: sense primer, 5′-CCT TGT AGT TGA GAA CCA GG-3′ and antisense primer, 5′-GGG CTT GGT ATA TAT GTG G-3′. The PCR amplicons were separated by agarose gel electrophoresis, stained with ethidium bromide, and photographed. The fraction of spliced XPB1 was then determined by densitometry. Levels of immunoglobulin-binding protein (BiP) mRNA were determined using quantitative PCR with TaqMan primers and TaqMan Universal PCR Mastermix (Applied Biosystems). Gene expression was quantitated using the comparative CT method (ΔΔCT method). Expression levels of BiP were first normalized to those of the glyceraldehyde-3-phosphate dehydrogenase internal control gene (ΔCT) and then to expression levels measured in cells transfected with empty vector (ΔΔCT). Results were expressed as fold changes calculated with the formula 2−ΔΔCT.
RESULTS
Modeling the effect of the p.L104P mutation.
Position 104 in human cationic trypsinogen corresponds to amino acid 99 in the conventional numbering of chymotrypsin-like serine proteases, and it usually harbors aliphatic amino acids such as the Leu found in human trypsins. This residue helps to shape the S2 subsite, which is also lined by His63 (chymotrypsin numbering His57) and Trp216 (chymotrypsin numbering Trp215). Although the S2 site is relatively hydrophobic, with the exception of Asp and Glu, it can accommodate a variety of residues with little selectivity (3, 11). The side chain of Leu104 can form van der Waals contacts with the P2 side chain of the bound substrate or inhibitor, as seen, for example, in the structure of the Michaelis complex of an inactive bovine trypsin mutant with the Pittsburgh variant of the α1-antitrypsin inhibitor (Fig. 1) (9). In addition, an H bond between the backbone amino and carbonyl groups of Leu104 and Asp100 (chymotrypsin numbering Asp95) contributes to the H bond network that stabilizes the loop between Pro97 (chymotrypsin numbering Pro92) and Asp107 (chymotrypsin numbering Asp102). Mutation of Leu104 to Pro should alter its interactions with the P2 side chain of substrates and inhibitors and also eliminates the H bond stabilizing the Pro97-Asp107 loop. These changes are expected to alter substrate and inhibitor binding.
Fig. 1.

Role of Leu104 in the S2 subsite of the trypsin substrate binding site. The Michaelis complex between a catalytically inactive bovine trypsin mutant and the Pittsburgh variant of α1-antitrypsin is shown (Protein Data Bank file 1OPH). The side chains contributing to the S2 subsite of trypsin are shown in red, and the P1 Arg and P2 Pro residues of the inhibitor are indicated in green. See text for details. The broken line indicates an H bond between the main chain atoms of positions 104 and 100 that stabilizes the 97–107 loop. The image was rendered by UCFS Chimera 1.10.2 (www.cgl.ucsf.edu/chimera).
Catalytic activity of p.L104P cationic trypsin.
Determination of the kinetic parameters of the mutant enzyme on small synthetic substrates N-CBZ-Gly-Pro-Arg-p-nitroanilide and N-CBZ-Gly-Pro-Arg-7-amido-4-methylcoumarin revealed an ∼10-fold decrease in the catalytic efficiency that was mostly attributable to an increase in the KM (Table 1). Larger protein substrates were also cleaved less efficiently by the mutant. Thus, the mutant enzyme activated human chymotrypsinogens B1, B2 (data not shown), and C ∼20-fold slower relative to wild-type cationic trypsin, whereas activation of human proCPA1 was reduced by about twofold (Fig. 2). Finally, mutant p.L104P digested bovine β-casein at a twofold decreased rate (Fig. 3).
Table 1.
Kinetic parameters of wild-type and mutant human cationic trypsin on the synthetic chromogenic substrate GPR-pNA and GPR-AMC
| kcat, s−1 | KM, μM | kcat/KM, M−1·s−1 | |
|---|---|---|---|
| GPR-pNA | |||
| Wild type | 135 ± 5.9 | 23.9 ± 4.1 | 5.7 × 106 |
| p.L104P | 64 ± 1.5 | 118.7 ± 6.4 | 5.4 × 105 |
| GPR-AMC | |||
| Wild type | 99.5 ± 1.3 | 22.7 ± 1.2 | 4.4 × 106 |
| p.L104P | 69.7 ± 1.3 | 208.5 ± 9 | 3.3 × 105 |
Values are means ± SD; n = 3 experiments.
kcat, Catalytic rate constant.
N-CBZ-Gly-Pro-Arg-p-nitroanilide (GPR-pNA) and fluorescent substrate N-CBZ-Gly-Pro-Arg-7-amido-4-methylcoumarin (GPR-AMC), measured in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20, at 22°C.
Fig. 2.

Effect of mutation p.L104P on the activation of pancreatic proenzymes by trypsin. A and B: wild-type and mutant human cationic trypsin (25 nM) were incubated with 2 μM human chymotrypsinogen B1 (CTRB1) or chymotrypsinogen C (CTRC) in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20 (final concentrations) at 37°C. At the indicated times, 2-μl aliquots were removed, and chymotrypsin activity was measured using 150 μM Suc-Ala-Ala-Pro-Phe-p-nitroanilide substrate. C: wild-type and mutant human cationic trypsin (25 nM) were incubated with 2 μM human procarboxypeptidase A1 (proCPA1) in 0.1 M Tris·HCl (pH 8.0) and 1 mM CaCl2 (final concentrations) at 37°C. At the indicated times, 100-μl aliquots were precipitated with 10% trichloroacetic acid, electrophoresed on 15% SDS-PAGE gels, and stained with Coomassie blue. CPA1, activated carboxypeptidase A1. Representative graphs and gel picture from two or three experiments are shown.
Fig. 3.

Effect of mutation p.L104P on the digestion of bovine β-casein with trypsin. Wild-type and mutant human cationic trypsin (2 nM) were incubated with β-casein (0.2 mg/ml) in 0.1 M Tris·HCl (pH 8.0) and 1 mM CaCl2 (final concentrations) at 37°C. At the indicated times, 100-μl aliquots were precipitated with 10% trichloroacetic acid, electrophoresed on 15% SDS-PAGE gels, and stained with Coomassie blue. A representative gel from two experiments is shown.
Autoactivation of p.L104P cationic trypsinogen.
Experiments described above indicate that the p.L104P trypsin mutant cleaves a variety of substrates at a reduced rate. Remarkably, however, this was not the case when trypsin-mediated trypsinogen activation (autoactivation) was tested. When measured at pH 8.0 in 1 mM calcium, the mutant trypsinogen autoactivated slightly faster than wild type (Fig. 4A). SDS-PAGE analysis of the autoactivation process confirmed more rapid conversion of the p.L04P trypsinogen band to trypsin compared with wild-type cationic trypsinogen (Fig. 4B). Interestingly, the typically seen proteolytic fragments resulting from cleavage of the Arg122-Val123 peptide bond were much less noticeable in case of the mutant enzyme, indicating that this autolytic site is poorly cleaved by p.L104P trypsin (Fig. 4B).
Fig. 4.

Effect of mutation p.L104P on autoactivation of trypsinogen. Wild-type and mutant human cationic trypsinogen (2 μM) were incubated with 10 nM initial trypsin in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20 (final concentrations) at 37°C. A: at the indicated times 2-μl aliquots were removed, and trypsin activity was measured with 150 μM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate as described in materials and methods. Trypsin activity was converted to active trypsin concentration using the maximal plateau activity as the 2 μM reference value. B: autoactivation was also followed by SDS-PAGE. At the indicated times, 100-μl aliquots were precipitated with 10% trichloroacetic acid, electrophoresed on 15% SDS-PAGE gels, and stained with Coomassie blue. *Two chains of trypsin cleaved at the Arg122 autolytic site. Representative graph and gel from three experiments are shown.
Autoactivation of human cationic trypsinogen is regulated by CTRC through specific proteolytic cleavages. When autoactivation of wild-type trypsinogen is measured in the presence of CTRC, the rate is slightly increased due to processing of the activation peptide to a shorter form, whereas trypsin levels are markedly reduced due to trypsinogen degradation (33). This latter process requires not only a CTRC-mediated cleavage after Leu81 in the calcium-binding loop but also an autolytic cleavage after Arg122. Compared with wild-type cationic trypsinogen, autoactivation of mutant p.L104P was less affected by CTRC, and the mutant autoactivated to higher trypsin levels (Fig. 5), e.g., in the presence of 50 nM CTRC, mutant activity reached ∼35%, whereas wild type plateaued at 15% of potential maximal activity. Visualizing the activation reaction on Coomassie blue-stained gels confirmed the stronger trypsin bands in the p.L104P mutant and the absence of the proteolytic fragments due to cleavage at Arg122 (Fig. 5C). Thus, the apparent resistance of p.L104P trypsinogen to CTRC-mediated degradation is due to the defective autolytic cleavage of the Arg122-Val123 peptide bond (see also Fig. 4B). This conclusion was supported by direct digestion experiments with CTRC that demonstrated no change in cleavage of the Leu81-Glu82 peptide bond in p.L104P trypsinogen relative to wild type (Fig. 6).
Fig. 5.

Effect of mutation p.L104P on autoactivation of trypsinogen in the presence of chymotrypsin C (CTRC). Wild-type (A) and mutant (B) human cationic trypsinogen (1 μM) were incubated in the absence or presence of the indicated concentrations of human CTRC and 10 nM cationic trypsin in 0.1 M Tris·HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20 (final concentrations) at 37°C. At the indicated times, 2-μl aliquots were removed, and trypsin activity was measured as described in materials and methods. Trypsin activity was expressed as percentage of the maximal activity measured in the absence of CTRC. C: autoactivation in the presence of 10 nM CTRC was also followed by SDS-PAGE. At the indicated times, 150-μl aliquots were precipitated with 10% trichloroacetic acid, electrophoresed on 15% SDS-PAGE gels, and stained with Coomassie blue. The faint proteolytic fragment observed in the wild-type sample at 15 kDa corresponds to the COOH-terminal chain of trypsin cleaved at the Arg122 autolytic site. Representative graph and gel from two experiments are shown.
Fig. 6.

Effect of mutation p.L104P on the cleavage of the Leu81-Glu82 peptide bond in trypsinogen (Tg) by CTRC. Wild-type and mutant human cationic trypsinogen (2 μM) were incubated with 25 nM human CTRC in 0.1 M Tris·HCl (pH 8.0) (final concentrations) at 37°C. A: at the indicated times, 100-μl reactions were precipitated with 10% trichloroacetic acid (final concentration) and analyzed by SDS-PAGE and Coomassie blue staining. B: densitometric evaluation of the intensity of the intact trypsinogen bands. Representative gel from three experiments is shown. Error bars were omitted for clarity; the error was within 10% of the mean.
Binding of SPINK1 trypsin inhibitor to p.L104P trypsin.
The physiological inhibitor SPINK1 binds with high affinity to cationic trypsin. In equilibrium-binding assays, we determined a KD value of 3.9 ± 0.4 (SE) pM (n = 3). In contrast, SPINK1 bound to mutant p.L104P with a KD of 284 ± 56 (SE) pM (n = 3), indicating a 70-fold decreased affinity as a result of less favorable S2–P2 interactions (Fig. 7). As a control experiment, we also measured inhibition by the small-molecule trypsin inhibitor benzamidine, which binds to the primary specificity pocket of trypsin and does not make extended subsite contacts. Wild-type and p.L104P mutant trypsin were inhibited by benzamidine with Ki values of 32 and 42 μM, respectively, indicating that mutation p.L104P has no significant effect on the primary binding pocket of trypsin (reviewed but not shown).
Fig. 7.
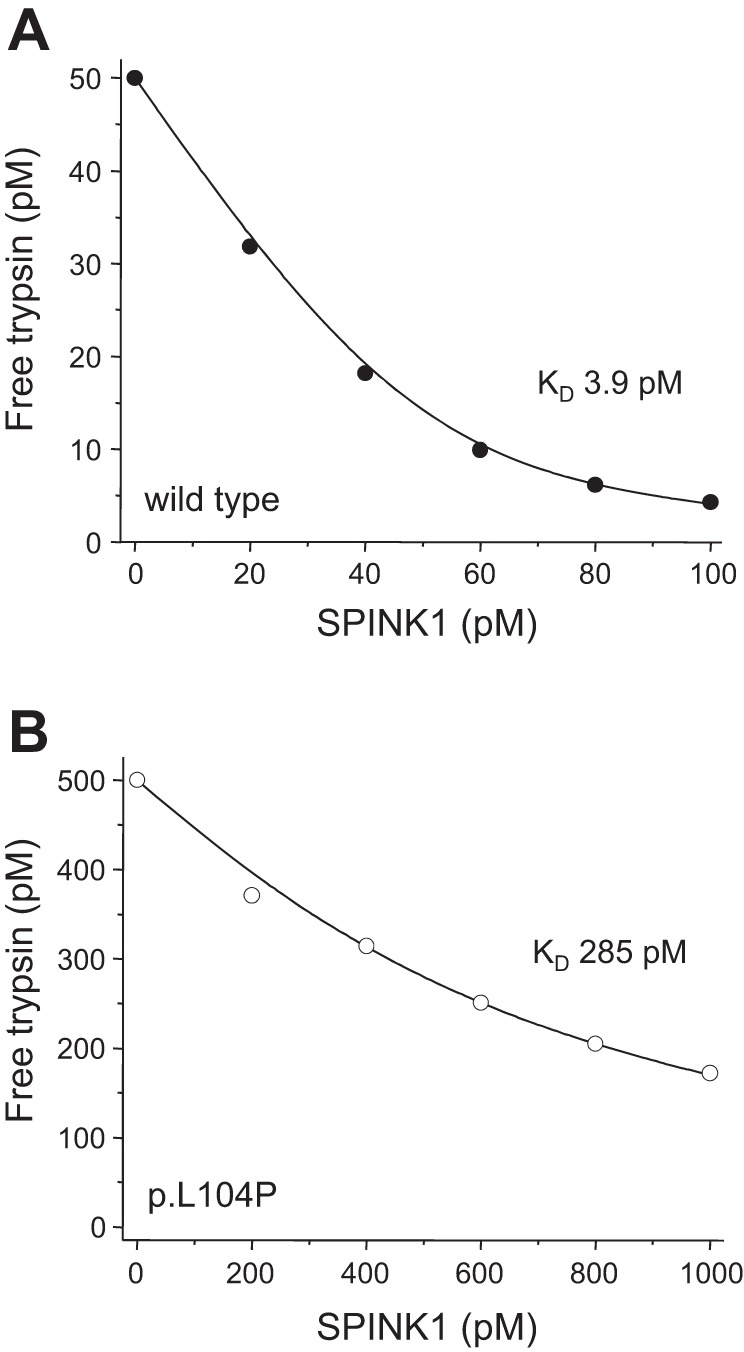
Effect of mutation p.L104P on the binding of serine protease inhibitor Kazal type 1 (SPINK1) to trypsin. Wild-type (50 pM, A) or mutant (500 pM, B) trypsin were incubated for 16 h with the indicated concentrations of SPINK1 inhibitor. Residual trypsin activity was measured, and KD values were calculated as described in materials and methods. Representative binding experiments are show. KD values were calculated from three experiments. See text for errors of the mean.
Secretion of p.L104P trypsinogen.
To study the effect of the p.L104P mutation on cellular secretion of trypsinogen, we transiently transfected HEK 293T cells with expression plasmids carrying wild-type and mutant cationic trypsinogen cDNA. Although 293T cells do not have a regulated secretory pathway as acinar cells, this cell line has been exceedingly useful to characterize mutation-induced misfolding and secretion defects, since these cells do not secrete endogenous proteins to a significant level and, unlike acinar cells, can be transfected using expression plasmids with high efficiency (14, 30). Secretion of cationic trypsinogen in the incubation medium was measured by SDS-PAGE, Western blot, and activity assays (Fig. 8). Remarkably, all three approaches indicated that secretion of the p.L104P mutant was markedly reduced. As judged by the trypsin activity measured in the medium after enteropeptidase activation, the extent of the secretion defect was about ninefold relative to wild type (Fig. 8C).
Fig. 8.

Effect of mutation p.L104P on cellular secretion of trypsinogen. HEK 293T cells were transiently transfected with expression plasmids for wild-type and mutant human cationic trypsinogen, and conditioned media were collected after 48 h. Trypsinogen protein levels were determined by Coomassie blue-stained SDS-polyacrylamide gels (A), by Western blots (B), and by measuring trypsin activity in the medium after activation with enteropeptidase (C). Trypsinogen protein levels secreted in the media were calculated from the enzyme activity (mean ± SD, n = 8, ****P < 0.0001). Representative gel and blot from three experiments are shown.
To address the cause of the secretion defect, we used Western blot to detect trypsinogen in cell lysates. The amount of intracellular trypsinogen was comparable in cells transfected with wild-type and p.L104P trypsinogen constructs, indicating that the mutant enzyme is translated normally but becomes intracellularly retained, most likely due to misfolding in the ER (Fig. 9A). To determine whether aggregation could play a role in intracellular retention, we separated the insoluble trypsinogen fraction by ultracentrifugation of cell lysates. Insoluble trypsinogen was recovered only in the pellet of lysates from cells expressing p.L104P mutant but not from cells expressing wild-type trypsinogen, demonstrating that the mutant enzyme is more prone to aggregation intracellularly (Fig. 9B).
Fig. 9.

Intracellular retention and aggregation of trypsinogen mutant p.L104P. A: lysates (20 μg total protein) of HEK 293T cells expressing wild-type or mutant human cationic trypsinogen were analyzed by Western blotting. B: cell lysates (20 μg) were centrifuged at 50,000 g for 15 min at 4°C. The distribution of trypsinogen between the supernatant and pellet was then analyzed by Western blotting. Representative blots from three experiments are shown.
ER stress in HEK 293T cells expressing p.L104P trypsinogen.
The apparent secretion defect and intracellular aggregation of mutant p.L104P suggest the possibility of misfolding-induced ER stress, as we observed previously with other mutant digestive enzymes (1, 30, 36, 39, 45). To test for ER stress, we examined mRNA levels of the ER chaperon BiP and analyzed the inositol-requiring enzyme 1 (IRE1)-mediated mRNA splicing of the transcription factor XBP1. Both ER stress markers were significantly elevated in cells expressing p.L104P trypsinogen (Fig. 10).
Fig. 10.

Endoplasmic reticulum stress markers in HEK 293T cells expressing wild-type or p.L104P mutant trypsinogen. A: levels of immunoglobulin-binding protein (BiP) mRNA were measured using quantitative reverse transcription-PCR. B and C: splicing of X-box-binding protein 1 (XBP1) mRNA was followed by reverse transcription-PCR using primers that amplify both the spliced and unspliced species. Levels of spliced mRNA (XBP1s) were determined by densitometric analysis. Mean values ± SD (n = 5, each performed in duplicate) are indicated. Significance was tested with 1-way analysis of variance (P < 0.0001) followed by Tukey-Kramer post hoc analysis (***P < 0.001). Representative agarose gel from five experiments is shown.
DISCUSSION
In the present study we investigated the functional effects of the p.L104P PRSS1 variant to determine whether this rare variant might play a pathogenic role in chronic pancreatitis. We focused our attention on this mutation because this is the only natural PRSS1 variant that affects the substrate binding site of trypsin. Specifically, the mutation alters the S2 subsite, a hydrophobic groove that imparts broad P2 specificity to trypsin (9, 11). In addition, the main chain N of position 104 forms an H bond with position 100 that stabilizes the loop between amino acids 97 and 107. Because Pro cannot act as an H donor, the p.L104P mutation may destabilize the Pro97-Asp107 loop and change the S2 site more profoundly and in a somewhat unpredictable manner. Consistent with our predictions of altered subsite specificity, we found that p.L104P trypsin cleaved peptide and protein substrates with reduced efficiency and bound the physiological trypsin inhibitor SPINK1 with decreased affinity. There was no clear relationship between the magnitude of the reduction in activity/binding and the physicochemical properties of the P2 side chains, e.g., both CTRB1 and proCPA1 contain a Ser residue in the P2 position with respect to the activating peptide bond, yet trypsin-mediated activation of CTRB1 was more severely impacted by the p.L104P mutation (20-fold decrease) than activation of proCPA1 (2-fold decrease). On the other hand, the p.L104P mutant exhibited slightly increased autoactivation, indicating that cleavage of the trypsinogen activation peptide by p.L104P trypsin was somewhat improved. The P2 position in the trypsinogen activation peptide corresponds to Asp22. Acidic residues, Asp in particular, are not tolerated well in the otherwise broadly specific S2 subsite of trypsin (21). Accordingly, Asp22 serves as a critical inhibitor of trypsinogen autoactivation, and mutation of Asp22 to Gly (p.D22G) or Ala (p.D22A) results in markedly accelerated autoactivation (13, 21). In case of the p.L104P mutant, the altered S2 subsite seems to accommodate the P2 Asp22 slightly better, resulting in the small increase in autoactivation.
Surprisingly, autoactivation of the p.L104P mutant was not suppressed well by CTRC-mediated degradation, and the mutant autoactivated to higher trypsin levels than wild-type trypsinogen. Trypsinogen degradation requires CTRC-mediated cleavage of the Leu81-Glu82 peptide bond in the calcium-binding loop and a trypsin-mediated autolytic cleavage of the Arg122-Val123 peptide bond (33, 38). The resistance of mutant p.L104P trypsinogen to degradation during autoactivation was due to slow cleavage at the Arg122 autolytic site, whereas CTRC cleavage after Leu81 was unchanged.
The increased autoactivation of mutant p.L104P in the presence of CTRC combined with its decreased inhibition by SPINK1 should allow this variant to activate sooner and to higher trypsin levels than its wild-type counterpart inside the pancreas. Although active p.L104P trypsin may not be as harmful because of its defective cleavage of most protein substrates, it can still promote autoactivation of wild-type cationic trypsinogen produced from the wild-type allele in heterozygous carriers. Thus, the biochemical basis for a potentially strong pathogenic effect of the p.L104P mutation has been identified. The caveat to this conclusion is that mutant p.L104P needs to be produced and secreted to normal levels, which turned out not to be the case.
Unexpectedly, we found that the p.L104P mutation caused intracellular retention and aggregation of trypsinogen in transfected 293T cells. Consistent with an interpretation of mutation-induced misfolding, we observed elevated ER stress markers in cells expressing the p.L104P mutant. Thus, the dominant effect of mutation p.L104P appears to be not biochemical but cell biological. In other words, the mutation belongs to the class of variants that act through the misfolding-dependent pathway rather than the trypsin-dependent pathway. Although the exact mechanism by which mutation p.L104P causes misfolding is not readily apparent, the loss of a potentially important H bond that stabilizes the Pro97-Asp107 loop is likely a contributing factor.
Mutation-induced misfolding and consequent perturbations of ER homeostasis have been conclusively linked with chronic pancreatitis. First, mutations p.R116C and p.C139S in PRSS1 were found to undergo misfolding with consequent ER stress, suggesting that some trypsinogen variants might exert their effects via a mechanism that is unrelated to trypsin activity (14). This conclusion was later supported by analysis of 13 rare PRSS1 variants presumed to cause pancreatitis (30). Among these, five variants exhibited strong (p.D100H, p.C139F) or moderate (p.K92N, p.S124F, p.G208A) secretion defects suggestive of misfolding, although ER stress has not been studied. It is interesting to note that misfolding PRSS1 variants were typically found in cases of sporadic chronic pancreatitis with no family history, suggesting a milder pathogenic effect relative to the trypsin-dependent high-penetrance mutations such as p.R122H. Additional evidence for the role of misfolding in chronic pancreatitis came from studies on the CPA1 gene that demonstrated that loss-of-function CPA1 variants are associated with early onset disease and represent strong risk factors (45). The majority of CPA1 variants found in patients exhibited severely reduced cellular secretion due to intracellular retention and degradation. With the use of the relatively more frequent p.N256K variant as a test case, strong ER stress was demonstrated in AR42J acinar cells expressing mutant CPA1 (45). Besides PRSS1 and CPA1 mutations, some CTRC variants were also shown to result in misfolding and ER stress (1, 39). However, in contrast to trypsinogen and procarboxypeptidase that are produced in high abundance by the pancreas, the lower expression levels of CTRC make it questionable whether this mechanism is relevant to the pathogenic action of CTRC variants. Similarly, misfolding SPINK1 variants are unlikely to elicit ER stress due to low expression levels (4, 5, 18). More recently, association of chronic pancreatitis with a novel duplicated hybrid allele of the CEL gene encoding carboxyl ester lipase was described (12). Although not demonstrated experimentally, it appears likely that this newly formed lipase molecule could also result in misfolding and ER stress. Finally, a rare pancreatic triglyceride lipase (PNLIP) variant was found in the homozygous state in two brothers with lipase deficiency (2). Functional analysis revealed that the p.T221M variant caused intracellular retention, loss of secretion, and strong ER stress (36). Although a diagnosis of chronic pancreatitis has not been established in the published carriers, signs of pancreatic insufficiency have been documented, suggesting that ER stress-related pathology may have contributed to their disease.
The mechanism by which ER stress increases risk for chronic pancreatitis is not completely understood. Chronic unresolved ER stress promotes apoptosis through upregulation of the proapoptotic transcription factor CHOP, and this pathway may contribute to parenchymal atrophy and subsequent fibrosis (41, 48). Proinflammatory effects of ER stress have been also documented. Thus, translational attenuation by PERK-dependent eIF2α phosphorylation results in a relative increase of NF-κB vs. its inhibitor IκB. Alternatively, NF-κB can be activated through degradation of IκB as a result of the recruitment and activation of IκB kinase by the complex of IRE1 and NF receptor-associated factor 2 (TRAF2). The IRE1-TRAF2 complex can also activate the c-Jun NH2-terminal kinase pathway leading to activator protein-1-mediated transcriptional activation of proinflammatory genes (41, 48). Whether or not these pathways are relevant to the development of chronic pancreatitis remains to be determined.
In conclusion, the observations presented here argue that the p.L104P variant of human cationic trypsinogen increases risk for chronic pancreatitis primarily through misfolding and ER stress. Although biochemical changes also indicate a strong propensity for increased trypsinogen activation, the secretion defect secondary to misfolding would diminish a trypsin-dependent pathogenic effect.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-058088, R01-DK-082412, and R01-DK-095753 to M. Sahin-Tóth. Work in the Szeged laboratory was supported by Hungarian Scientific Research Fund (OTKA) Grant K116634.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.B., P.H., and M.S.-T. conception and design of research; A.B. performed experiments; A.B. and M.S.-T. analyzed data; A.B., P.H., and M.S.-T. interpreted results of experiments; A.B. and M.S.-T. prepared figures; A.B. and M.S.-T. drafted manuscript; A.B., P.H., and M.S.-T. edited and revised manuscript; A.B., P.H., and M.S.-T. approved final version of manuscript.
ACKNOWLEDGMENTS
András Szabó is acknowledged for help, advice, and reagents.
REFERENCES
- 1.Beer S, Zhou J, Szabó A, Keiles S, Chandak GR, Witt H, Sahin-Tóth M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 62: 1616–1624, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behar DM, Basel-Vanagaite L, Glaser F, Kaplan M, Tzur S, Magal N, Eidlitz-Markus T, Haimi-Cohen Y, Sarig G, Bormans C, Shohat M, Zeharia A. Identification of a novel mutation in the PNLIP gene in two brothers with congenital pancreatic lipase deficiency. J Lipid Res 55: 307–312, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchini EP, Louvain VB, Marque PE, Juliano MA, Juliano L, Le Bonniec BF. Mapping of the catalytic groove preferences of factor Xa reveals an inadequate selectivity for its macromolecule substrates. J Biol Chem 277: 20527–20534, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Boulling A, Keiles S, Masson E, Chen JM, Férec C. Functional analysis of eight missense mutations in the SPINK1 gene. Pancreas 41: 329–330, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Boulling A, Le Maréchal C, Trouvé P, Raguénès O, Chen JM, Férec C. Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. Eur J Hum Genet 15: 936–942, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Chang YT, Wei SC, LPC, Tien YW, Jan IS, Su YN, Wong JM, Chang MC. Association and differential role of PRSS1 and SPINK1 mutation in early-onset and late-onset idiopathic chronic pancreatitis in Chinese subjects. Gut 58: 885, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Chen JM, Férec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet 10: 63–87, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 339: 653–658, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Dementiev A, Simonovic M, Volz K, Gettins PGW. Canonical inhibitor-like interactions explain reactivity of alpha1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J Biol Chem 278: 37881–37887, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Derikx MH, Kovacs P, Scholz M, Masson E, Chen JM, Ruffert C, Lichtner P, Te Morsche RHM, Cavestro GM, PanEuropean Working group on Alcoholic Chronic Pancreatitis members and collaborators, Férec C, Drenth JPH, Witt H, Rosendahl J. Polymorphisms at PRSS1-PRSS2 and CLDN2-MORC4 loci associate with alcoholic and non-alcoholic chronic pancreatitis in a European replication study. Gut 64: 1426–1433, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Djie MZ, Le Bonniec BF, Hopkins PC, Hipler K, Stone SR. Role of the P2 residue in determining the specificity of serpins. Biochemistry 35: 11461–11469, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Fjeld K, Weiss FU, Lasher D, Rosendahl J, Chen JM, Johansson BB, Kirsten H, Ruffert C, Masson E, Steine SJ, Bugert P, Cnop M, Grützmann R, Mayerle J, Mössner J, Ringdal M, Schulz HU, Sendler M, Simon P, Sztromwasser P, Torsvik J, Scholz M, Tjora E, Férec C, Witt H, Lerch MM, Njølstad PR, Johansson S, Molven A. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat Genet 47: 518–522, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geisz A, Hegyi P, Sahin-Tóth M. Robust autoactivation, chymotrypsin C independence and diminished secretion define a subset of hereditary pancreatitis-associated cationic trypsinogen mutants. FEBS J 280: 2888–2899, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kereszturi E, Szmola R, Kukor Z, Simon P, Weiss FU, Lerch MM, Sahin-Tóth M. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum Mutat 30: 575–582, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Király O, Boulling A, Witt H, Maréchal CL, Chen JM, Rosendahl J, Battaggia C, Wartmann T, Sahin-Tóth M, Férec C. Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary Pancreatitis. Hum Mutat 28: 469–476, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Király O, Guan L, Sahin-Tóth M. Expression of recombinant proteins with uniform N-termini. Methods Mol Biol 705: 175–194, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Király O, Guan L, Szepessy E, Tóth M, Kukor Z, Sahin-Tóth M. Expression of human cationic trypsinogen with an authentic N terminus using intein-mediated splicing in aminopeptidase P deficient Escherichia coli. Protein Expr Purif 48: 104–111, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Király O, Wartmann T, Sahin-Tóth M. Missense mutations in pancreatic secretory trypsin inhibitor (SPINK1) cause intracellular retention and degradation. Gut 56: 1433–1438, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lengyel Z, Pál G, Sahin-Tóth M. Affinity purification of recombinant trypsinogen using immobilized ecotin. Protein Expr Purif 12: 291–294, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Németh BC, Sahin-Tóth M. Human cationic trypsinogen (PRSS1) variants and chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 306: G466–G473, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemoda Z, Sahin-Tóth M. The tetra-aspartate motif in the activation peptide of human cationic trypsinogen is essential for autoactivation control but not for enteropeptidase recognition. J Biol Chem 280: 29645–29652, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemoda Z, Sahin-Tóth M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J Biol Chem 281: 11879–11886, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pál G, Szilágyi L, Gráf L. Stable monomeric form of an originally dimeric serine proteinase inhibitor, ecotin, was constructed via site directed mutagenesis. FEBS Lett 385: 165–170, 1996. [DOI] [PubMed] [Google Scholar]
- 24.Pallagi P, Venglovecz V, Rakonczay Z, Borka K, Korompay A, Ozsvári B, Judák L, Sahin-Tóth M, Geisz A, Schnúr A, Maléth J, Takács T, Gray MA, Argent BE, Mayerle J, Lerch MM, Wittmann T, Hegyi P. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl− channels and luminal anion exchangers. Gastroenterology 141: 2228–2239, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosendahl J, Landt O, Bernadova J, Kovacs P, Teich N, Bödeker H, Keim V, Ruffert C, Mössner J, Kage A, Stumvoll M, Groneberg D, Krüger R, Luck W, Treiber M, Becker M, Witt H. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut 62: 582–592, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvári B, Landt O, Schulz HU, Gress TM, Pfützer R, Löhr M, Kovacs P, Blüher M, Stumvoll M, Choudhuri G, Hegyi P, te Morsche RHM, Drenth JPH, Truninger K, Macek M, Puhl G, Witt U, Schmidt H, Büning C, Ockenga J, Kage A, Groneberg DA, Nickel R, Berg T, Wiedenmann B, Bödeker H, Keim V, Mössner J, Teich N, Sahin-Tóth M. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 40: 78–82, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahin-Tóth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem 275: 22750–22755, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Sahin-Tóth M, Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 278: 286–289, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27: 157–162, 1967. [DOI] [PubMed] [Google Scholar]
- 30.Schnúr A, Beer S, Witt H, Hegyi P, Sahin-Tóth M. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut 63: 337–343, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, Braganza J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med 339: 645–652, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Szabó A, Héja D, Szakács D, Zboray K, Kékesi KA, Radisky ES, Sahin-Tóth M, Pál G. High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4' acidic residues. J Biol Chem 286: 22535–22545, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabó A, Sahin-Tóth M. Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J Biol Chem 287: 20701–20710, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szabó A, Sahin-Tóth M. Determinants of chymotrypsin C cleavage specificity in the calcium-binding loop of human cationic trypsinogen. FEBS J 279: 4283–4292, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabó A, Salameh MA, Ludwig M, Radisky ES, Sahin-Tóth M. Tyrosine sulfation of human trypsin steers S2' subsite selectivity towards basic amino acids. PloS One 9: e102063, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szabó A, Xiao X, Haughney M, Spector A, Sahin-Tóth M, Lowe ME. A novel mutation in PNLIP causes pancreatic triglyceride lipase deficiency through protein misfolding. Biochim Biophys Acta 1852: 1372–1379, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szmola R, Bence M, Carpentieri A, Szabó A, Costello CE, Samuelson J, Sahin-Tóth M. Chymotrypsin C is a co-activator of human pancreatic procarboxypeptidases A1 and A2. J Biol Chem 286: 1819–1827, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szmola R, Sahin-Tóth M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: Identity with Rinderknecht's enzyme Y. Proc Natl Acad Sci USA 104: 11227–11232, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szmola R, Sahin-Tóth M. Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut 59: 365–372, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teich N, Bauer N, Mössner J, Keim V. Mutational screening of patients with nonalcoholic chronic pancreatitis: identification of further trypsinogen variants. Am J Gastroenterol 97: 341–346, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett 332: 249–264, 2013. [DOI] [PubMed] [Google Scholar]
- 42.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14: 141–145, 1996. [DOI] [PubMed] [Google Scholar]
- 43.Whitcomb DC, LaRusch J, Krasinskas AM, Klei L, Smith JP, Brand RE, Neoptolemos JP, Lerch MM, Tector M, Sandhu BS, Guda NM, Orlichenko L, Alzheimer's Disease Genetics Consortium, Alkaade S, Amann S.T, Anderson MA, Baillie J, Banks PA, Conwell D, Coté GA, Cotton PB, DiSario J, Farrer LA, Forsmark CE, Johnstone M, Gardner TB, Gelrud A, Greenhalf W, Haines JL, Hartman DJ, Hawes RA, Lawrence C, Lewis M, Mayerle J, Mayeux R, Melhem NM, Money ME, Muniraj T, Papachristou GI, Pericak-Vance MA, Romagnuolo J, Schellenberg GD, Sherman S, Simon P, Singh VP, Slivka A, Stolz D, Sutton R, Weiss FU, Wilcox CM, Zarnescu NO, Wisniewski SR, O'Connell MR, Kienholz ML, Roeder K, Barmada MM, Yadav D, Devlin B. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis Nat Genet 44: 1349–1354, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 132: 1557–1573, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Witt H, Beer S, Rosendahl J, Chen JM, Chandak GR, Masamune A, Bence M, Szmola R, Oracz G, Macek M, Bhatia E, Steigenberger S, Lasher D, Bühler F, Delaporte C, Tebbing J, Ludwig M, Pilsak C, Saum K, Bugert P, Masson E, Paliwal S, Bhaskar S, Sobczynska-Tomaszewska A, Bak D, Balascak I, Choudhuri G, Nageshwar Reddy D, Rao GV, Thomas V, Kume K, Nakano E, Kakuta Y, Shimosegawa T, Durko L, Szabó A, Schnúr A, Hegyi P, Rakonczay Z, Pfützer R, Schneider A, Groneberg DA, Braun M, Schmidt H, Witt U, Friess H, Algül H, Landt O, Schuelke M, Krüger R, Wiedenmann B, Schmidt F, Zimmer KP, Kovacs P, Stumvoll M, Blüher M, Müller T, Janecke A, Teich N, Grützmann R, Schulz HU, Mössner J, Keim V, Löhr M, Férec C, Sahin-Tóth M. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet 45: 1216–1220, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witt H, Luck W, Hennies HC, Claßen M, Kage A, Laß U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 25: 213–216, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Witt H, Sahin-Tóth M, Landt O, Chen JM, Kähne T, Drenth JP, Kukor Z, Szepessy E, Halangk W, Dahm S, Rohde K, Schulz HU, Le Maréchal C, Akar N, Ammann RW, Truninger K, Bargetzi M, Bhatia E, Castellani C, Cavestro GM, Cerny M, Destro-Bisol G, Spedini G, Eiberg H, Jansen JBMJ, Koudova M, Rausova E, Macek M, Malats N, Real FX, Menzel HJ, Moral P, Galavotti R, Pignatti PF, Rickards O, Spicak J, Zarnescu NO, Böck W, Gress TM, Friess H, Ockenga J, Schmidt H, Pfützer R, Löhr M, Simon P, Weiss FU, Lerch MM, Teich N, Keim V, Berg T, Wiedenmann B, Luck W, Groneberg DA, Becker M, Keil T, Kage A, Bernardova J, Braun M, Güldner C, Halangk J, Rosendahl J, Witt U, Treiber M, Nickel R, Férec C. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet 38: 668–673, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 454: 455–462, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]