

Abstract
Although significant research data exist on the pathophysiology of nonalcoholic steatohepatitis (NASH), finding an efficient treatment regimen for it remains elusive. The present study used sparstolonin B (SsnB), a novel TLR4 antagonist derived from the Chinese herb Sparganium stoloniferum, as a possible drug to mitigate early inflammation in NASH. This study used an early steatohepatitic injury model in high-fat-fed mice with CYP2E1-mediated oxidative stress as a second hit. SsnB was administered for 1 wk along with bromodichloromethane (BDCM), an inducer of CYP2E1-mediated oxidative stress. Results showed that SsnB administration attenuated inflammatory morphology and decreased elevation of the liver enzyme alanine aminotransferase (ALT). Mice administered SsnB also showed decreased mRNA expression of proinflammatory cytokines TNF-α, IFN-γ, IL-1β, and IL-23, while protein levels of both TNF-α and IL-1β were significantly decreased. SsnB significantly decreased Kupffer cell activation as evidenced by reduction in CD68 and monocyte chemoattractant protein-1 (MCP1) mRNA and protein levels with concomitant inhibition of macrophage infiltration in the injured liver. Mechanistically, SsnB decreased TLR4 trafficking to the lipid rafts, a phenomenon described by the colocalization of TLR4 and lipid raft marker flotillin in tissues and immortalized Kupffer cells. Since we have shown previously that NADPH oxidase drives TLR4 trafficking in NASH, we studied the role of SsnB in modulating this pathway. SsnB prevented NADPH oxidase activation in vivo and in vitro as indicated by decreased peroxynitrite formation. In summary, the present study reports a novel use of the TLR4 antagonist SsnB in mitigating inflammation in NASH and in parallel shows a unique molecular mechanism of decreasing nitrative stress.
Keywords: SsnB, inflammation, NADPH oxidase, p47phox, peroxynitrite
nonalcoholic steatohepatitis (NASH), a hepatic manifestation of metabolic syndrome, arises from an underlying condition of obesity (22).
After nearly three decades there remains a void of drugs approved for the treatment of NASH. Although sufficient progress in the understanding of pathophysiology, diagnosis, and stages of the disease has been made, a treatment regimen to cure this metabolic disease remains a challenge. Several treatment modalities have been tried, yet no single approach has been found to be effective in treating NASH (10). Lipid-lowering drugs like fibrates and statins have some effect, while insulin sensitizers (metformin, thiazolidinediones), endocannabinoid receptor agents, ursodeoxycholic acid, and green tea polyphenols have mixed effects (12). Results from both preclinical and clinical trials distinctly point to a combinatorial approach (caloric restriction, anti-inflammatory, and antioxidant) as a therapeutic remedy for NASH (33, 40, 49). Therefore there is a dire need for sustained research and drug discovery efforts that aim at identifying new targets for therapy and eventually combine those that target concomitant and synergistic pathways of NASH pathogenesis.
As for alcoholic steatohepatitis, Toll-like receptors (TLRs) have been shown to be involved in the pathogenesis of NASH, as reviewed recently (36, 50). A pioneering study by Farhadi et al. (16) reported endotoxemia resulting from possible gut leakiness as a cause for NASH. Later, many reports emerged about the involvement of endotoxin from the gut as a cause for activation of the TLR4 receptors and the downstream inflammatory pathways (1, 16, 18–20, 45, 51, 54, 56, 57, 59, 60, 63, 64). We have recently shown (8) that peroxynitrite, a nitrative species formed from the reaction between superoxide and nitric oxide, is responsible for TLR4 recruitment in lipid rafts. Lipid raft trafficking of TLR4 remains a significant step in TLR4 signaling and inflammation (37, 62). Upon binding to the TLR4 ligand, it is rapidly assembled in the lipid-containing domains, an event that is facilitated by NADPH oxidase (62). Our previously published observations (8) point to the role of peroxynitrite in contributing to the trafficking of TLR4 in rafts primarily following NADPH oxidase activation in NASH livers. Abrogation of TLR4 trafficking significantly decreased inflammation in NASH models and had improved histological outcomes (8). Several research reports in the past have described TLR4 antagonists and their effectiveness in attenuating TLR4-based inflammation in disease models (44). Unfortunately, none could be recommended for therapeutic use in humans. However, the search for an effective TLR4 antagonist continues owing to the importance of the TLR4 pathway in a myriad of diseases.
Extending this endeavor in search of an effective TLR4 antagonist, our collaborators have recently published several research studies that report the effective TLR4-antagonizing role of sparstolonin B (SsnB), a natural product derivative (2, 24, 26–31). SsnB, an isocoumarin, was isolated from a Chinese herb, Sparganium stoloniferum; its structure was determined by NMR spectroscopy and X-ray crystallography (27). In light of the above evidence of SsnB being a potent antagonist for TLR4-induced inflammation and tissue injury, we tested the hypothesis that administration of SsnB in murine models of NASH and immortalized Kupffer cells will attenuate early inflammation and tissue injury induced by TLR4 activation. In the present study we used a rodent model of NASH in which oxidative stress mediated by CYP2E1, lipotoxicity, and innate immune mediators were used as “second/multiple hits” to cause NASH. Results showed that SsnB significantly attenuated TLR4 trafficking to the lipid rafts, a significant event in TLR4 activation. The effects of SsnB on TLR4 trafficking were also correlated with a significant inhibition of NADPH oxidase activation and peroxynitrite formation, an event that was reported by us to be upstream of TLR4 activation in NASH though TLR4-induced NADPH oxidase activation.
MATERIALS AND METHODS
Cell Culture
A rat immortalized Kupffer cell line (Applied Biological Materials) was maintained in complete Dulbecco's modified Eagle's medium (DMEM; Corning) supplemented with 10% fetal bovine serum (FBS; Atlas Biologicals) and 1× penicillin-streptomycin solution (GIBCO, Life Technologies) and grown at 37°C in a humidified incubator with 5% CO2. HBSS buffer and tissue culture plastic wares were purchased from Corning. SsnB compound was provided by D. Fan. A stock solution of SsnB was prepared in dimethyl sulfoxide (DMSO). The final concentration of SsnB on cells was 100 μg/ml, 0.1% DMSO. The cells were plated on 12-well plates (100,000/well) with DMEM supplemented with 10% FBS. Before the cells were treated with lipopolysaccharide (LPS, 50 ng/ml; Sigma), leptin (100 ng/ml; BioVision), and SsnB (100 μg/ml), cells were serum starved (DMEM with 0.25% FBS) overnight. All treatments were given for 24 h in 0.25% FBS-containing DMEM medium. Cells treated with 0.1% DMSO were used as control. Cells treated with leptin and LPS also had 0.1% DMSO. The cell supernatant was collected for nitiric oxide quantification. Cells were plated on coverslips by putting the coverslips on each well of 12-well plates maintaining the aforementioned conditions, and the cells adhered on coverslips were used for immunofluorescence dual-labeling staining after completion of the treatment. Primary mouse (C57BL/6) hepatic macrophages (ScienCell Research Lab, Carlsbad, CA) were cultured in macrophage-specific MaM medium supplemented with macrophage growth supplement (MaGS), FBS, and penicillin-streptomycin solution (P/S) (ScienCell Research Lab), as instructed by the manufacturer, on poly-l-lysine (ScienCell Research Lab)-coated 12-well plates and incubated at 37°C in a humidified incubator with 5% CO2 to initiate the culture. The cell culture medium was replaced with fresh medium the next morning, and two different cell treatment groups were treated with LPS or LPS+SsnB, respectively, maintaining the same concentration and incubation time described above. Cells treated with 0.1% DMSO were used as control.
Mouse Models
Pathogen-free male C57BL/6J background mice (purchased from Jackson Laboratories, Bar Harbor, ME) were housed one per cage with a 12:12-h light-dark cycle at 23–24°C and ab libitum access to food and water. These mice were fed with 60% kcal high-fat diet (purchased from Research Diets, New Brunswick, NJ) from 6 wk until 16 wk to generate a model of steatosis (S group). Another group of these high-fat-diet-fed mice were administered bromodichloromethane (BDCM; 1 mmol/kg, diluted in corn oil) intraperitoneally twice a week for 1 wk to generate a toxin-induced early steatohepatitic injury model of mice (SH group). BDCM and corn oil were purchased from Sigma-Aldrich (St. Louis, MO). A set of high-fat-diet-fed, BDCM-treated mice were administered SsnB (3 mg/kg body wt) intraperitoneally twice a week for 1 wk (SH+SsnB group). A group of steatotic mice were also administered SsnB (3 mg/kg body wt) intraperitoneally twice a week for 1 wk (S+SsnB group). Age-matched lean control mice (LC group) were fed chow diet having 10% kcal fat. Another group of chow diet-fed lean mice were treated with the same dosage of BDCM as above for 1 wk (LC+BDCM group). All animals were treated in strict accordance with the US Public Health Service Policy for Humane Care and Use of Laboratory Animals and local IACUC standards. All experiments were approved by the institutional review boards at National Institute of Environmental Health Sciences, Duke University, and the University of South Carolina at Columbia. After completion of the treatment, mice belonging to all groups were euthanized for liver tissues and serum samples for further experiments.
Laboratory Analysis
Immunohistochemistry.
Mouse livers were collected and fixed in 10% neutral buffered formalin (Sigma-Aldrich). These formalin-fixed tissues were paraffin embedded and cut in 5-μm-thick sections. With a standard protocol (53), these liver sections were deparaffinized. The deparaffinized sections were subjected to epitope retrieval, which was performed with an epitope retrieval solution and steamer (IHC World, Woodstock, MD) according to the manufacturer's instructions. The endogenous peroxidases were blocked by 3% H2O2. Anti-mouse primary antibodies such as anti-IL-1β, anti-TNF-α, anti-monocyte chemoattractant protein-1 (MCP1), anti-F4/80, and anti-3-nitrotyrosine (Abcam, Cambridge, MA) were used in recommended dilutions. According to manufacturer's protocols (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA), biotinylated conjugated secondary antibody (species specific) and streptavidin conjugated with HRP were used to perform antigen-specific immunohistochemistry. 3,3′-Diaminobenzidine (Sigma-Aldrich) was used as chromogenic substrate. Mayer's hematoxylin (Sigma-Aldrich) was used as counterstain. phosphate-buffered saline was used for washing three times between the steps. Finally, the sections were mounted in Simpo-Mount (GBI Laboratories, Mukilteo, WA) and observed under a ×20 objective with an Olympus BX51 microscope (Olympus America, Center Valley, PA). Morphometric analysis was done with cellSens software from Olympus America.
Hematoxylin and eosin staining.
Formalin-fixed tissues were paraffin embedded. Liver tissue sections were stained with hematoxylin and eosin by the Instrumentation Resource Facility, University of South Carolina School of Medicine.
Serum alanine aminotransferase.
Serum samples collected from mice were subjected to alanine aminotransferase (ALT) analysis (Discovery Life Sciences) according to manufacturer's instructions.
Nitric oxide quantification assay.
After the rat Kupffer cells were treated with LPS and LPS+SsnB (described above), the cell supernatants were collected. The supernatant collected from the wells, where cells were not treated with LPS or LPS+SsnB, served as a control. Nitrite was measured in these samples with the Griess Reagent System (Promega) according to the manufacturer's instructions.
ELISA.
After the primary mouse hepatic macrophages were treated with LPS and LPS+SsnB (described above), the cell supernatants were collected. The supernatant collected from the wells, where cells were not treated with LPS or LPS+SsnB, served as a control. TNF-α concentration was measured in these samples with a mouse TNF ELISA kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions.
Immunofluorescence dual labeling.
IN VIVO.
Formalin-fixed, paraffin-embedded tissue sections were subjected to deparaffinization according to standard instructions. Epitope retrieval of the deparaffinized sections was done with an epitope retrieval solution and steamer (IHC World) according to the manufacturer's protocol. The primary antibodies anti-gp-91phox, anti-p47phox, flottilin1, and TLR4 (purchased from Santa Cruz Biotechnology, Santa Cruz, CA, and Abcam) and used at recommended dilutions. Species-specific anti-IgG secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen) were used against anti-p47phox, anti-flottilin1, and Alexa Fluor 633 (Invitrogen) was used against anti-gp91phox and TLR4. The sections were mounted in a ProLong Gold antifade reagent with DAPI (Life Technologies, Carlsbad, CA). Images were taken under ×20 and ×60 oil objectives with an Olympus BX51 microscope.
IN VITRO.
After completion of the treatments under serum-starved conditions as in Cell Culture, cells attached on coverslips were fixed with 10% neutral buffered saline. After the cells were washed with PBS containing 0.1% Triton X (Sigma), they were blocked with 3% BSA, 0.2% Tween (Fisher), 10% FBS in PBS. Cells were incubated with primary antibodies anti-gp91phox, anti-p47phox, anti-TLR4 (described above) and anti-flotillin 2 (Abcam), followed by species-specific Alexa Fluor 633 and 488 (described above), for immunofluorescence dual-labeling staining. Alexa Fluor 633 was used against anti-TLR4 and anti-gp91phox. Alexa Fluor 488 was used against anti-flottilin2 and anti-p47phox. The stained cells attached on the coverslips were mounted on slides with ProLong Gold antifade reagent with DAPI (Life Technologies) and viewed under ×40 and ×60 oil objectives with an Olympus BX51 microscope.
Quantitative real-time polymerase chain reaction.
Gene expression (mRNA) levels in the mouse liver tissue samples and mouse primary hepatic macrophages were measured by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR). Total RNA was isolated from liver tissues and mouse primary hepatic macrophages with TRIzol reagent (Life Technologies). TRIzol reagent was used to lyse the tissue; RNA extraction and purification were carried out with RNeasy mini kit columns (Qiagen, Valencia, CA) according to the manufacturer's protocol. One microgram of purified RNA was converted to cDNA with an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). qRT-PCR was performed with gene-specific primers, a CFX96 thermal cycler (Bio-Rad, Hercules, CA), and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Threshold cycle (Ct) values for genes of interest were normalized against 18S (internal control) values in the same sample. Reaction was carried out in triplicate for each gene and each tissue sample. The relative fold change was calculated by the 2−ΔΔCt method. The sequences for the primers used for real-time PCR are provided in Table 1.
Table 1.
Mouse-specific primers
| Gene | Primer Sequence |
|---|---|
| IL-1β | Sense: CCTCGGCCAAGACAGGTCGC |
| Antisense: TGCCCATCAGAGGCAAGGAGGA | |
| TNF-α | Sense: CAACGCCCTCCTGGCCAACG |
| Antisense: TCGGGGCAGCCTTGTCCCTT | |
| IFN-γ | Sense: TGCGGGGTTGTATCTGGGGGT |
| Antisense: GCGCTGGCCCGGAGTGTAG | |
| IL-23 | Sense: AAAGGATCCGCCAAGGTCTG |
| Antisense: GCAGGCTCCCCTTTGAAGAT | |
| CD68 | Sense: GCTACATGGCGGTGGAGTACAA |
| Antisense: ATGATGAGAGGCAGCAAGATGG | |
| MCP1 | Sense: CACAGTTGCCGGGCTGGAGCAT |
| Antisense: GTAGCAGCAGGTGAGTGGGGC | |
| p47phox | Sense: GGTCGACCATCCGCAACGCA |
| Antisense: TGTGCCATCCGTGCTCAGCG | |
| TLR4 | Sense: GGAGTGCCCCGCTTTCACCTC |
| Antisense: ACCTTCCGGCTCTTGTGGAAGC |
MCP1, monocyte chemoattractant protein-1; TLR4, Toll-like receptor 4.
MicroRNA21 expression levels in liver tissues.
Liver tissues were homogenized in QIAzol reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions to isolate the total microRNA (miRNA). The purification was done with miRNeasy mini kit columns (Qiagen). One hundred nanograms of purified miRNA was converted to cDNA with the miScript cDNA synthesis kit (Qiagen) according to the manufacturer's protocol. qRT-PCR was performed with miRNA-specific primers (Qiagen), miScript SYBR Green PCR Master Mix (Qiagen), and a CFX96 thermal cycler (Bio-Rad Laboratories, Hercules, CA). Ct values for the selected gene were normalized against RNU6-2 (internal miRNA expression control) values in the same sample.
Western blotting.
In the presence of phosphatase and protease inhibitors (Pierce, Rockford, IL), 30 mg of each liver tissue was homogenized in 500 μl of RIPA buffer (Sigma-Aldrich) with dounce homogenizer. The homogenate was centrifuged, and the supernatant was collected for further experiments. Twenty-five micrograms of each protein sample was loaded on 4–12% Bis-Tris gradient gel (Invitrogen) for SDS PAGE. With precut nitrocellulose/filter paper sandwiches (Bio-Rad Laboratories) and the Trans-Blot Turbo transfer system (Bio-Rad), proteins were transferred to nitrocellulose membrane. Blots were blocked with 5% nonfat milk solution. Primary antibodies against phosphatase and tensin homolog (PTEN), β-actin (purchased from Abcam), and TLR4 (Santa Cruz Biotechnology) were used at recommended dilutions and incubated overnight at 4°C. Species-specific secondary antibodies conjugated with HRP (Abcam) were used, and the blots were incubated at room temperature for 2 h. Pierce ECL Western Blotting substrate (Thermo Fisher Scientific, Rockford, IL) was used. The blot was developed with BioMax MS Films and cassettes (with intensifying screen; Kodak). Densitometry analysis of the images was done with Lab Image 2006 Professional 1D gel analysis software from KAPELAN Bio-Imaging Solutions (Liepzig, Germany).
Statistical Analyses
Statistical analysis was performed by analysis of variance (ANOVA), which was followed by Bonferroni post hoc correction for intergroup comparisons. P < 0.05 and P < 0.01 are considered statistically significant.
RESULTS
SsnB Administration Attenuates Early Steatohepatitic Injury in Obese Mice
Feeding a high-fat diet to C57BL6/J mice for 4 wk starting from 8 wk of postgestational age causes insulin and leptin resistance and steatosis but does not cause inflammation, tissue injury, and fibrosis (5). NASH can be a result of multiple hits that may include oxidative stress, increased CYP2E1 activity, cytokine release, and/or altered metabolism (58). We used a toxin-induced steatohepatitis and lipotoxicity model in mice to show the therapeutic effects of SsnB (53). Results showed that SsnB administration markedly decreased necrosis, ballooning, Mallory-Denk bodies, and inflammatory foci in the steatohepatitis (SH) group (Fig. 1A). SsnB administration significantly decreased the levels of serum ALT compared with the SH group, but levels were significantly higher than in the steatosis (S) group (Fig. 1B) (P < 0.05).
Fig. 1.

Sparstolonin B (SsnB) administration attenuates early steatohepatitic injury in obese mice. A: hematoxylin and eosin-stained paraffin-embedded liver tissue sections of steatosis (S; i), steatohepatitic injury (SH; ii), and SH+SsnB (iii) mice. Images were taken at ×10 magnification. B: serum levels of alanine aminotransferase (ALT) from S, SH, and SH+SsnB groups. *P < 0.05.
NASH is known to have a significant proinflammatory phase primarily arising from activation of pattern recognition receptors (PRRs) in the liver primarily from higher endotoxin levels, HMGB1, or other endogenous ligands (4, 45). To study the effect of SsnB administration to SH mice, liver proinflammatory cytokine gene and protein levels were analyzed by qRT-PCR and immunohistochemistry. Results showed that, as expected, mRNA expression of TNF-α, IFN-γ, and IL-1β, which are proinflammatory in nature, was significantly elevated in the SH group compared with the S group (Fig. 2A) (P < 0.05). IL-23 is known as a marker for M1 polarization (38, 52). Results showed that mRNA expression of IL-23 also was significantly upregulated in the SH group compared with the S group (Fig. 2A) (P < 0.05). SsnB administration significantly decreased the mRNA expression of proinflammatory markers TNF-α, IFN-γ, and IL-1β and macrophage polarization marker IL-23 compared with the SH group (Fig. 2A) (P < 0.05). Since TLR4 activation has been shown to be a major event in progression of NASH, we studied the downstream proinflammatory molecules for this important pathway. Results showed that there was a significant increase in TNF-α and IL-1β immunoreactivity in liver slices from SH mice compared with the S group (Fig. 2, B–D) (P < 0.05). The localization of the cytokines was unevenly distributed, with TNF-α localized in the centrilobular regions and IL-1β at both zone 1 and zone 3 (Fig. 2B). SsnB administration significantly decreased the immunoreactivity of both TNF-α and IL-1β in liver tissue compared with SH group mice (Fig. 2, B–D) (P < 0.05).
Fig. 2.

SsnB administration to SH mice ameliorates proinflammatory cytokines and decreases M1 polarization. A: qRT-PCR analysis of mRNA expression of TNF-α, IFN-γ, IL-1β, and IL-23 from liver homogenates of S, SH, and SH+SsnB mice, normalized against S (*P < 0.05). B: TNF-α (top) and IL-1β (bottom) immunoreactivity as shown by immunohistochemistry in liver slices from S (i), SH (ii), and SH+SsnB (iii) mouse samples. Images were taken at ×20 magnification. C: morphometric analysis of TNF-α immunoreactivity in S, SH, and SH+SsnB groups (*P < 0.05). D: morphometric analysis of IL-1β immunoreactivity in S, SH, and SH+SsnB groups (*P < 0.05).
SsnB Administration Modulates Kupffer Cell Activation in Early Steatohepatitic Injury
NASH is associated with activation of the resident macrophages in the liver (5). Kupffer cells have been shown to migrate more toward zone 3 in liver injury, generate reactive oxygen species, release proinflammatory cytokines and chemokines, and play a significant role in inflammatory mechanisms in NASH (61). We examined the role of SsnB in modulating Kupffer cell activation in vivo by studying the mRNA and immunoreactivity of Kupffer cell activation marker CD68 and MCP1 in the injured livers. Results showed that SsnB administration significantly decreased mRNA expression of both CD68 and MCP1 (decrease of >12-fold for CD68) (Fig. 3A) (P < 0.05). Immunoreactivity to MCP1 was also significantly decreased in SH mice that were administered SsnB compared with SH-alone mice, suggesting a possibility of low leukocyte infiltration in the liver lobule (Fig. 3B, top; Fig. 3C, morphometric analysis) (P < 0.05). To examine the extent of leukocyte infiltration in the liver lobule, immunoreactivity to pan-macrophage marker F4/80 was studied with immunohistochemistry. Results showed that there were significant increases in F4/80 immunoreactivity in the injured liver compared with the S group while administration of SsnB significantly attenuated the leukocyte infiltration as evidenced by decreased immunoreactivity to F4/80 in the liver (Fig. 3B, bottom; Fig. 3D, morphometry) (P < 0.05).
Fig. 3.

SsnB administration modulates Kupffer cell activation in early steatohepatitic injury. A: qRT-PCR analysis of mRNA expression of CD68 and monocyte chemoattractant protein-1 (MCP1) from liver homogenates of S, SH, and SH+SsnB mice, normalized against S (*P < 0.05). B: MCP1 (top) and F4/80 (bottom) immunoreactivity as shown by immunohistochemistry in liver slices from S (i), SH (ii), and SH+SsnB (iii) mouse samples. Images were taken at ×20 magnification. C: morphometric analysis of MCP1 immunoreactivity in S, SH, and SH+SsnB groups (*P < 0.05). D: morphometric analysis of F4/80 immunoreactivity in S, SH, and SH+SsnB groups (*P < 0.05).
SsnB Administration Attenuates microRNA21 Expression and Repression of PTEN
We have shown previously that steatohepatitic injury following obesity causes a marked increase in expression of microRNA21 (miR21), a noncoding RNA that has significant regulatory role in liver inflammation (9, 46). Higher miR21 levels led to decreased GRHL3 and SMAD7 proteins in both human NASH and rodent models of obesity (9, 46). Since TLR4 has been shown to play a role in miR21 induction, we studied the effects of SsnB, a probable TLR4 antagonist, in TLR4-induced miR21 pathway. Results showed that SH mice had a significant increase in miR21 expression while administration of SsnB markedly decreased its expression in the murine obese liver (>3-fold decrease) (Fig. 4A) (P < 0.05). SsnB administration also decreased the levels of PTEN, a direct target of miR21 in the liver, as shown by Western blot analysis compared with the SH group (Fig. 4, B and C) (P < 0.05). The results suggested that SsnB might have a role in decreasing miR21-induced repression of PTEN primarily by blocking TLR4 activation; however, the interpretation of a SsnB role in blocking TLR is speculative at this time.
Fig. 4.
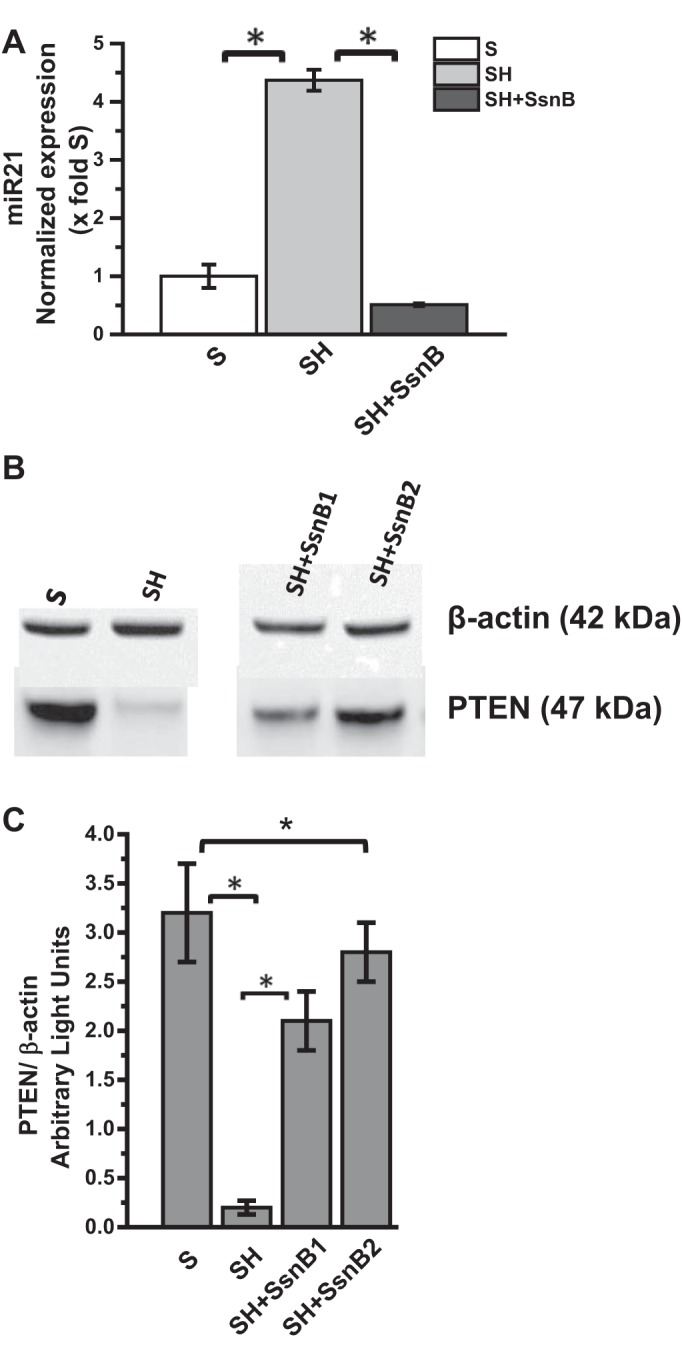
SsnB administration attenuates microRNA 21 (miR21) expression and repression of phosphatase and tensin homolog (PTEN). A: qRT-PCR analysis of miR21 expression of S, SH, and SH+SsnB mouse samples normalized against S (*P < 0.05). B: Western blot analysis of β-actin and PTEN protein levels of S, SH, and SH+SsnB sample 1 and sample 2. C: immunoreactive band analysis of PTEN normalized against β-actin. y-Axis depicts the PTEN-to-actin ratio from S, SH, and SH+SsnB sample1 and sample2 groups (*P < 0.05).
SsnB Targets TLR4 Trafficking to Lipid Rafts
Many studies, including ours, have shown a direct role of TLR4 activation in both alcoholic and nonalcoholic steatohepatitis (8, 36, 62). One of the key aspects of TLR4 activation is its trafficking to lipid-rich domains on the plasma membrane (62). Upon binding to a specific TLR4 ligand, which can include LPS, HMGB1, or free fatty acids, TLR4 protein is translocated to lipid-containing rafts where the association to adaptor proteins and membrane assembly take place. Our collaborators in this study have shown via direct evidence that SsnB disrupts TLR4 activation and can be a potent TLR4 antagonist. To show the mechanistic insight of SsnB action in steatohepatitic liver injury, we studied the TLR4 trafficking into lipid rafts and its disruption by SsnB. Results showed that SsnB administration significantly decreased the colocalization of TLR4 (red) and lipid raft component flotillin (green) in the plasma membrane of SH mice (Fig. 5A). The number of colocalization events/microscopic field was significantly decreased in the SsnB-treated group compared with SH mouse livers (Fig. 5B) (P < 0.05). The results suggested that SsnB has a significant role in TLR4 trafficking in early steatohepatitic injury and the modulation of TLR4 trafficking might be contributing in part to the attenuation of inflammation seen with SsnB administration. Since resident macrophages play a clear role in liver inflammation in NASH, we studied the TLR4 trafficking in immortalized Kupffer cells after their stimulation with LPS, the classical ligand of TLR4. Results showed that incubation of Kupffer cells with LPS (50 ng/ml) significantly increased colocalization of TLR4 with lipid raft marker flotillin while coincubation with SsnB (100 μg/ml) significantly attenuated the TLR4 translocation to the rafts (Fig. 6, A and B). SsnB coincubation resulted in a threefold decrease in the colocalization events (Fig. 6B) (P < 0.05); the colocalization is also shown in higher magnification in Fig. 6C.
Fig. 5.

SsnB targets TLR4 trafficking to lipid rafts. A: immunofluorescence dual labeling depicting TLR4 (red)-flottilin1 (green) colocalization (yellow) in S (i) SH (ii), and SH+SsnB (iii) liver samples, taken at ×20 magnification. B: morphometric analysis of colocalization events in A, shown as colocalization events per 3× field (*P < 0.05).
Fig. 6.

SsnB targets LPS-induced TLR4 trafficking in Kupffer cells. A: immunofluorescence laser scanning images for colocalization of TLR4 (red) and flottilin2 (green) in rat Kupffer cells of the control (i–iv), LPS-treated (v–viii), and LPS+SsnB-treated (ix–xii) groups. Images in overlay panels on right depict colocalizations of TLR4-flottilin2, as revealed by the yellow regions. Images were taken at ×40 magnification. B: morphometric analysis of colocalization events in A, shown as colocalization events per 100 cells (*P < 0.05). C: immunofluorescence dual labeling of LPS-treated rat Kupffer cell sample depicting TLR4 (red)-flottilin2 (green) colocalization (yellow) taken at ×60 (oil) magnification.
SsnB Targets Leptin-Induced TLR4 Trafficking in Kupffer Cells
We have shown previously that proinflammatory adipokine leptin regulates Kupffer cell activation via reactive oxygen species generation (5). Leptin increased peroxynitrite generation by increasing NADPH oxidase activation that resulted in higher inflammatory mediation. Since hepatic leptin resistance is a common phenomenon in NASH patients and selective leptin signaling observed in hepatic stellate cells, we studied the role of leptin in TLR4 trafficking to the lipid rafts in vitro (11, 23). Results showed that leptin administration to immortalized Kupffer cell culture significantly increased the TLR4-flotillin colocalization as evidenced by yellow spots on the membranes of the incubated cells (Fig. 7, A and C, ×40 and ×60 oil images showing clusters of cells with colocalization in the membranes). Analysis of the number of colocalization events showed a significant increase in the leptin-treated group compared with cell-only controls (Fig. 7, A and B) (P < 0.05), with a fourfold increase recorded in this group. SsnB coincubation with leptin significantly decreased the colocalization events in these cells (Fig. 7, A and B) (P < 0.05). The results suggested that, similar to LPS, TLR4 trafficking to the lipid rafts was also influenced by leptin, a proinflammatory adipokine with known effects in NASH. Although the mechanism of leptin action via TLR4 ligand binding is highly unlikely in these cells, SsnB modulation of leptin might be an upstream event and can be a common target for SsnB apart from being a strong antagonist for TLR4 activation.
Fig. 7.

SsnB targets leptin-induced TLR4 trafficking in Kupffer cells. A: immunofluorescence laser scanning images for colocalization of TLR4 (red) and flottilin2 (green) in rat Kupffer cells of the Control (i–iv), leptin-treated (v–viii), and leptin+SsnB-treated (ix–xii) groups. Images in overlay panels on right depict colocalizations of TLR4-flottilin2, as revealed by the yellow regions. Images were taken at ×40 magnification. B: morphometric analysis of colocalization events in A, shown as colocalization events per 100 cells (*P < 0.05). C: immunofluorescence dual labeling of leptin-treated rat Kupffer cell sample depicting TLR4 (red)-flottilin2 (green) colocalization (yellow) taken at ×60 (oil) magnification.
SsnB Attenuates NADPH Oxidase Activation and Peroxynitrite Generation
Since SsnB administration significantly decreased TLR4-induced hepatic inflammation in obesity in response to both LPS and leptin, we argued for the presence of a significant target upstream of the TLR4 pathway that might be common and conspicuous for both leptin- and LPS-mediated signaling. We recently showed that TLR4-lipid raft trafficking in NASH was mediated by peroxynitrite generated by the activation of NADPH oxidase (8). To show that SsnB modulated NADPH oxidase activation and peroxynitrite generation, events that are upstream of TLR4 activation, colocalization analysis of NADPH oxidase membrane subunits in the membranes of liver tissue were performed by immunofluorescence microscopy. Results showed that p47phox (green), a cytoplasmic subunit required for NADPH oxidase activation, colocalized with gp91 (red), a membrane subunit in liver tissue (Fig. 8, A and C, ×20 and ×60 oil images showing colocalization clusters). Analysis of the number of colocalization events in the liver tissue showed a significant increase in the SH-treated group while SsnB-treated mouse liver showed a significant decrease in the colocalization events, suggesting a decreased NADPH oxidase activation (Fig. 8B) (P < 0.05). Kupffer cells treated with both leptin and LPS showed a marked increase in the gp91/p47phox colocalization events, while administration of SsnB (100 μg/ml) resulted in a significant decrease in the events (Fig. 8D). Since NADPH oxidase activation resulted in generation of peroxynitrite in NASH, we examined the formation of 3-nitrotyrosine, a marker for peroxynitrite generation in the diseased liver and immortalized Kupffer cells. Results showed that 3-nitrotyrosine immunoreactivity as analyzed by immunohistochemistry was significantly increased in SH mouse livers compared with the S group (Fig. 9, A and B) (P < 0.05). SsnB-administered mouse livers showed a significant decrease in 3-nitrotyrosine immunoreactivity compared with SH mouse livers (Fig. 9, A and B) (P < 0.05). Since peroxynitrite generation requires both O2·− and NO∙ reactivity in equal diffusion-controlled rates, we studied the release of NO∙ from the Kupffer cell supernatants incubated with leptin, LPS, and SsnB. Results showed that LPS and leptin significantly increased NO∙ release while coincubation with SsnB significantly attenuated the release of NO∙ analyzed by Greiss assay (Fig. 9C) (P < 0.05). The results suggested that apart from the strong TLR4 pathway antagonism function of SsnB, it also plays a significant role in NADPH oxidase activation and NO∙ release in inflammation. The SsnB attenuation of TLR4 pathway might be in part related to its effect on NADPH oxidase activation that is upstream of TLR4 trafficking to lipid rafts.
Fig. 8.

SsnB attenuates NADPH oxidase activation. A: immunofluorescence dual labeling depicting gp91phox (red)-p47phox (green) colocalization (yellow) in S (i), SH (ii), and SH+SsnB (iii) liver samples, taken at ×20 magnification. B: morphometric analysis of colocalization events in A, shown as colocalization events per 3× field (*P < 0.05). C: immunofluorescence dual labeling of SH liver sample depicting gp91phox (red)-p47phox (green) colocalization (yellow) taken at ×60 (oil) magnification. D: immunofluorescence dual labeling of control (i), LPS (ii), LPS+SsnB (iii), control (iv), leptin (v), and leptin+SsnB (vi) rat Kupffer cell samples depicting gp91phox (red)-p47phox (green) colocalization (yellow), taken at ×40 magnification.
Fig. 9.

SsnB attenuates NADPH oxidase activation and peroxynitrite generation. A: 3-nitrotyrosine immunoreactivity as shown by immunohistochemistry in liver slices from S (i), SH (ii), and SH+SsnB (iii) mouse samples. Images were taken at ×20 magnification. B: morphometric analysis of 3-nitrotyrosine immunoreactivity in S, SH, and SH+SsnB groups (*P < 0.05). C: nitric oxide (nitrite) concentration measured by nitric oxide colorimetric assay in the supernatant collected from control, LPS-treated, and LPS+SsnB-treated rat Kupffer cells (*P < 0.05).
SsnB Attenuates TLR Ligand-Induced Gene Expression of Inflammatory Cytokines, p47phox, CD68, and Cytokine Release in Mouse Primary Hepatic Macrophages
To see the direct effect of SsnB on mouse primary hepatic macrophages, qRT-PCR was performed to measure the proinflammatory cytokine gene expression levels. Results showed that, as expected, IL-1β, MCP1, and TNF-α proinflammatory cytokine gene expression were significantly elevated in LPS-treated cells compared with untreated control cells. However, coincubation of LPS-treated cells with SsnB decreases the expression of these genes significantly compared with LPS only-treated cells (Fig. 10A). In another set of experiments, after mouse primary hepatic macrophages were treated with SsnB and LPS for 24 h, the supernatant was collected for measurement of TNF-α cytokine concentration by ELISA. As shown in Fig. 10B, the TNF-α level in the control medium was almost undetectable. LPS (50 ng/ml) treatment dramatically increased the TNF-α cytokine level. However, coincubation with SsnB significantly reduced the TNF-α cytokine level (Fig. 10B). We showed above in Fig. 8 and Fig. 9 that SsnB significantly reduces the formation of peroxynitrite and corresponding inflammation and Kupffer cell activation by decreasing the colocalization of gp91/p47phox and thus attenuates NADPH oxidase activation. To see whether SsnB has any effect in attenuating p47phox gene expression, a qRT-PCR was performed to measure p47phox gene expression levels. Results showed that mRNA expression of p47 phox gene expression was significantly elevated in LPS-treated cells compared with untreated control cells (Fig. 10C). However, coincubation with SsnB decreases the expression of these genes significantly compared with LPS only-treated cells.
Fig. 10.

SsnB attenuates TLR ligand-induced macrophage activation, gene expression of P47phox, and gene expression and release of inflammatory cytokines in mouse primary hepatic macrophages. A: qRT-PCR analysis of mRNA expression of IL-1β, MCP1, and TNF-α from control (untreated), LPS-treated, and LPS+SsnB-treated mouse primary hepatic macrophages, normalized against control (*P < 0.05). B: TNF-α cytokine levels measured by ELISA from the supernatant collected from control, LPS-treated, and LPS+SsnB-treated mouse primary hepatic macrophages, normalized against control (*P < 0.05). C: qRT-PCR analysis of mRNA expression of p47phox from control (untreated), LPS-treated, and LPS+SsnB-treated mouse primary hepatic macrophages, normalized against control (*P < 0.05).
SsnB Treatment Does Not Reduce TLR4 Gene Expression or Protein Concentration
To address the question of whether SsnB acts by decreasing TLR4 mRNA expression, qRT-PCR experiments were performed to quantify TLR4 mRNA expression both in vivo and in vitro. Our data show that SsnB administration may have slightly attenuated the upregulation of TLR4 mRNA levels compared with the SH group (Fig. 11A). Coincubation of mouse primary hepatic macrophages with SsnB also resulted in a faint reduction of TLR4 mRNA expression compared with LPS only-treated cells (Fig. 11B). Liang et al. (27) observed a similar incident and described it as a secondary event to the suppression of proinflammatory signaling pathways. A Western blot experiment was also performed with mouse liver homogenates to see whether TLR4 protein expression is reduced by SsnB administration. Our data clearly indicates that SsnB administration did not cause any reduction in total TLR4 protein level (Fig. 11, C and D). However, it has already been shown by Liang et al. (27) that SsnB possibly acts by attenuating the interaction of MyD88 and TLR4 Toll/interleukin-1 receptor (TIR) domain. Our findings also suggest that SsnB decreases TLR4 trafficking in lipid rafts by preventing NADPH oxidase activation and reducing nitrative stress. Thus there is a possibility that SsnB-induced TLR4 signaling attenuation may not be dependent on the total TLR4 protein concentration.
Fig. 11.

SsnB treatment does not reduce TLR4 gene expression or protein concentration. A: qRT-PCR analysis of mRNA expression of TLR4 from liver homogenates of S, SH, and SH+SsnB mice, normalized against S (*P < 0.05). B: qRT-PCR analysis of mRNA expression of TLR4 from control (untreated), LPS-treated, and LPS+SsnB-treated mouse primary hepatic macrophages, normalized against control (*P < 0.05). C: Western blot analysis of β-actin and TLR4 protein levels in the liver homogenates of lean control (LC), LC+bromodichloromethane (BDCM), S, S+SsnB, SH, and SH+SsnB groups. D: immunoreactive band analysis of TLR4 normalized against β-actin. y-Axis depicts the TLR4-to-β-actin ratio for LC, LC+BDCM, S, S+SsnB, SH, and SH+SsnB groups.
DISCUSSION
The present study reports a novel role and mechanistic insight of the TLR4 antagonist SsnB. Using a rodent model of early steatohepatitic injury, we show that SsnB attenuated inflammation in the liver by blocking TLR4 trafficking to the lipid rafts. Furthermore, we deciphered that SsnB could block TLR4 trafficking into the lipid rafts by decreasing NADPH oxidase activation and peroxynitrite generation. The role of TLR4 in NASH and its consequences are being pursued vigorously across the scientific community (36, 48, 50). The potent role of TLR4 activation and its downstream signaling has been reported in both alcoholic and nonalcoholic steatohepatitis (50). In alcoholic steatohepatitis, portal endotoxemia from gut leaching has been accepted as a source for the TLR4 ligand in the liver (34, 56). Various studies have reported free fatty acids and HMGB1 as ligands in TLR4 activation in NASH (6, 13, 62). We recently reported (8) that peroxynitrite generation activates TLR4 signaling by modulating its trafficking into the lipid rafts primarily by an upstream NADPH oxidase activation. The results in that study also confirmed an earlier report showing NADPH oxidase as a prime regulator of TLR4 trafficking but could not provide a molecular mediator for its actions (62). Interestingly, TLR4 downstream signaling is also known to produce NADPH oxidase activation (15). Our results in this report show a novel role of SsnB, a natural product derivative, in modulating TLR4 trafficking into lipid rafts, a mechanism that had remained elusive before. SsnB has been shown by our collaborators in numerous in vivo and in vitro studies to be a portent TLR4 antagonist (26–31). SsnB structural analysis and its characterization in terms of mechanism of action showed that it reduced the association of MyD88 with TLR4 and TLR2 but not with TLR9 (27). Inflammatory cytokine signaling emanating from the TLR4 signaling in vitro was specifically blocked by SsnB, while LPS-induced septic shock was reversed by its administration in vivo (28). Having known the earlier-reported mechanisms of SsnB action, we explored its role in NASH-induced inflammatory injury. Our rationale was based on the strong inflammatory phase in NASH that is appreciated in both rodent models of NASH and clinical phases (3). Our results showed that SsnB administration in vivo reversed inflammatory pathophysiology primarily evidenced by reduced leukocyte infiltration, decreased necrosis in zone 3, and lower levels of proinflammatory cytokines IL-1β and TNF-α. Since Kupffer cell activation is a prime inflammatory event in NASH that also has been shown to play a vital role in activating stellate cells, we examined the levels of CD68 and MCP1. Results showed a dramatic decrease in the levels of the chemokines and surface expression of CD68. The results were in line with earlier reports from our lab (5) and others (48) that Kupffer cell activation reversal can have a beneficial effect on NASH progression and treatment at least in rodent models of NASH. The studies reported here, however, have not explored the role of SsnB in reversal of Kupffer cell activation since we knew that SsnB might target TLR4 that has been shown previously to activate Kupffer cells (14). Although Kupffer cells respond to LPS-mediated TLR4 activation, there are reports of TLR4 expression in hepatocytes, endothelial cells, and stellate cells (45, 50). Interestingly, Kupffer cells are primary foci for generating an inflammatory response and SsnB action in these cells might mediate the TLR4 activation. The SsnB role in preventing TLR4 signaling in Kupffer cells might produce a stalling effect on the initial inflammatory phase that might eventually block stellate cell activation and extracellular matrix formation. For the purposes discussed above we used immortalized Kupffer cells to illustrate the mechanism of SsnB deactivation of TLR4 pathway.
We have recently shown (9, 46) that NASH is characterized by an increase in miR21-induced repression of GRHL3 and SMAD7, two key proteins that have a pronounced role in sinusoidal endothelial injury and extracellular matrix formation. Interestingly, miR21 is a TLR4-inducible noncoding RNA, and our results in this report of a significant decrease in miR21-induced repression of PTEN confirmed the SsnB targeting of TLR4 pathway (55). It will be of interest in later studies to find out how SsnB may help in alleviating the miR21-induced repressions of GRHL3 and SMAD7 in NASH.
To mechanistically explain the role of SsnB in TLR4 deactivation response we studied the trafficking of TLR4 into lipid rafts, which is a prime event in TLR4 activation and downstream signaling (62). Lipid rafts are lipid-rich domains within the plasma membrane that help in assembly of many transmembrane receptors including TLR4 (62). This myriad of molecular events and assembly starts with the binding of an appropriate ligand to TLR4. We observed that SsnB administration significantly attenuated the trafficking of TLR4 into lipid rafts as shown by the decreased number of colocalization events (TLR4 and flotillin) in NASH livers. Immortalized rat Kupffer cells were also used to show the trafficking process and its attenuation by SsnB following TLR stimulation by LPS. Our results of significant decrease in colocalization events of TLR4 and flotillin in Kupffer cells show that SsnB could exert its TLR4 antagonism by stalling the lipid raft recruitment process. Although SsnB has been shown by our collaborators to interfere in the association of TLR4 with its adaptor MyD88, it can be speculated that SsnB could competitively bind to TLR4, thus restricting the classical ligand LPS to exert its effects (27). However, this observation is highly speculative at this point since SsnB binding to TLR4 in NASH has not been studied. Interestingly, TLR4 trafficking to rafts occurs on binding of the ligand to TLR4, and it would be interesting to see the molecular aspects of SsnB binding to TLR4 or its restrictive capacity to alter TLR4 ligand binding in future studies.
We have shown previously (9, 46) that the proinflammatory adipokine leptin mediates the progression of NASH by activating Kupffer cells, upregulation of noncoding RNAs, and activating PRR P2X7r. Leptin has been shown recently to upregulate TLR2 expression in monocytes (21). Extending the studies of leptin-induced proinflammatory action in Kupffer cells, we studied their role in activating TLR4 trafficking in these cells. Results showed that leptin activated TLR4 signaling via promoting the TLR4 trafficking to lipid rafts, an event that was attenuated by SsnB coincubation. These results were significant since they showed that SsnB can target an upstream molecular event in lipid raft trafficking of TLR4, considering the fact that leptin is not a ligand for TLR4 and has not been shown to have structural similarity to a classical ligand of TLR4. We also have evidence from previous published reports from our lab (5, 8) that 1) leptin targets NADPH oxidase activation and 2) NADPH oxidase drives TLR4 trafficking via peroxynitrite generation. Based on these arguments we conducted studies to show the role of SsnB in NADPH oxidase activation. Our results of a significant increase in colocalization of gp91 (membrane subunit) and p47phox (cytoplasmic subunit) in SH groups and its subsequent decrease in these events following SsnB administration indicated a novel role of SsnB apart from its proven role as an TLR4 antagonist. Parallel to the in vivo data, Kupffer cells also showed significant attenuation of NADPH oxidase activation following SsnB administration in response to both leptin and LPS. However, it needs to be considered that NADPH oxidase activation primarily depends on membrane assembly of a number of subunits, p47phox being one of them, a crucial element for NOX2-type NADPH oxidases (41, 42). Finally, SsnB-induced decrease in peroxynitrite marker 3-nitrotyrosine in vivo and nitric oxide release in vitro showed a clear mechanistic connection between NADPH oxidation, increased nitrative stress, and TLR4 activation following SsnB administration. It is worth mentioning here that peroxynitrite-induced release of HMGB1, a TLR4 activator, has been shown (32). A decrease in peroxynitrite formation following NADPH oxidase inhibition by SsnB might be a parallel mechanism of SsnB amelioration of TLR4 pathway and subsequent decrease in early steatohepatitic injury and inflammation in the liver.
Taken together, our data describe a novel mechanistic role of SsnB in resolving NASH-induced inflammation primarily by blocking TLR4 lipid raft trafficking and NADPH oxidase activation. Future studies featuring SsnB binding kinetics to TLR4 or pathways unique to deactivation of NADPH oxidase might provide new light on probable therapeutic benefits of SsnB.
GRANTS
This work has been supported by National Institutes of Health (NIH) Pathway to Independence Award, Grant R00 ES-019875, and Grant P01 AT-003961 to S. Chatterjee; NIH Grant R01 DK-053792 to A. M. Diehl; and NIH Grants P01 AT-003961, P20 GM-103641, R01 AT-006888, R01 ES-019313, and R01 MH-094755 and Department of Veterans Affairs Merit Award BX001357 to M. Nagarkatti and P. Nagarkatti.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.D., R.K.S., and S.C. conception and design of research; D.D., R.K.S., S.D., F.A., V.C., and S.C. performed experiments; D.D., R.K.S., G.A.M., D.F., M.N., A.M.D., and S.C. analyzed data; D.D., R.K.S., G.A.M., D.F., M.N., P.N., A.M.D., and S.C. interpreted results of experiments; D.D., R.K.S., and S.C. prepared figures; G.A.M., M.N., A.M.D., and S.C. edited and revised manuscript; D.F., M.N., P.N., A.M.D., and S.C. drafted manuscript; D.F., M.N., P.N., A.M.D., and S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the technical services of Benny Davidson at the Instrumentation Resource Facility (IRF), University of South Carolina School of Medicine, and AML Labs (Baltimore, MD). We also thank the IRF at the University of South Carolina for equipment usage and consulting services.
REFERENCES
- 1.Ajamieh H, Farrell G, Wong HJ, Yu J, Chu E, Chen J, Teoh N. Atorvastatin protects obese mice against hepatic ischemia-reperfusion injury by Toll-like receptor-4 suppression and endothelial nitric oxide synthase activation. J Gastroenterol Hepatol 27: 1353–1361, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Bateman HR, Liang Q, Fan D, Rodriguez V, Lessner SM. Sparstolonin B inhibits pro-angiogenic functions and blocks cell cycle progression in endothelial cells. PloS One 8: e70500, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohinc BN, Diehl AM. Mechanisms of disease progression in NASH: new paradigms. Clin Liver Dis 16: 549–565, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee S, Das S. P2X7 receptor as a key player in oxidative stress-driven cell fate in nonalcoholic steatohepatitis. Oxid Med Cell Longev 2015: 172493, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee S, Ganini D, Tokar EJ, Kumar A, Das S, Corbett J, Kadiiska M, Waalkes M, Diehl AM, Mason RP. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental nonalcoholic steatohepatitis. J Hepatol 58: 778–784, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen M, Huang W, Wang C, Nie H, Li G, Sun T, Yang F, Zhang Y, Shu K, Wang C, Gong Q. High-mobility group box 1 exacerbates CCl4-induced acute liver injury in mice. Clin Immunol 153: 56–63, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones E, Szabo G. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol 300: G433–G441, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das S, Alhasson F, Dattaroy D, Pourhoseini S, Seth RK, Nagarkatti M, Nagarkatti PS, Michelotti GA, Diehl AM, Kalyanaraman B, Chatterjee S. NADPH oxidase-derived peroxynitrite drives inflammation in mice and human nonalcoholic steatohepatitis via TLR4-lipid raft recruitment. Am J Pathol 185: 1944–1957, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M, Michelotti GA, Diehl AM, Chatterjee S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 308: G298–G312, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day CP. Clinical spectrum and therapy of non-alcoholic steatohepatitis. Dig Dis 30, Suppl 1: 69–73, 2012. [DOI] [PubMed] [Google Scholar]
- 11.Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol 166: 1655–1669, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durazzo M, Belci P, Collo A, Grisoglio E, Bo S. Focus on therapeutic strategies of nonalcoholic fatty liver disease. Int J Hepatol 2012: 464706, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, Tobe K, Arai H, Kadowaki T, Nagai R. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 15: 518–533, 2012. [DOI] [PubMed] [Google Scholar]
- 14.El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Harris JK, Lovell MA, Finegold MJ, Sokol RJ. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology 55: 1518–1528, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest 112: 1234–1243, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarzian A. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int 28: 1026–1033, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr 35: 14S–20S, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol 18: 2609–2618, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, Kato S, Mawatari H, Shibata W, Kitani H, Ikejima K, Kirikoshi H, Nakajima N, Saito S, Maeyama S, Watanabe S, Wada K, Nakajima A. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab 16: 44–54, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Jaedicke KM, Roythorne A, Padget K, Todryk S, Preshaw PM, Taylor JJ. Leptin up-regulates TLR2 in human monocytes. J Leukoc Biol 93: 561–571, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Kim CH, Younossi ZM. Nonalcoholic fatty liver disease: a manifestation of the metabolic syndrome. Cleve Clin J Med 75: 721–728, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab 16: 144–152, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Fan D, Dipette DJ, Singh US. Sparstolonin B, a novel plant derived compound, arrests cell cycle and induces apoptosis in N-myc amplified and N-myc nonamplified neuroblastoma cells. PloS One 9: e96343, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larrain S, Rinella ME. A myriad of pathways to NASH. Clin Liver Dis 16: 525–548, 2012. [DOI] [PubMed] [Google Scholar]
- 26.Liang Q, Dong S, Lei L, Liu J, Zhang J, Li J, Duan J, Fan D. Protective effects of Sparstolonin B, a selective TLR2 and TLR4 antagonist, on mouse endotoxin shock. Cytokine 75: 302–309, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang Q, Wu Q, Jiang J, Duan J, Wang C, Smith MD, Lu H, Wang Q, Nagarkatti P, Fan D. Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J Biol Chem 286: 26470–26479, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang Q, Yu F, Cui X, Duan J, Wu Q, Nagarkatti P, Fan D. Sparstolonin B suppresses lipopolysaccharide-induced inflammation in human umbilical vein endothelial cells. Arch Pharm Res 36: 890–896, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Li J, Jubair S, Wang D, Luo Y, Fan D, Janicki JS. Sparstolonin B attenuates hypoxia-induced apoptosis, necrosis and inflammation in cultured rat left ventricular tissue slices. Cardiovasc Drugs Ther 28: 433–439, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Q, Li J, Liang Q, Wang D, Luo Y, Yu F, Janicki JS, Fan D. Sparstolonin B suppresses rat vascular smooth muscle cell proliferation, migration, inflammatory response and lipid accumulation. Vascul Pharmacol 67–69: 59–66, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Wang J, Liang Q, Wang D, Luo Y, Li J, Janicki JS, Fan D. Sparstolonin B attenuates hypoxia-reoxygenation-induced cardiomyocyte inflammation. Exp Biol Med (Maywood) 239: 376–384, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Loukili N, Rosenblatt-Velin N, Li J, Clerc S, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc Res 89: 586–594, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma YY, Li L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol 19: 6911–6918, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 32: 1008–1017, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Mehal WZ. The inflammasome in liver injury and non-alcoholic fatty liver disease. Dig Dis 32: 507–515, 2014. [DOI] [PubMed] [Google Scholar]
- 36.Miura K, Ohnishi H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol 20: 7381–7391, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Wang X, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203: 2377–2389, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nebiker CA, Han J, Eppenberger-Castori S, Iezzi G, Hirt C, Amicarella F, Cremonesi E, Huber X, Padovan E, Angrisani B, Droeser RA, Rosso R, Bolli M, Oertli D, von Holzen U, Adamina M, Muraro MG, Mengus C, Zajac P, Sconocchia G, Zuber M, Tornillo L, Terracciano L, Spagnoli GC. GM-CSF production by tumor cells is associated with improved survival in colorectal cancer. Clin Cancer Res 20: 3094–3106, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Pacana T, Sanyal AJ. Recent advances in understanding/management of non-alcoholic steatohepatitis. F1000Prime Rep 7: 28, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pacana T, Sanyal AJ. Vitamin E and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care 15: 641–648, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paik YH, Brenner DA. NADPH oxidase mediated oxidative stress in hepatic fibrogenesis. Korean J Hepatol 17: 251–257, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal 20: 2854–2872, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasarin M, La Mura V, Gracia-Sancho J, Garcia-Caldero H, Rodriguez-Vilarrupla A, Garcia-Pagan JC, Bosch J, Abraldes JG. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PloS One 7: e32785, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peri F, Piazza M. Therapeutic targeting of innate immunity with Toll-like receptor 4 (TLR4) antagonists. Biotechnol Adv 30: 251–260, 2012. [DOI] [PubMed] [Google Scholar]
- 45.Petrasek J, Csak T, Szabo G. Toll-like receptors in liver disease. Adv Clin Chem 59: 155–201, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Pourhoseini S, Seth RK, Das S, Dattaroy D, Kadiiska MB, Xie G, Michelotti GA, Nagarkatti M, Diehl AM, Chatterjee S. Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PloS One 10: e0116780, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ratziu V. Pharmacological agents for NASH. Nat Rev Gastroenterol Hepatol 10: 676–685, 2013. [DOI] [PubMed] [Google Scholar]
- 48.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 47: 571–579, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez B, Torres DM, Harrison SA. Physical activity: an essential component of lifestyle modification in NAFLD. Nat Rev Gastroenterol Hepatol 9: 726–731, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol 28, Suppl 1: 38–42, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K. Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet 26: 30–46, 2011. [DOI] [PubMed] [Google Scholar]
- 52.Seth RK, Das S, Pourhoseini S, Dattaroy D, Igwe S, Ray JB, Fan D, Michelotti GA, Diehl AM, Chatterjee S. M1 polarization bias and subsequent nonalcoholic steatohepatitis progression is attenuated by nitric oxide donor DETA NONOate via inhibition of CYP2E1-induced oxidative stress in obese mice. J Pharmacol Exp Ther 352: 77–89, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM, Chatterjee S. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol Sci 134: 291–303, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shanab AA, Scully P, Crosbie O, Buckley M, O'Mahony L, Shanahan F, Gazareen S, Murphy E, Quigley EM. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci 56: 1524–1534, 2011. [DOI] [PubMed] [Google Scholar]
- 55.Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q, Johnson DS, Chen Y, O'Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 11: 141–147, 2010. [DOI] [PubMed] [Google Scholar]
- 56.Siebler J, Galle PR, Weber MM. The gut-liver-axis: endotoxemia, inflammation, insulin resistance and NASH. J Hepatol 48: 1032–1034, 2008. [DOI] [PubMed] [Google Scholar]
- 57.Singh R, Bullard J, Kalra M, Assefa S, Kaul AK, Vonfeldt K, Strom SC, Conrad RS, Sharp HL, Kaul R. Status of bacterial colonization, Toll-like receptor expression and nuclear factor-kappa B activation in normal and diseased human livers. Clin Immunol 138: 41–49, 2011. [DOI] [PubMed] [Google Scholar]
- 58.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52: 1836–1846, 2010. [DOI] [PubMed] [Google Scholar]
- 59.Tsujimoto T, Kawaratani H, Kitazawa T, Uemura M, Fukui H. Innate immune reactivity of the ileum-liver axis in nonalcoholic steatohepatitis. Dig Dis Sci 57: 1144–1151, 2012. [DOI] [PubMed] [Google Scholar]
- 60.Velayudham A, Dolganiuc A, Ellis M, Petrasek J, Kodys K, Mandrekar P, Szabo G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 49: 989–997, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, Brigstock D, George J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 137: 713–723, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem 284: 27384–27392, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y, Man K, Lo CM, Li X, Xu A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 61: 1058–1067, 2012. [DOI] [PubMed] [Google Scholar]
- 64.Zheng Z, Xu X, Zhang X, Wang A, Zhang C, Huttemann M, Grossman LI, Chen LC, Rajagopalan S, Sun Q, Zhang K. Exposure to ambient particulate matter induces a NASH-like phenotype and impairs hepatic glucose metabolism in an animal model. J Hepatol 58: 148–154, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]