

SUMMARY
Ring NTPases are a class of ubiquitous molecular motors involved in basic biological partitioning processes. dsDNA viruses encode ring ATPases that translocate their genomes to near-crystalline densities within pre-assembled viral capsids. Here, X-ray crystallography, cryoEM, and biochemical analyses of the dsDNA packaging motor in bacteriophage phi29 show how individual subunits are arranged in a pentameric ATPase ring, and suggest how their activities are coordinated to translocate dsDNA. The resulting pseudo-atomic structure of the motor and accompanying functional analyses show how ATP is bound in the ATPase active site; identify two DNA contacts, including a potential DNA translocating loop; demonstrate that a trans-acting arginine finger is involved in coordinating hydrolysis around the ring; and suggest a functional coupling between the arginine finger and the DNA translocating loop. The ability to visualize the motor in action illuminates how the different motor components interact with each other and with their DNA substrate.
INTRODUCTION
The ability to interconvert various forms of energy is an essential feature of living systems. Biological molecular motors accomplish this task by coupling the making and breaking of high-energy covalent bonds to conformational changes in large macromolecules. Among these, the homomeric ring NTPases are a sub-group of the large ASCE (Additional Strand Catalytic E (glutamate)) NTPase superfamily whose members are involved in numerous macromolecular force-generating tasks including chromosome segregation, DNA recombination/strand separation/conjugation, protein degradation, and the generation and maintenance of concentration gradients and electrostatic potentials (Burroughs et al., 2007; Mitchell et al., 2002; Singleton et al., 2007; Thomsen and Berger, 2008). In these motors, several energy-generating NTPase subunits are arranged as a ring, and coordinated hydrolysis of NTP molecules in the ring induces conformational changes in the motor that are coupled to the translocation of a polymeric substrate. Understanding the mechanisms by which these motors operate will illuminate the general mechanistic principles of molecular partitioning in biology as well as provide insight into the fundamental question of how chemical energy is converted to mechanical work in biological systems.
Double-stranded DNA viruses, including herpesviruses and tailed bacteriophages, encode for homomeric ASCE ring ATPases that they use to package their genomes into preformed protein shells (capsids) (Mitchell et al., 2002; Morais, 2012). The process of genome encapsidation is remarkable since considerable entropic, electrostatic, and DNA bending energies must be overcome to package DNA to near-crystalline densities within the confines of the capsid. Given the high forces involved in DNA compaction, packaging motors must work against substantially higher resisting forces than other ASCE motors. Indeed, viral DNA packaging motors are among the most powerful biological motors known, capable of generating forces greater than 60 piconewtons (Rickgauer et al., 2008; Smith et al., 2001). Thus, insights gained from the study of viral packaging motors will not only shed light on the basic mechanistic principles of a broad class of macromolecular motors, but can also illuminate how these principles have been adapted by viruses to generate and control the large molecular forces necessary for genome encapsidation.
Bacteriophage phi29 is an excellent model system for mechanistic studies of genome packaging since a highly efficient in vitro DNA packaging system has been developed, which has allowed packaging to be probed via multiple experimental approaches (Grimes et al., 2002; Guo et al., 1986; Morais, 2012). Extensive genetic, biochemical, and structural studies have shown that the motor consists of three macromolecular components (Figure 1A) (Morais, 2012; Morais et al., 2008): 1) a dodecameric connector protein (gene product 10 (gp10)) (Simpson et al., 2000), termed the portal protein in other phage systems; 2) a pentameric ring of a phage encoded structural RNA molecule (pRNA) (Cao et al., 2014; Ding et al., 2011; Guo et al., 1987; Morais et al., 2001; Simpson et al., 2000); and 3) a pentameric ASCE ATPase ring (gene product 16 (gp16)), analogous to the large terminases in other phage systems, which provides the energy for packaging (Koti et al., 2008; Morais et al., 2008; Simpson et al., 2000). These three components are arranged as three stacked rings, and the dsDNA genome is translocated through a continuous channel along their shared central axis into the phage capsid (Figure 1A). Of note, the macromolecular components that constitute the phi29 packaging motor are relatively small compared to other phages; in phi29, the connector/portal and ATPase motor proteins that are common to all phage packaging motors are ~ 60% of the size of their counterparts in other phages, suggesting that they represent the essential minimum for motor operation.
Figure 1.

The bacteriophage phi29 dsDNA packaging motor. A) Cut-away side view of the bacteriophage phi29 dsDNA packaging motor as determined by cryoEM. Molecular envelopes of the connector, pRNA, and ATPase are shown in green, magenta, and blue, respectively. B) Model of the mechano-chemical cycle of the packaging motor as determined by single molecule experiments (Chistol et al., 2012). The top and bottom halves of the panel show the chemical and mechanical transitions in the dwell phase and burst phases, respectively. Selected points of the mechano-chemical cycle that are specifically referred to in the text are marked by asterisks.
More recently, biochemical and single molecule studies have shown that the genome packaging mechanism utilized by the motor is complex and highly coordinated (Chistol et al., 2012; Liu et al., 2014a; Liu et al., 2014b; Moffitt et al., 2009). It was shown that the mechano-chemical cycle is separated into a dwell phase and a burst phase (Figure 1B) (Moffitt et al., 2009). During the dwell phase, no DNA translocation occurs while all five gp16 subunits release ADP from the previous hydrolysis cycle and load ATP in an interlaced fashion. During the burst phase, sequential ATP hydrolysis and release of inorganic phosphate (Pi) by four gp16 subunits is coupled to the translocation of 10 bp of DNA in four 2.5 bp steps. Rather than translocating DNA, hydrolysis in the fifth subunit has been proposed to play a functionally unique regulatory role in the motor (Chistol et al., 2012; Liu et al., 2014b). Additional single molecule experiments using altered DNA substrates show that the motor makes two types of contacts during packaging (Aathavan et al., 2009). During the dwell phase, the motor makes strong specific electrostatic contacts with the DNA phosphate backbone while the nucleotide state of the motor is reset. During burst phase, the motor employs non-specific contacts to drive translocation of the DNA into the capsid. Recently, it has also been shown that DNA rotation occurs during packaging, likely to maintain motor/substrate alignment, and that the magnitude of this rotation is coupled to the changing step-size of the motor as the head fills (Liu et al., 2014b).
Despite having developed a comprehensive description of the kinetic cycle of the motor, the structural changes in the motor that drive DNA packaging have yet to be determined. Here, an atomic resolution structure of the ATPase domain of gp16 crystallized in the presence of the substrate analog AMP-PNP has been determined to 1.94 Å by X-ray crystallography. This structure, along with atomic structures of the connector and the pRNA, was fitted into a 5-fold averaged 12 Å cryoEM reconstruction of phi29 particles stalled during packaging in a distinct mechano-chemical state, resulting in the first nearly complete atomic model of a packaging motor that includes the DNA substrate, and thus the first atomic model of an actively packaging motor. Finally, biochemical analysis of gp16 mutants was used to characterize the roles of critical residues in ATP hydrolysis, DNA translocation, and inter-subunit communication, including demonstration of a trans-acting arginine finger that suggests a role for this element in coordinating motor activity.
RESULTS
Solution of the X-ray crystallographic structures of the ATPase domain of gp16
The domain structure of gp16 is relatively simple: the 39 Kd 332 amino acid protein consists of only 2 domains (Morais, 2012). The N-terminal domain is approximately 200 amino acids long, and contains the conserved ASCE ATPase domain. The last ~100 amino acids are predicted to adopt an oligonucleotide/oligosacharide binding fold (OB-fold), implying a possible role in either pRNA or DNA binding (Morais, 2012). Although attempts to crystallize the full length protein were unsuccessful, crystals of the ATPase domain (residues 1-208) grew in the presence of the substrate analog AMP-PNP (Mao et al, in preparation). These crystals belong to space group P212121 with a= 33.1 b = 36.8 and c = 139.1 Å, and were solved to 1.94 Å resolution using single-wavelength anomalous dispersion data from a seleno-methionine substituted protein (Figure 2; Table 1). There is only a single polypeptide in the crystallographic asymmetric unit, and application of crystallographic symmetry does not generate rings or any other point group symmetry, indicating that the protein crystallized as a monomer.
Figure 2.

X-ray crystal structure of the N-terminal ATPase domain of gp16. A) Two views of a ribbon diagram of gp16. β-sheets and α-helices on the conserved ASCE core are shown in green and orange, respectively, and are numbered in the right image of the panel. Additional strands and helices characteristic of the HerA/FtsK clade of the ASCE superfamily are shown in pink and blue, respectively, and a blue oval indicates the position of the C-terminal OB domain. B) Topology diagram of gp16; colors and symbols are the same as in (A). C) The proposed adenine binding motif is shown in blue, the Walker A motif in magenta; and the Walker B motif in red. The proposed phosphosensor, catalytic glutamate, and arginine finger are shown as stick figures colored green, red and cyan, respectively. The substrate analog AMP-PNP is shown as a stick figure; atoms are colored by element except for carbon, which is colored yellow to facilitate viewing. D) stereo-diagram of active site residues in gp16; some backbone atoms have been removed to facilitate viewing. Magnesium and waters are shown as green and red spheres, respectively.
Table 1.
| Data Collection | |
| Protein | SeMet gp16 (a.a. 1-207) |
| X-ray source | CAMD |
| Wavelength (Å) | 0.97941 |
| Resolution (Å) | 1.941 |
| Space group | P212121 |
| Unit cell (Å) | a = 33.1, b = 36.8, c = 139.1 |
| Unique reflections | 12,985 |
| Average redundancy | 4.4 |
| I/σa | 31.5 (5.4) |
| Completeness (%) | 98.19 (72.6) |
| Rmerge (%)b | 14.5 (32.0) |
| Phasing powerc | 1.021(anomalous) |
| Refinement | |
| Resolution (Å) | 50-1.94 |
| Rworking (%)d | 20.4 |
| Rfree (%)e | 24.7 |
| Average B factor (Å2) | 22.371 |
| Rmsd bonds (Å) | 0.007 |
| Rmsd angels (°) | 1.093 |
Values in parentheses throughout the table correspond to the last shell.
Rmerge = Σ| I – < I > | / Σ I, where I is measured intensity for reflections with indices hkl.
Phasing power = [ | Fh(calc) | / phase-integrated lack of closure ].
Rworking = Σ ∥ Fobs | – | Fcalc∥ / Σ | Fobs |.
Rfree has the same formula as Rworking except that calculation was made with the structure factors from the test set.
Since a truncated construct was crystallized, the oligomeric state of the full-length protein in solution was also investigated. In contrast to previously reported results (Koti et al., 2008), analytical ultracentrifugation (AUC) sedimentation velocity experiments on the codon-optimized construct indicate that even at concentrations above 3 mg/ml (near the saturation limit of the protein), the full-length protein is more than 85% monomeric under the buffer conditions used for protein purification (data not shown). Thus, although ASCE proteins often function as oligomeric rings, gp16 did not form any sort of higher order structure in the absence of the capsid and other motor components.
Key functional features of the gp16 ATPase domain structure
The N-terminal ATPase domain of gp16 consists of a nine-stranded beta-sheet that is sandwiched between alpha-helices (Figure 2A and B). As expected, the central part of the sheet adopts the canonical ASCE fold (Burroughs et al., 2007; Morais, 2012), which itself is an elaboration of the ubiquitous Rossmann fold (Rossmann et al., 1974), consisting of five parallel beta-strands interspersed by four alpha-helices. This arrangement results in the C-terminal ends of each strand forming one edge of a β-sheet with α-helices on either face of the sheet. Mapping the ASCE functional motifs onto the structure indicates that a proposed adenine-binding domain (Figure 2C; blue), the Walker A motif (Figure 2C; magenta), the proposed catalytic glutamate at the end of the Walker B motif (Figure 2C; red), and the putative phosphate sensor (N158; Figure 2C; green) all reside on the same side of the β-sheet on loops that connect β-strands to downstream helices. In contrast, the putative arginine finger (R146; Figure 2C; cyan) maps to a loop on the opposite side of the sheet.
Docking AMP-PNP in the structure indicates how the conserved ASCE motifs in gp16 likely function during ATP hydrolysis (Figures 2C and D). The backbone amides of the glycine residues in the Walker A motif (G29 and G27, located between strand S1 and helix H1) coordinate the phosphate groups of the AMP-PNP, the ε-amino group of the conserved lysine K30 forms ion pairs with the β- and γ-phosphates (Figure 2D). A conserved tyrosine located immediately upstream of the Walker A motif (Y32), helps orient the ATP substrate via π-stacking interactions with the adenine ring. The hydroxyl group of the Walker A serine S31 helps bind a critical catalytic Mg2+ ion that coordinates the β- and γ-phosphates of ATP and helps position a water molecule that is in the same approximate position as water molecules in other phage DNA packaging motors and ASCE ATPases that are postulated to act as the activated nucleophile during hydrolysis of ATP at the γ-phopshate (Figure 2D) (Hilbert et al., 2015; Sun et al., 2007; Thomsen and Berger, 2008; Zhao et al., 2013). The Walker B motif is located between strand S3 and helix H3 and includes a conserved aspartate D118 that provides another coordinating ligand for the catalytic Mg2+ that positions both the ATP substrate and the water nucleophile for in-line attack of the activated water molecule at the γ-phosphate. The catalytic glutamate E119 immediately follows the Walker B motif, and, by analogy with other ASCE NTPases (Thomsen and Berger, 2008), is positioned to act as a general base that helps activate the water molecule for nucleophilic attack on the γ-phosphate of ATP (figure 2D). Mutational switching of the D118 and E119 (D118E/E119D double mutant) residues yields a gp16 that is both ATPase and packaging defective, supporting this assignment (Table 2) (Schwartz et al., 2013; Sun et al., 2007). In the ATPase from bacteriophage T4, an ‘adenine binding motif’ interacts with N6 and N7 nitrogens of the adenine ring and thus helps bind and position the ATP substrate (Mitchell et al., 2002; Sun et al., 2007). However, in gp16, the equivalent motif is located upstream of the Walker A lysine in gp16 (residues 9-10; Figure 2C; blue), and does not interact with the nitrogenous base in phi29 (Figure 2C; blue).
Table 2.
| ATPase and Packaging Activities of Select ATPase Mutants | ||
|---|---|---|
| Mutant tested | ATPase Activity (% of Wt) | DNA Packaging Activity (% of Wt) |
| D/E | 4.0 ± 0.7 | 0 |
| R146K | 6.9 ± 1.4 | 0 |
| R146A | 4.4 ± 1.2 | 0 |
| R122A | 66.2 ± 14.0 | 0 |
| K124A | 113.3 ± 11.7 | 41.3 ± 2.5 |
| N158A | 2.8 ± 0.8 | 0 |
| negative control | 3.3 ± 1.2 | 0 |
The conserved ‘γ-phosphate sensor’, asparagine N158 in gp16, is positioned at the C-terminal end of strand S4 (Figures 2C and D). In other members of the ASCE superfamily, this residue is proposed to play a role in transmitting conformational changes to other parts of the protein upon ATP binding/hydrolysis by ‘sensing’ the position of the γ-phosphate (Iyer et al., 2004a; Iyer et al., 2004b; Story and Steitz, 1992; Yoshida and Amano, 1995). In gp16, the proposed phosphate sensor N158 is approximately 3 Å from the γ-phosphate of the AMP-PNP, and is thus well positioned to interact with the phosphate leaving group as it is released during hydrolysis of ATP. Mutation of N158 to alanine abolished both ATPase and DNA packaging activities (Table 2), supporting its assignment as the gp16 phosphate sensor.
Many ASCE ATPases are proposed to utilize an ‘arginine finger’ to coordinate ATP hydrolysis between adjacent subunits (Burroughs et al., 2007; Morais, 2012; Thomsen and Berger, 2008; Yoshida and Amano, 1995). Typically, hydrolysis in one subunit is believed to be coupled to a conformational change that results in the insertion of its arginine finger into the active site of a neighbor. This promotes ATP hydrolysis in the adjacent subunit; the positively charged guanidinium group of the arginine is thought to stabilize the pentavalent phospho-anhydride transition state that forms during hydrolysis and promote separation of the phosphate leaving group (Braig et al., 2000; Nadanaciva et al., 1999; Ogura et al., 2004). In gp16, the predicted arginine finger, residue 146 (Burroughs et al., 2007), is located immediately upstream of strand S4 on the opposite edge of the central β-sheet where the other ASCE motifs reside (Figure 2C-D). Although only a monomer of the ATPase domain crystallized here, previous cryoEM reconstructions of phi29 show that gp16 forms a pentameric ring when the motor is assembled on a prohead (Koti et al., 2008; Morais et al., 2008; Simpson et al., 2000), consistent with in trans activation of adjacent subunits by an arginine finger (see also below, arrangement of gp16 in the phi29 dsDNA packaging motor.)
Structural homology with other ASCE family members
Although it is clear that the N-terminal domain of gp16 belongs to the ASCE superfamily, there has been some debate regarding exactly where gp16 resides in the ASCE family tree (Burroughs et al., 2007; Mitchell et al., 2002; Morais, 2012; Schwartz et al., 2013; Yu et al., 2010). Given its functional homology with other phage packaging enzymes, it would seem that gp16 should be grouped with the dsDNA bacteriophage Terminase Large Subunit (TLS) family (Mitchell et al., 2002). However, based on the lack of a nuclease function in its C-terminal domain, the predicted topology of a small insertion domain between S2 and H2, and the position of the putative arginine finger (R146) immediately upstream of β-strand S4, gp16 was also proposed to belong to the HerA/FtsK branch of the ASCE superfamily (Figure 2a) (Burroughs et al., 2007). Alternatively, a homology model of gp16 based on the packaging NTPase from a dsRNA cystovirus belonging to the RecA branch of the family was also proposed (Yu et al., 2010). It has also been asserted that the N-terminal domain of gp16 is a member of the AAA+ branch of the ASCE superfamily (Schwartz et al., 2013).
Based on the structure reported here, the N-terminal domain of gp16 has the highest structural homology with members of the TLS superfamily (Sun et al., 2007; Zhao et al., 2013), as judged by the lowest RMSD over the largest number of atoms (Table 3). However, gp16 also has considerable homology to both the bacterial translocase FtsK (Massey et al., 2006) and the P4 (Mancini et al., 2004) packaging protein from the cystovirus phi12. Thus, mechanistic features of these three branches of the ASCE superfamily may be present in gp16 (see below). However, structural homology with the AAA+ branch of the superfamily is considerably worse than with the other three branches, likely due to the reduced number of strands in the central sheet, the very different position and topology of the insertion domain, and the different location of the putative arginine finger. Hence, mechanistic models based on structural homology with the AAA+ branch of the superfamily (Schwartz et al., 2013) seem unlikely.
Table 3.
| Structural Superpositions of gp16 N-terminal domain (a.a. 1-207) with Other ATPases | ||||
|---|---|---|---|---|
| Protein | Function | PDB Accession Number | Number of Equivalent Cα Atoms | Rmsd (Å) |
| T4 gp17 | Phage large terminase | 2O0H | 167 | 3.4 |
| Sf6 gp2 | Phage large terminase | 4IEE | 167 | 3.1 |
| FtsK | DNA translocase | 2IUU | 131 | 4.1 |
| ϕ12 P4 | dsRNA phage packaging motor | 1W44 | 113 | 3.7 |
Arrangement of gp16 subunits in the phi29 dsDNA packaging motor
In order to develop a comprehensive mechanistic model of the packaging motor, it is necessary to not only know the structures of each separate motor component, but also how the individual motor components are arranged with respect to one another. Presently, since it is not possible to assemble the motor complex in the absence of the prohead, the motor was imaged in the context of the entire prohead. Previously, cryoEM analysis of the entire motor complex was used to determine the approximate molecular envelopes of the connector, the pRNA, and the ATPase, and indicated that the ATPase forms a pentameric ring at the distal ends of the A-helices of the pRNA ring (Koti et al., 2008; Morais et al., 2008). More recently, an asymmetric sub-nanometer cryoEM reconstruction of the prohead-pRNA complex allowed fitting the atomic structures of the connector, and the prohead-binding domain and the essential UCCA bulge of pRNA into their corresponding densities, as well as modeling the remainder of the A-helix (Cao et al., 2014). The result was a pseudo-atomic structure of the connector-pRNA complex. Although this structure provided insight into the structure and assembly of the pRNA-connector complex, there has been little information regarding the structure of the ATPase, and virtually no information regarding the structure of an actively packaging ATPase motor engaged with the DNA substrate.
Here we imaged particles that were stalled during packaging with γ-S-ATP and generated an approximately 12 Å cryo-electron microscopy 3D reconstruction (Figure 3, Supplemental Figure S1). Based on the current model of the phi29 DNA packaging mechanism derived from biochemical and single-molecule analysis (Chistol et al., 2012; Liu et al., 2014a; Moffitt et al., 2009), this reconstruction likely represents the state of the motor just prior to the burst phase, where all five ATPase subunits are in the ATP bound state, and one holds DNA (Figure 1B, *).
Figure 3.

CryoEM reconstruction of packaging particles stalled via the addition of substrate analog γ-S-ATP. A) Cut-away side view; density corresponding to the capsid, DNA, connector, pRNA and ATPase is colored grey, red, yellow, magenta, and blue, respectively. B) Close-up cut-away side view of the motor with atomic resolution structures of the connector (yellow), pRNA (magenta), ATPase (blue), and DNA (red) fitted into their corresponding densities. The C-terminus of the ASCE domain of gp16 is indicated and is shown as purple van der Waal spheres. AMP-PNP is shown as van der Waal spheres colored as in Figure 2, the putative DNA translocating loop is colored green, density for the C-terminal OB-domain is labelled ‘CTD’, and two observed DNA contacts are indicated. C) End-on views of the fitted pRNA and N-terminal ATPase domain of gp16 (contact one) viewed from outside the phage looking in (left) and inside the phage looking out (right). D) End-on views of the fitted pRNA and the OB-domain of gp16 (contact two) viewed from outside the phage looking in (left) and inside the phage looking out (right).
Consistent with previous results (Koti et al., 2008; Morais et al., 2008), density corresponding to the ATPase is clearly visible as a ring structure at the distal ends of the pRNA spokes (Figure 3). In this 5-fold averaged reconstruction, adjacent subunits in the pentameric ring are close together, with significant interactions between them that create the functional interface between ATPase subunits (Figure 3C). In addition, density corresponding to the DNA is also clearly visible (Figure 3A and B). A longitudinal section of the reconstruction shows that the motor interacts with the DNA at two distinct locations: 1) at the lumen of the distal portion of the ATPase ring, and 2) below the connector, between the pRNA ring and the DNA (Figure 3B). The observation of two motor-DNA contacts may have critical implications for the mechanism of DNA packaging (see below). At very low contour levels, additional connections are observed between the connector and the DNA; however, since these connections are only visible at < 0.2 standard deviations above the mean, it is difficult to distinguish these features from noise. Thus, although several packaging models feature extensive interactions between the connector and the DNA, the data presented here does not support these models (Guo et al., 2013; Lebedev et al., 2007).
Fitting atomic structures of motor components into cryoEM maps of packaging intermediates
The connector and the pRNA structures can be unambiguously docked into their corresponding densities of our cryoEM reconstructions (Figure 3B). However, density corresponding to gp16 is somewhat featureless and globular in shape, as is the atomic structure of the ATPase domain of gp16; thus, it is difficult to obtain a unique fit for the five individual copies of gp16. Although the density is not featured enough to reliably fit individual gp16 subunits, it can be used as a filter to examine whether various models of oligomeric rings of gp16 agree with the cryoEM data. Three models of pentameric rings of gp16 were generated based on the ring structures of TLS, HerA/FtsK, and RecA ATPase members of the ASCE superfamilies, respectively. For the TLS-based model, the ATPase domain of gp16 was simply superimposed on the five equivalent domains of the pentameric model of the bacteriophage T4 ATPase (Sun et al., 2008). For the HerA/FtsK-based model, one subunit of gp16 was first superimposed on one subunit of the FtsK ring (Massey et al., 2006) and then, to accommodate the differing symmetries of gp16 and FtsK (5 vs 6), subsequent gp16 monomers were added with a 72° rotation around the central ring axis. This ring was then fitted to density corresponding to gp16 in the cryoEM map, and Individual subunits were then translated radially inwards to maximize agreement with the cryoEM density and minimize steric clashing between adjacent subunits (Figure 3B and C). A similar procedure was used for the RecA-like model using the hexameric structure of the packaging NTPase P4 from the RNA phage phi12 (Mancini et al., 2004).
Despite the fact that individual subunits of gp16 are structurally most similar to the TLS family members (Table 3), the gp16 pentameric ring based on the ASCE-domain ring of the T4 pentamer had the poorest fit to the observed cryoEM density since the radius of the T4-motor structure is considerably larger than the density observed for gp16 (Supplemental Figure S2). However, as stated above, the model of the T4 packaging motor was determined in the absence of ATP and DNA. Interestingly, the diameter of the part of the ring formed by C-terminal nuclease domain of the T4 motor is approximately equal to the diameter of the ASCE part of the ring in phi29. Furthermore, the distance between the top of the connector and the bottom of the ASCE ring in phi29 is approximately equal to the distance between the top of the connector and the bottom of the nuclease part of the ring in T4. The procedure for generating HerA/FtsK- and RecA-based models resulted in similar ring structures (Supplemental Figure S2), both of which fit the cryoEM density considerably better than the TLS-based model. Of the two, the FtsK-based model fit the observed density slightly better than the P4-based model, and subsequent discussion will refer to this model (Figure 3B and C).
Position of the C-terminus
Having fit the N-terminal ATPase domain of gp16 into its corresponding density, a nearly complete pseudo-atomic structure of an actively packaging motor engaged with the DNA substrate has been generated. The last component needed to complete this model is the C-terminal OB-domain of gp16. The only density in the reconstruction of stalled particles that is not accounted for corresponds to the second DNA contact point (Figure 3B, DNA contact 2, and Figure 3D). This volume of density is unlikely to be pRNA since all bases in this region of the pRNA are already accounted for in the model. Similarly, density for the connector and the DNA have also been identified. Thus, simply by the process of elimination, this density corresponds to the C-terminal OB-domain of gp16. This assignment is supported by the presence of weak density connecting this region to density corresponding to the N-terminal ATPase domain (Figures 3A and B); indeed, the C-terminus of the fitted ATPase domain is positioned at the beginning of this putative linker region (Figure 3B). Therefore, based on this interpretation, both domains of gp16 contact the DNA during packaging. Furthermore, if full-length FtsK monomers are superimposed on the fitted gp16, the C-terminal domain of FtsK is positioned in density assigned to the C-terminal domain of gp16 (Supplemental Figure S3). Although the two domains do not share any obvious sequence homology, they are both approximately the same size, consist primarily of beta-structure, and are similarly positioned relative to their N-terminal ASCE domains. The mechanistic implications of this structural conservation are discussed below. In contrast, the relative arrangements of the N- and C-terminal ATPase domains in phi29 is somewhat different than the proposed arrangement in the T4 ATPase; in particular, the relative orientation of the N- and C-terminal domains to the prohead reported for T4 is inverted compared to phi29 (compare Figure S1 to figures 3A,B and S3)
DNA translocation: identification of lumenal loops
In the model of the gp16 ring presented here, a lumenal loop is positioned to interact with the DNA (residues 120-130; Figures 3, 4, and 5; green). This loop has two positively charged residues (K122, R124) that could promote favorable interactions with the negatively charged phosphate backbone of dsDNA (Figure 4A). Such interactions are required given the demonstration of a critical, load-bearing contact between the motor and DNA in the dwell phase (Aathavan et al., 2009). Furthermore, the lumenal loop superimposes on the RNA translocating lever in the P4 packaging motor from the RNA phage phi12 (Mancini et al., 2004) (Supplemental Figure S4). In contrast, a different arginine was reported to function as the DNA translocating lever in a model of the ring ATPase from the thermophilic phage P74-26 (R101) (Hilbert et al., 2015). In our model of the phi29 ATPase ring, the nearest equivalent positively charged residues (K81 and R83) are situated near the outer radius of the ring, and are thus unlikely to play a role in DNA translocation.
Figure 4.

Identification of the trans-acting arginine finger R146. A) Ribbon diagram of gp16; mechanistically important residues, including R146, are labelled. The AMP-PNP substrate is shown as a stick figure, and the Mg2+ ion as a green sphere. The walker A and putative DNA translocating loops are colored magenta and green, respectively; the α-helix connecting the DNA translocating loop to the arginine finger R146 is shown in orange. B) SDA page gels showing the DNA packaging activity of gp16 mutants targeting arginines near R146. Arginine residues that cluster near the putative arginine finger R146 were mutated to lysine and assayed for the ability to support DNA packaging. To assess packaging, the packaged DNA that was protected from DNase treatment was extracted from particles and analyzed by agarose gel electrophoresis. Lanes were rearranged, indicated by a space between lanes, to facilitate viewing. C) Complementation experiments demonstrating that R146 is a trans-acting arginine finger. ATPase activity was measured for proheads complexed with mixtures of the catalytically inactive D/E mutant and the arginine finger mutant R146A. When mixed together, complementation occurs since the D/E mutant can contribute its arginine finger in-trans to an adjacent R146A subunit (n=4, error bars are SD). In a 50% mixing ratio, perfect complementation between a D/E adjacent to an R146 subunit would be 25%. Observed complementation between the mutants was 13.7% ± 1.6%. Similar results are obtained with the R146K mutant.
Figure 5.
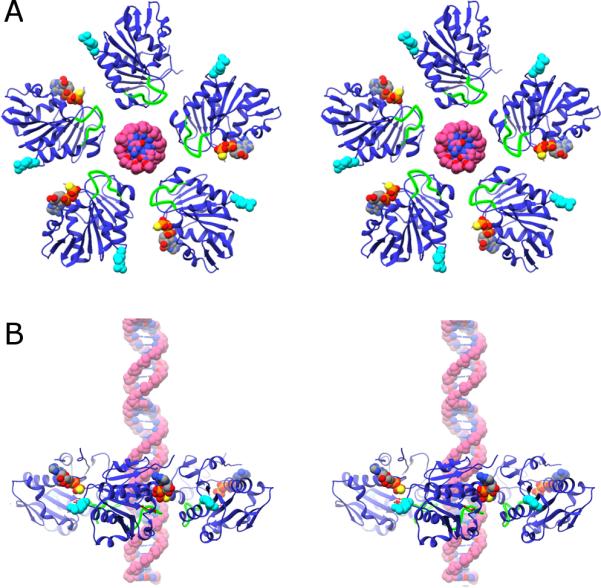
Ring structure of the gp16 and DNA. A) Cross-eyed stereo diagram of the ring structure of gp16 based on fitting the X-ray structure of the monomer into its corresponding cryoEM density in a 5-fold averaged reconstruction of particles stalled during packaging via the addition of γ-S-ATP. The viewing direction is from outside the phage looking toward the interior. The putative DNA translocating loop is shown in green, the arginine finger R146 is shown as cyan van der Waal spheres, and the substrate analog AMP-PNP and DNA as van der Waal spheres colored by element. B) Stereo-diagram of a side-view of the gp16 ring, colored as in (A); the capsid would be above the ring, and the back-half of the ring has been left out to facilitate viewing.
To test the roles of this loop in packaging, the positively charged residues R122 and K124 in the loop were mutated and the resulting proteins assayed for their ability to support DNA packaging and ATP hydrolysis. The mutants R122A and K124A had altered packaging phenotypes (Table 2); R122A did not support DNA packaging, and packaging in K124 was reduced ~2.5-fold compared to wild-type. To assess if these mutations impacted ATP hydrolysis, the mutants were assayed for ATPase activity as prohead/motor complexes (prohead/pRNA/gp16) in a DNA-free ATPase assay. Both of these mutants support ATPase activity in the absence of DNA at levels similar to wild type, suggesting that these mutations do not affect the ability to hydrolyze ATP. In contrast, the mutants described above that targeted conserved catalytic residues were null for both packaging and ATP hydrolysis (Table 2; key functional features of the gp16 ATPase domain structure). Thus, mutation of these charged loop residues impacted DNA movement, but not the production of energy necessary to power that movement.
Inter-subunit coordination: confirmation of the arginine finger
In the oligomeric arrangement of gp16 described above, the ATP binding site is located near the interface between two adjacent subunits (Figure 5). A similar arrangement is seen in other members of the ASCE superfamily, such as the FtsK DNA translocating motor and the RecA-like RNA packaging motor P4 from phage phi12 (Mancini et al., 2004; Massey et al., 2006). In these and other ASCE family members, it is proposed that ATP hydrolysis results in a conformational change that causes the arginine finger of one subunit to move into the active site of its neighbor, where it interacts with the negatively charged oxygens on the γ-phosphate of ATP.
Although R139 was recently reported to function as the arginine finger in the thermophilic phage P74-26 (Hilbert et al., 2015), there is no arginine near the equivalent position in gp16. In contrast, there are three arginines in gp16 (R146, R148, and R150) that are in the vicinity of the arginine finger in FtsK; of these, only R146 is strictly conserved in all phi29 relatives. However, R148 superimposes more directly on the FtsK arginine finger than does R146, and based on the model above, is closer to the ATP substrate in the neighboring subunit. Thus, identification of the gp16 arginine finger required examination of multiple arginine residues in this loop. Lysine substitutions were made for all three of these arginines, and each was assessed for its ability to support DNA packaging in vitro (Table 2; Figure 4B). Of the three mutants tested, only R146K was unable to support packaging, indicating that R146 is the arginine-finger in gp16.
To test whether R146 acts in cis or in trans, we conducted complementation experiments to test for restoration of ATPase activity by mixing an arginine finger mutant with a catalytically inactive mutant (Figure 4). Since mutation of the arginine finger to a lysine in some other ASCE NTPases resulted in low levels of residual NTPase activity (Wittinghofer et al., 1997), we used an R146A mutant in these experiments to ensure ATPase activity was completely abolished (Table 2; Figure 4B). Experiments were conducted by mixing the R146A with the catalytically-inactive Walker B D/E double mutant. Neither the D/E nor the R146A mutant supports ATP hydrolysis in the DNA-free ATPase assay as prohead/motor complexes (Table 2). We predicted that if the R146 arginine finger acts in trans then mixing these two mutants would result in partial restoration of ATPase activity; the D/E mutant would contribute its wild type arginine finger to a neighboring R146A mutant, thus stimulating its functional ATPase active site. As predicted, proheads mixed with combinations of the R146A and the D/E inversion mutants yielded considerably higher ATPase activity in DNA-free ATPase assays than either of these null mutants alone (Figure 4C). These results support a role for a trans-acting arginine finger, R146, in a DNA-independent mode of communication and coordination of the gp16 motor ATPase.
DISCUSSION
Mechanism of DNA packaging
ATP hydrolysis
To understand how the packaging motor functions, three operational levels of the packaging process must be addressed: 1) how ATP is hydrolyzed in an individual subunit; 2) how DNA is translocated; and 3) how trans-acting structural elements coordinate hydrolysis/translocation events between neighboring subunits. The structural and biochemical data presented here provide insight into each of these aspects of motor function. Regarding ATP hydrolysis, residues from the Walker A and Walker B motif interact with the ATP substrate such that is positioned for nucleophilic attack by the catalytic water molecule (Figures 2C and D). This water is bound to the catalytic glutamate (E119) immediately downstream of the Walker B motif, and is well aligned for in-line SN1 nucleophilic attack at the gamma phosphate of ATP. In other members of the ASCE NTPase superfamily, it has been proposed that an equivalent water molecule (Thomsen and Berger, 2008) is activated for hydrolysis via its interaction with the carboxylate of the catalytic glutamate, which would help extract a proton, and with the positively charged Mg+ that would stabilize the resulting hydroxide ion (Thomsen and Berger, 2008). The rate of hydrolysis may be accelerated by the insertion of the positively charged arginine finger from a neighboring molecule that acts in trans to stabilize the negative charge that develops on the γ-phosphate oxygens in the transition state of ATP hydrolysis. The phosphosensor N158 is positioned to bind the γ-phosphate leaving group. In addition to further accelerating hydrolysis by stabilizing the leaving group, the phosphosensor has also been proposed to couple ATP hydrolysis to DNA translocation in other ASCE family members (Iyer et al., 2004a; Iyer et al., 2004b; Story and Steitz, 1992; Yoshida and Amano, 1995), though the mechanism of this coupling is unknown.
DNA translocation
In our structural model of the packaging motor, five loops line the lumen of the pentameric ATPase ring (Figures 3 and 4). The proximity of these lumenal loops to the dsDNA substrate suggests a direct role in DNA translocation. Furthermore, this loop superimposes with the proposed translocating levers in the phi12 (Supplemental Figure S4) and HerA nucleic acid translocases (Mancini et al., 2004; Rzechorzek et al., 2014), suggesting that these loops might be involved in DNA translocation. Indeed, the loop mutants R122A and K124A have decreased packaging activities yet retain near wild-type ATP hydrolysis capabilities as prohead/motor complexes, providing experimental evidence that these loops are involved in DNA translocation (Table 2). Single molecule experiments show that the phi29 motor makes specific electrostatic contacts with the DNA phosphate backbone during the dwell phase of packaging, whereas in the burst phase, the motor employs non-specific contacts to drive translocation of the DNA into the capsid (Aathavan et al., 2009). These data suggest specific roles for different residues in the loop during translocation as suggested by single molecule experiments. In the dwell phase, one or both of the positively charged residues in the loop are likely involved in the prolonged electrostatic contact between the motor and the negatively charged phosphate backbone of the DNA helix. In contrast, one of the bulkier side chains within the loop may function as a steric paddle that pushes DNA into the head upon ATP hydrolysis; Y129 is a candidate for the steric paddle based on its position in the loop (Figure 4A) and its conservation in all phi29 relatives. A similar bulky residue has been identified in the related unfoldase Clpx, and is believed to interact with the polypeptide substrate in a non-specific fashion (Glynn et al., 2009). It is not clear exactly how ATP hydrolysis is coupled to movement of the lumenal loop, but since the loop is adjacent to the active site (the catalytic glutamate E119 is the first residue of the loop) and close to the putative phosphsensor N158 (Figures 2C and 5A), it is well positioned to respond to the state of the bound nucleotide.
The role of the C-terminal contact in the DNA translocating model presented above is not entirely clear, but it also may be involved in holding the DNA in place during the dwell phase. Alternatively, this observed second contact may play a more active role in DNA translocation, as in the inchworm-type mechanism proposed for FtsK (Massey et al., 2006). In this type of mechanism, ATP hydrolysis causes a DNA-bound domain within a subunit to move with respect to the other domain, resulting in translocation of the DNA into the capsid. For gp16, the two contacts may be the lumenal loop in the N-terminal ATPase domain and the contact observed for the C-terminal domain. Similar inchworm-type packaging models for bacteriophages T4 and SF6 were proposed based on two observed conformations of their ATPases (Sun et al., 2008; Zhao et al., 2013); conformational switching between these two states, driven by ATP binding, hydrolysis and release, would provide the power stroke for a rotary inchworm-type mechanism. In contrast to the FtsK inchworm model where two domains in a subunit contact the DNA, only the C-terminal nuclease domain the T4/SF6 large terminase is proposed to contact the DNA (albeit via entirely different sides of the domain) (Sun et al., 2008; Zhao et al., 2013). Thus while it is clear that two DNA contacts are present in the dwell phase, it is not yet known if two contacts within a subunit are part of the translocation mechanism during the burst phase. Future studies targeting visualization of the motor in different mechano-chemical states will provide the structural snapshots required to document how different motor components interact with each other and with the DNA.
In yet another model for packaging, DNA translocation is proposed to be driven by transitions between A- and B-form DNA (Dixit et al., 2012; Dixit et al., 2013; Harvey, 2015; Ray et al., 2010). In one version of this model, the ATPase applies a force directly on the DNA causing it to compress into the shorter A-form DNA (Dixit et al., 2012; Dixit et al., 2013; Ray et al., 2010). Based on the structural data presented here, a translocation cycle could begin such that the DNA contact points in the N- and C-terminal domains have both engaged DNA. During ATP hydrolysis, the lower contact would move closer to the upper contact, compressing the DNA in the process. The upper contact point would then release DNA such that it can re-extend only in the capsid direction, thus endowing the process with directionality. In an alternate version of this model, the transition between A- and B-form DNA is driven by sequentially dehydrating and rehydrating the DNA rather than by direct application of force (Harvey, 2015). This model requires a DNA-dehydrating component in between two DNA gripping points; gripping and release of the DNA substrate is coordinated with de- and re-hydration such that the DNA translocates into the capsid (Harvey, 2015). Based on the structural data presented here, the connector and the N-terminal domain of the ATPase could provide the two DNA grips, and the C-terminal domain would function as the dehydrator. The proposed gripping and releasing requires additional levels of coordination between various motor components that extend beyond coordination of the timing of hydrolysis at different positions around the ring.
Motor coordination and communication
Within a single ATPase subunit, the lumenal loop and the arginine finger R146 reside at opposite ends of the same α-helix (Figures 2C, 4, and 5A); given the relative rigidity of α-helices, the movement of one of these features may be coupled to the movement of the other. Indeed, normal mode analysis of the structure of the N-terminal ATPase domain of gp16 indicates that movement of the lumenal loop and the arginine finger are coupled (see Supplemental Figure S5, for an example of one of the many major vibrational modes that couples the movement of these two structural elements). Hence, as one subunit translocates DNA via the lumenal loop, it might also position its arginine finger into the active site of the next subunit to trigger hydrolysis and DNA translocation and/or sense the nucleotide-bound state of a neighbor in the ring. It is also likely that additional elements, including the DNA substrate itself, have a role in communication/coordination in the motor.
Single molecule studies on the phi29 motor demonstrated that the activities of individual subunits around the ATPase ring are highly coordinated (Chistol et al., 2012; Liu et al., 2014a; Moffitt et al., 2009). Sequential interlaced ADP release/ATP binding during the dwell and subsequent firing/stepping during the burst require precisely timed communication around the ring. The structural data and functional analysis presented suggest how molecular events are coupled within and between ATPase subunits. In particular, our results indicate that adjacent subunits in the ring operate as functional pairs to hydrolyze ATP and translocate DNA. The structural data and DE/R146A complementation experiments described above, along with sequence and structural homology to other ASCE family members (Burroughs et al., 2007), suggest that R146 functions in trans, triggering (or sensing) hydrolysis of ATP in the adjacent subunit; pairwise interaction is implicit with in trans-activation. Together, these results indicate that the functional unit of the phi29 packaging motor consists of elements from two adjacent ATPase monomers. This functional pairing exists not only in other ring forming members of the ASCE superfamily, but also in the monomeric ATPases such RecA and SF3 helicases. Although these motor proteins are single polypeptides, the ASCE fold is duplicated within their sequences such that NTP binding site is positioned between these covalently linked ASCE domains and hydrolysis is triggered via pseudo in trans activation of one domain via an arginine finger originating from the other (Burroughs et al., 2007; Thomsen and Berger, 2008)
Integrating these results with the mechano-chemical cycle of the motor determined from single molecule studies (Chistol et al., 2012) suggests the following model for coordinated DNA translocation. A mechano-chemical cycle begins with ATP bound to all five subunits (figure 1B; *). Once hydrolysis occurs in the first subunit to initiate a burst (figure 1B; **), the motor operates as four pairs of subunits. Hydrolysis in the first subunit triggers hydrolysis in the next via the trans-acting arginine finger, resulting in DNA translocation (possibly via the proposed coupling of the DNA translocating loop and arginine finger) followed by passing DNA to the next subunit. These events are propagated around the ring until all subunits have hydrolyzed ATP (figure 1B, ***). The motor must now enter a dwell phase to initiate coordinated ADP-ATP exchange, and thus the fifth subunit and the first cannot continue this pairwise interaction. As a consequence, hydrolysis in the first subunit at the beginning of the burst (figure 1B, going from * to **) cannot be triggered by this same arginine finger-mediated pair-wise mechanism since the subunit preceding it has not yet hydrolyzed its ATP (figure 1B, *). Instead, the first hydrolysis event must either occur stochastically, or must be triggered by some other more subtle aspect of motor operation such as completion of ADP/ATP exchange to form a symmetric ATP-bound pentameric ring. Hence, although there are five subunits, only four sets of coordinated pairs operate in the motor to translocate DNA since the first hydrolysis in the ATP-loaded ring is not functionally paired in the same manner with the preceding subunit. Indeed, pairwise operation of the motor provides a mechanistic basis for the observation that although five hydrolysis events occur during the burst, there are only four DNA translocation steps (Moffitt et al., 2009). Furthermore, the exclusion of one subunit from a functionally paired interface in the pentameric ring provides a simple explanation for the origin of the “special” subunit that holds the DNA during the dwell and initiates the burst phase. Similarly, the interlaced ADP release and ATP binding that occurs in the dwell phase presents the same challenge in that all subunits are occupied by ADP at the end of a burst (figure 1B, **). Hence, one subunit must go first without a trigger from its neighbor, whereas the remaining four are coordinated as equivalent pairs in the pentameric ring. Again, this points to the essential role of a unique subunit that initiates both the ATP hydrolysis cascade and the ADP release sequence during the burst and dwell, respectively.
METHODS
Production of Packaging Components
Production of proheads
Proheads were produced by infection of Bacillus subtilis 12A (sup-) with the phi29 mutant sus 8.5(900)-16(300)-14(1241) (defective in head fibers and the packaging ATPase) and purified by sucrose gradients as previously described (Grimes and Anderson, 1997; Zhao et al., 2008).
Production of pRNA
120b pRNA was produced from the plasmid pRT71 by in vitro transcription using T7 RNA polymerase and purified by denaturing urea polyacrylamide gel electrophoresis as previously described (Reid et al., 1994).
Prodution of RNA-free proheads reconstituted with pRNA
Purified proheads were treated with RNase A and re-purified as previously described (Zhao et al., 2008). The RNA-free proheads were reconstituted with 120b as previously described (Zhao et al., 2008).
Production of genomic DNA for packaging assays
DNA-gp3 was extracted from phage and isolated on a CsCl gradient as previously described (Grimes and Anderson, 1997).
Production of gp16 used for packaging and activity assays
To facilitate rapid site-directed mutagenesis, gene 16 was cloned into the SUMOpro plasmid (LifeSensors) and expressed in E. coli. A SacI site was introduced downstream of the C-terminal glycine-glycine codons of the SUMO protein moiety in the SUMOpro plasmid to make pSUMO-SacI-XhoI. A SacI-gp16-XhoI PCR product was amplified from the pSAC-gp16 plasmid (Koti et al., 2008) and ligated into the pSUMO-SacI-XhoI plasmid. PCR was used to remove the six-nucleotide SacI site, changing the first three nucleotides to a serine codon to improve SUMO protease digestion and the second three nucleotides to the native aspartate codon corresponding to the second residue in gp16. The plasmid was transformed into competent Rosetta™ (DE3) cells (Novagen) and selected for by growth in LB supplemented with kanamycin (50μg/ml) and chloroamphenicol (34μg/ml). The sequence of the SUMO-gp16 fusion construct was verified by DNA sequencing. The SUMO-gp16 plasmid was used as a template for creating gp16 mutants by site-directed mutagenesis using the inverse PCR based on the method of Wang and Wilkinson (Wang and Wilkinson, 2000, 2001). Plasmid DNA was sequenced to verify the presence of the desired mutations.
Purification of wild-type and mutant gp16 for packaging and ATPase assays
Rosetta cells containing SUMO-gp16 plasmid were grown overnight in LB medium containing 50μg/ml kanamycin and 34μg/ml chloroamphenicol at 37°C. The next morning the culture was then diluted 1/100 in LB medium containing 50μg/ml kanamycin and 34μg/ml chloroamphenicol at 37°C to a density of OD600=0.5. IPTG was added to 0.3mM and the culture incubated at 37°C for 5 min and then shifted to 18°C for protein expression. After overnight induction, the cells were collected by centrifugation and concentrated 15-fold in buffer containing 50 mM Tris–HCl, pH 8, 500 mM NaCl, 5% glycerol, and 1 mM Tris(2-carboxyethyl)phosphine (TCEP) and lysed by passage through a French press. MgCl2 was added to 2.5mM and DNase to 5μg/ml and the sample incubated at room temp for 15 min. The lysate was clarified in the SS34 rotor at 10,000 rpm for 40 min at 4°C. The supernatant was incubated with Talon resin (Clontech) that had been equilibrated in wash buffer (50 mM Tris–HCl, pH 8, 400 mM NaCl, 5% glycerol, and 1 mM TCEP) at 4°C for 60 min. The resin with SUMO-gp16 bound was then washed with 10 volumes wash buffer. The SUMO-gp16 fusion protein was eluted in 5 volumes elution buffer (50 mM Tris–HCl, pH 8, 400 mM NaCl, 5% glycerol, 100mM Imidazole (pH 8) and 1 mM TCEP) and 0.5ml fractions were collected. The fractions were analyzed by SDS-PAGE to identify peak fractions. To cleave the SUMO tag and recover native gp16 protein, 100μl of SUMO-gp16 fusion protein was mixed with 200μl wash buffer and 2.5 units of SUMO protease (Life Technologies) and incubated at 4°C overnight. Ni-Sepharose™ 6 Fast Flow resin (GE Healthcare) was equilibrated with wash buffer and then incubated with the cleaved protein mixture for 60 min at 4°C. The resin containing the bound SUMO tag was pelleted in the microfuge at 2000×g for 5 min and the supernatant containing the native gp16 was recovered for use in biological assays.
DNA packaging assay
The in vitro DNA packaging assay is based on a DNase protection assay and was performed as described previously (Grimes and Anderson, 1997; Zhao et al., 2008). Briefly, proheads (8.3 nM), DNA-gp3 (4.2 nM),and either wild-type or mutant gp16 (166 to 208 nM) were mixed together in 0.5× TMS buffer in 20 μl and incubated for 5 min at room temperature. ATP was then added to 0.5 mM to initiate packaging. After 15 min incubation, the mixture was treated with DNase I (1 μg/ml final) and incubated for 10 min to digest the unpackaged DNA. An EDTA/Proteinase K mix was then added to the reaction mixture (25mM and 500 μg/ml final concentration, respectively) and incubated for 30 min at 65°C to inactivate the DNase I and release the protected, packaged DNA from particles. The packaged DNA was analyzed by agarose gel electrophoresis. Packaging efficiency was calculated by densitometry using a UVP Gel Documentation System.
Production of DNA Packaging Intermediates for cryoEM
To generate the stalled DNA packaging intermediates for cryoEM reconstruction, packaging reactions consisting of DNA-gp3, RNA-free proheads reconstituted with 120b pRNA and gp16 were assembled at 2× concentration (see above) and ATP was added to initiate DNA packaging. Three minutes post-ATP addition, gamma-S-ATP was added to 100μM (Roche) (ATP concentration is 500μM) and incubated for 2 minutes. 1 unit of RQ DNase I (Promega) was added and incubated at room temperature for 10 minutes. The sample was placed on ice until grid preparation for cryoEM imaging. A prohead:DNA ratio of 2:1 was used to maximize packaging efficiency. Three minutes packaging corresponds to ~75% head-full..
ATPase assays
ATPase activity of prohead/gp16 (wt or mutant) motor complexes was determined by measuring production of inorganic phosphate by the EnzChek Phosphate Assay Kit (Life Technologies) as described previously (Harjes et al., 2012). Briefly, a reaction mixture containing reaction buffer (either kit buffer [50mM Tris, pH 7.5, 1mM MgCl2, 0.1mM sodium azide] or TM buffer [25 mM Tris, pH7.6, 5 mM MgCl2]), 0.2 mM of MESG (2-amino-6-mercapto-7-methylpurine riboside) with proheads (4.2nM) and gp16 (125nM) in 90μl was preincubated at room temperature for 10 min in the presence of PNP (purine nucleoside phosphorylase, 0.1 unit). ATP was added to 1mM to initiate the reaction and production of Pi measured in the spectrophotometer at 360 absorbance for 10 min. For the complementation assay, the D/E and R146A mutants were mixed in the ratios indicated to yield the 125nM gp16 concentration.
Expression and purification of the N-terminal ATPase domain of gp16 for X-ray crystallography
Cloning, expression, purification, and crystallization of the N-terminal ATPase domain of gp16 will be described elsewhere (Mao et al. unpublished). Briefly, a codon-optimized (DNA2.0), seleno-methionine substituted, recombinant gp16 construct (a.a. 1-207) was overexpressed in E.coli and purified using Ni2+-affinity chromatography and size-exclusion chromatography. Purified protein was used for crystallization trials in 96-well plates (ARI Intelliplate) using a Phoenix crystallization robot (Art Robbins Instruments). Small plate-like crystals grew 2.8 M sodium acetate trihydrate, pH7.0. A Complete dataset to 1.8 Å resolution was collected at the Gulf Coast Protein Crystallography Consortium (GCPCC) protein crystallography beam line at the Center for Advanced Microstructure and Devices (CAMD) synchrotron at Baton Rouge, Louisiana, USA. Data were indexed, integrated, and scaled using the HKL 3000 (Minor et al., 2006); the Phenix software suite was used for subsequent structure solution and refinement (Adams et al., 2010). Data collection and processing statistics are listed in Table 1. The X-ray structure was deposited under PDB accession number 5HD9
Cryo-electron microscopy
Approximately 3 μl of prohead particles stalled during packaging (see above production of packaging intermediates for cryoEM) were applied to grids prior to plunge freezing. Samples were manually flash-frozen on holey carbon grids. Images were recorded at 60K magnification using a 200 KV JEOL 2200FS transmission electron microscope equipped with an in-column omega energy filter and a 4k by 4k Gatan CCD camera, and with electron dose levels of approximately 25e−/Å2 (Suppemental Figure S1). These micrographs have a pixel size of 1.94 Å. Defocus ranges of ~0.74 to 3.0 μm were used for data collection. Image processing was performed with EMAN (Ludtke et al., 1999). Briefly, particles were picked using a semi-automated boxing procedure followed by manual culling of the boxed particle set. CTF parameters were determined semi-automatically for particles from each micrograph; particles were then phase flipped prior to subsequent classification and alignment. Equally spaced angular projections of a modified phi29 map from the EM-database (EMD–1419) (Morais et al., 2005) were used for initial particle alignment and classification, and the resulting map used for the next refinement cycle. This iterative procedure was repeated until convergence. A total of 12,093 particles were incorporated into the final reconstructions of the packaging intermediates. The resolution of the reconstruction was determined by the Fourier shell correlation (FSC) method using a correlation coefficient of 0.5 between two independent half data sets as the cutoff criterion. The cryo-EM structure was deposited under EMDB accession number EMD-6560.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by Public Health Service grant GM095516 (to S.G. and M.C.M.) and GM059604 (to S.G.) from the National Institutes of Health. We would also like to acknowledge the Sealy Center for Structural Biology and Molecular Biophysics (SCSB) for support of the UTMB X-ray and cryoEM core facilities; the Texas Advanced Computing Center (TACC) for providing computational resources; Henry Bellamy and the Center for Advanced Microstructures and Devices (CAMD) for assistance with X-ray data collection; and Jiao Xie, Wei Zhao, and Bora Kuyomba Faulkner for technical assistance with the project
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.M. solved the X-ray structure of gp16; M.S. determined all cryoEM reconstructions; E.R. helped with crystallization of gp16; M.B.S. and M.W. helped with cryoEM data collection; R.A. helped with design and production of gp16 mutants; S.G. helped with design and interpretation of DNA translocating loops; P.J.J. over saw biochemical and genetic experiments; and M.C.M collected cryoEM data and oversaw all structural experiments. S.G., P.J.J., and M.C.M. wrote the manuscript.
References
- Aathavan K, Politzer AT, Kaplan A, Moffitt JR, Chemla YR, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Substrate interactions and promiscuity in a viral DNA packaging motor. Nature. 2009;461:669–673. doi: 10.1038/nature08443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig K, Menz RI, Montgomery MG, Leslie AG, Walker JE. Structure of bovine mitochondrial F(1)-ATPase inhibited by Mg(2+) ADP and aluminium fluoride. Structure. 2000;8:567–573. doi: 10.1016/s0969-2126(00)00145-3. [DOI] [PubMed] [Google Scholar]
- Burroughs AM, Iyer LM, Aravind L. Comparative genomics and evolutionary trajectories of viral ATP dependent DNA-packaging systems. Genome Dyn. 2007;3:48–65. doi: 10.1159/000107603. [DOI] [PubMed] [Google Scholar]
- Cao S, Saha M, Zhao W, Jardine PJ, Zhang W, Grimes S, Morais MC. Insights into the structure and assembly of the bacteriophage 29 double-stranded DNA packaging motor. J Virol. 2014;88:3986–3996. doi: 10.1128/JVI.03203-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistol G, Liu S, Hetherington CL, Moffitt JR, Grimes S, Jardine PJ, Bustamante C. High degree of coordination and division of labor among subunits in a homomeric ring ATPase. Cell. 2012;151:1017–1028. doi: 10.1016/j.cell.2012.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Lu C, Zhao W, Rajashankar KR, Anderson DL, Jardine PJ, Grimes S, Ke A. Structure and assembly of the essential RNA ring component of a viral DNA packaging motor. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1016690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit AB, Ray K, Black LW. Compression of the DNA substrate by a viral packaging motor is supported by removal of intercalating dye during translocation. Proc Natl Acad Sci U S A. 2012;109:20419–20424. doi: 10.1073/pnas.1214318109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit AB, Ray K, Thomas JA, Black LW. The C-terminal domain of the bacteriophage T4 terminase docks on the prohead portal clip region during DNA packaging. Virology. 2013;446:293–302. doi: 10.1016/j.virol.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes S, Anderson D. The bacteriophage phi29 packaging proteins supercoil the DNA ends. J Mol Biol. 1997;266:901–914. doi: 10.1006/jmbi.1996.0843. [DOI] [PubMed] [Google Scholar]
- Grimes S, Jardine PJ, Anderson D. Bacteriophage phi 29 DNA packaging. Adv Virus Res. 2002;58:255–294. doi: 10.1016/s0065-3527(02)58007-6. [DOI] [PubMed] [Google Scholar]
- Guo P, Grimes S, Anderson D. A defined system for in vitro packaging of DNA-gp3 of the Bacillus subtilis bacteriophage phi 29. Proc Natl Acad Sci U S A. 1986;83:3505–3509. doi: 10.1073/pnas.83.10.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Schwartz C, Haak J, Zhao Z. Discovery of a new motion mechanism of biomotors similar to the earth revolving around the sun without rotation. Virology. 2013;446:133–143. doi: 10.1016/j.virol.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo PX, Erickson S, Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage phi 29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- Harjes E, Kitamura A, Zhao W, Morais MC, Jardine PJ, Grimes S, Matsuo H. Structure of the RNA claw of the DNA packaging motor of bacteriophage Phi29. Nucleic Acids Res. 2012;40:9953–9963. doi: 10.1093/nar/gks724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC. The scrunchworm hypothesis: transitions between A-DNA and B-DNA provide the driving force for genome packaging in double-stranded DNA bacteriophages. J Struct Biol. 2015;189:1–8. doi: 10.1016/j.jsb.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbert BJ, Hayes JA, Stone NP, Duffy CM, Sankaran B, Kelch BA. Structure and mechanism of the ATPase that powers viral genome packaging. Proc Natl Acad Sci U S A. 2015;112:E3792–3799. doi: 10.1073/pnas.1506951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004a;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Makarova KS, Koonin EV, Aravind L. Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res. 2004b;32:5260–5279. doi: 10.1093/nar/gkh828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koti JS, Morais MC, Rajagopal R, Owen BA, McMurray CT, Anderson DL. DNA packaging motor assembly intermediate of bacteriophage phi29. J Mol Biol. 2008;381:1114–1132. doi: 10.1016/j.jmb.2008.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. Structural framework for DNA translocation via the viral portal protein. EMBO J. 2007;26:1984–1994. doi: 10.1038/sj.emboj.7601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chistol G, Bustamante C. Mechanical operation and intersubunit coordination of ring-shaped molecular motors: insights from single-molecule studies. Biophys J. 2014a;106:1844–1858. doi: 10.1016/j.bpj.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chistol G, Hetherington CL, Tafoya S, Aathavan K, Schnitzbauer J, Grimes S, Jardine PJ, Bustamante C. A viral packaging motor varies its DNA rotation and step size to preserve subunit coordination as the capsid fills. Cell. 2014b;157:702–713. doi: 10.1016/j.cell.2014.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Mancini EJ, Kainov DE, Grimes JM, Tuma R, Bamford DH, Stuart DI. Atomic snapshots of an RNA packaging motor reveal conformational changes linking ATP hydrolysis to RNA translocation. Cell. 2004;118:743–755. doi: 10.1016/j.cell.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Massey TH, Mercogliano CP, Yates J, Sherratt DJ, Lowe J. Double-stranded DNA translocation: structure and mechanism of hexameric FtsK. Mol Cell. 2006;23:457–469. doi: 10.1016/j.molcel.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Mitchell MS, Matsuzaki S, Imai S, Rao VB. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 2002;30:4009–4021. doi: 10.1093/nar/gkf524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Aathavan K, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Intersubunit coordination in a homomeric ring ATPase. Nature. 2009;457:446–450. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais MC. The dsDNA packaging motor in bacteriophage o29. Advances in experimental medicine and biology. 2012;726:511–547. doi: 10.1007/978-1-4614-0980-9_23. [DOI] [PubMed] [Google Scholar]
- Morais MC, Choi KH, Koti JS, Chipman PR, Anderson DL, Rossmann MG. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of phi29. Mol Cell. 2005;18:149–159. doi: 10.1016/j.molcel.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Morais MC, Koti JS, Bowman VD, Reyes-Aldrete E, Anderson DL, Rossmann MG. Defining molecular and domain boundaries in the bacteriophage phi29 DNA packaging motor. Structure. 2008;16:1267–1274. doi: 10.1016/j.str.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais MC, Tao Y, Olson NH, Grimes S, Jardine PJ, Anderson DL, Baker TS, Rossmann MG. Cryoelectron-microscopy image reconstruction of symmetry mismatches in bacteriophage phi29. J Struct Biol. 2001;135:38–46. doi: 10.1006/jsbi.2001.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadanaciva S, Weber J, Wilke-Mounts S, Senior AE. Importance of F1-ATPase residue alpha-Arg-376 for catalytic transition state stabilization. Biochemistry. 1999;38:15493–15499. doi: 10.1021/bi9917683. [DOI] [PubMed] [Google Scholar]
- Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 2004;146:106–112. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Ray K, Sabanayagam CR, Lakowicz JR, Black LW. DNA crunching by a viral packaging motor: Compression of a procapsid-portal stalled Y-DNA substrate. Virology. 2010;398:224–232. doi: 10.1016/j.virol.2009.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid RJ, Bodley JW, Anderson D. Characterization of the prohead-pRNA interaction of bacteriophage phi 29. J Biol Chem. 1994;269:5157–5162. [PubMed] [Google Scholar]
- Rickgauer JP, Fuller DN, Grimes S, Jardine PJ, Anderson DL, Smith DE. Portal motor velocity and internal force resisting viral DNA packaging in bacteriophage phi29. Biophys J. 2008;94:159–167. doi: 10.1529/biophysj.107.104612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann MG, Moras D, Olsen KW. Chemical and biological evolution of nucleotide-binding protein. Nature. 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- Rzechorzek NJ, Blackwood JK, Bray SM, Maman JD, Pellegrini L, Robinson NP. Structure of the hexameric HerA ATPase reveals a mechanism of translocation-coupled DNA-end processing in archaea. Nat Commun. 2014;5:5506. doi: 10.1038/ncomms6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz C, De Donatis GM, Fang H, Guo P. The ATPase of the phi29 DNA packaging motor is a member of the hexameric AAA+ superfamily. Virology. 2013;443:20–27. doi: 10.1016/j.virol.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature. 2000;408:745–750. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature. 2001;413:748–752. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- Story RM, Steitz TA. Structure of the recA protein-ADP complex. Nature. 1992;355:374–376. doi: 10.1038/355374a0. [DOI] [PubMed] [Google Scholar]
- Sun S, Kondabagil K, Draper B, Alam TI, Bowman VD, Zhang Z, Hegde S, Fokine A, Rossmann MG, Rao VB. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell. 2008;135:1251–1262. doi: 10.1016/j.cell.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Kondabagil K, Gentz PM, Rossmann MG, Rao VB. The structure of the ATPase that powers DNA packaging into bacteriophage T4 procapsids. Mol Cell. 2007;25:943–949. doi: 10.1016/j.molcel.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Thomsen ND, Berger JM. Structural frameworks for considering microbial protein-and nucleic acid-dependent motor ATPases. Mol Microbiol. 2008;69:1071–1090. doi: 10.1111/j.1365-2958.2008.06364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wilkinson MF. Site-directed mutagenesis of large (13-kb) plasmids in a single-PCR procedure. BioTechniques. 2000;29:976–978. doi: 10.2144/00295bm09. [DOI] [PubMed] [Google Scholar]
- Wang J, Wilkinson MF. Deletion mutagenesis of large (12-kb) plasmids by a one-step PCR protocol. BioTechniques. 2001;31:722–724. doi: 10.2144/01314bm01. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, Scheffzek K, Ahmadian MR. The interaction of Ras with GTPase-activating proteins. FEBS Lett. 1997;410:63–67. doi: 10.1016/s0014-5793(97)00321-9. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Amano T. A common topology of proteins catalyzing ATP-triggered reactions. FEBS Lett. 1995;359:1–5. doi: 10.1016/0014-5793(94)01438-7. [DOI] [PubMed] [Google Scholar]
- Yu J, Moffitt J, Hetherington CL, Bustamante C, Oster G. Mechanochemistry of a viral DNA packaging motor. J Mol Biol. 2010;400:186–203. doi: 10.1016/j.jmb.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Zhao H, Christensen TE, Kamau YN, Tang L. Structures of the phage Sf6 large terminase provide new insights into DNA translocation and cleavage. Proc Natl Acad Sci U S A. 2013;110:8075–8080. doi: 10.1073/pnas.1301133110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Morais MC, Anderson DL, Jardine PJ, Grimes S. Role of the CCA bulge of prohead RNA of bacteriophage o29 in DNA packaging. J Mol Biol. 2008;383:520–528. doi: 10.1016/j.jmb.2008.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.