

Summary
Porphyromonas gingivalis is a keystone pathogen in the development and progression of periodontal disease. Obstacles to the development of saturated transposon libraries have previously limited transposon mutant-based screens as well as essential gene studies. We have developed a system for efficient transposon mutagenesis of P. gingivalis using a modified mariner transposon. Tn-seq is a technique that allows for quantitative assessment of individual mutants within a transposon mutant library by sequencing the transposon-genome junctions and then compiling mutant presence by mapping to a base genome. Using Tn-seq, it is possible to quickly define all the insertional mutants in a library and thus identify non-essential genes under the conditions in which the library was produced. Identification of fitness of individual mutants under specific conditions can be performed by exposing the library to selective pressures.
Keywords: Porphyromonas gingivalis, transposon mutagenesis, essential genes, Mariner, Tn-seq
1. Introduction
Porphyromonas gingivalis, a Gram-negative, anaerobic, asaccharolytic, black-pigmenting bacterium, is a keystone pathogen in the development and progression of periodontal disease (1–3). While many important virulence factors of P. gingivalis have been identified, tools for high-throughput screening of genic and intergenic function have been limited until recently (4–7). Transposon mutagenesis, an important tool for creating inactivation mutants, utilizes a mobile genetic element to generate insertional mutations. The value of transposon mutagenesis is based on the ability of the transposable element to randomly insert into sites throughout the host genome, while giving only one insertion per strain. While transposon mutagenesis has been used to great effect in many bacterial species, early attempts to create transposon mutant libraries of P. gingivalis using transposons based on Tn4435 and Tn4400 were limited by preferential insertion into ‘hot-spots’, cryptic vector insertions, and a limited range of P. gingivalis strains that could be mutagenized (8–10). The Mariner transposon, first identified and described in Drosophila melanogaster, has been highly effective for in vitro bacterial mutagenesis in phylogenetically, physiologically, and metabolically diverse species(11–13). The only well documented constraint or preference for Mariner transposition is preferential insertion into ‘TA’ nucleotide sequences, which are abundant throughout genomes even in the face of GC-content skewing. A recent advance in analysis of bacterial transposon libraries has been the application of massively parallel sequencing technology to quantitatively identify the location of the transposon insertions in a library pool by sequencing of the transposon-genome junctions (14, 15). These strategies have been variously named Tn-seq, HITS, or INSeq (16–18). In this chapter, we describe modifications in the processes of transposon library creation and screening by Tn-seq that we developed to optimize the systems for use with P. gingivalis (19). Steps involved include library creation, testing of library complexity by nested semi-random PCR, preparation of the library for high-throughput screening, and bioinformatic analysis of outputs from Tn-seq.
2. Materials
2.1 Transposon Mutagenesis (16, 19)
- Bacterial strains and plasmids
- P. gingivalis
- E. coli S17-1 λpir
- Plasmids pSAM and pSAM_Bt
- Media
-
P. gingivalis solid media:Blood agar plates (BAPHK)
-
▪Trypticase soy agar
-
▪Defibrinated sheep’s blood (5.0% vol/vol)
-
▪Hemin (5.0 μg/ml)
-
▪Vitamin K (0.5 μg/ml)
-
▪
-
P. gingivalis liquid culture:Brain-Heart Infusion Broth (BHIHKSbcStgC)
-
▪Brain-Heart infusion
-
▪Yeast extract (1.0 mg/ml)
-
▪Hemin (5.0 μg/ml)
-
▪Vitamin K (0.5 μg/ml)
-
▪Sodium bicarbonate (1.0 mg/ml)
-
▪Sodium thioglycolate (0.25 mg/ml)
-
▪Cysteine (0.5 mg/ml)
-
▪
- P. gingivalis antibiotics Gentamicin (25–50 μg/ml) and Erythromycin (2–10 μg/ml)
- E. coli Luria broth base (LB) or Luria agar
- E. coli antibiotic Carbenicillin (50 μg/ml)
-
GasPak™ EZ Anaerobe Pouch System (BD Biosciences) (http://www.bdbiosciences.com/)
2.2 Nested Semi-Random PCR
DNeasy (Qiagen) (http://www.qiagen.com/)
Nuclease-free water (Ambion) (Invitrogen) (http://www.lifetechnologies.com/)
GoTaq DNA Polymerase (Promega) (http://www.promega.com/)
2.3 Library Preparation for Tn-seq (18–20)
2 mL round-bottom microfuge tubes
Branson 450 sonifier
High intensity cup horn
Circulating water bath
TdT enzyme (Promega) (http://www.promega.com/)
5× TdT reaction buffer (Promega) (http://www.promega.com/)
dideoxy CTP (Affymetrix) (www.affymetrix.com/)
dCTP
dNTP mix
Easy-A DNA polymerase mix (Agilent) (www.agilent.com/)
10× Easy-A DNA polymerase reaction buffer (Agilent) (www.agilent.com/)
Performa gel filtration cartridge (Edge Biosystems) (http://www.edgebio.com/)
Thermocycler (BioRad) (http://www.bio-rad.com/)
Nanodrop/spectrophotometer (Thermo Scientific) (http://www.thermofisher.com/)
ChemiDoc XRS+ imaging system (BioRad) (http://www.bio-rad.com/)
Illumina Genome Analyzer II (Illumina) (http://www.illumina.com/)
ABI 3130XL DNA sequencer (Invitrogen) (http://www.lifetechnologies.com/)
3. Methods
Media and culture conditions (19)
Wild type (WT) Porphyromonas gingivalis strain ATCC 33277 [Note1] was grown and maintained at 37°C under anaerobic conditions using the GasPak™ EZ Anaerobe Pouch System [Note5]. Blood agar plates (BAPHK) containing trypticase soy agar supplemented with defibrinated sheep’s blood (5% vol/vol), hemin (5 μg/ml), and menadione (0.5 μg/ml) as well as brain-heart infusion broth (BHIHKSbcStgC) containing brain-heart infusion, yeast extract (1 mg/ml), hemin (5 μg/ml), and menadione (0.5 μg/ml), sodium bicarbonate (1 mg/ml), sodium thioglycolate (0.25 mg/ml), and cysteine (0.5 mg/ml) were used for solid and liquid culture of P. gingivalis, respectively [Note3]. Gentamicin (25–50 μg/ml) and erythromycin (2–10 μg/ml) were used when appropriate for prevention of contamination as well as isolation and maintenance of P. gingivalis mutants [Note4].
Escherichia coli strain S17-1 λpir/pSAM_Bt was grown at 37°C under aerobic conditions in Luria broth base (LB) and Luria agar [Note2]. Carbenicillin (50 μg/ml) was added for plasmid maintenance and to prevent contamination.
Transposon mutagenesis
WT P. gingivalis (strain ATCC 33277) was inoculated into brain-heart infusion broth (BHIHKSbcStgC) without antibiotics. Broth cultures were grown to optical densities (OD600) between 0.50 and 1.00. Escherichia coli strain S17-1 λpir containing the pSAM_Bt plasmid was grown to optical densities OD 0.50–1.00. Broth cultures were set up such that between a 5:1 and 10:1 ratio of P. gingivalis (recipient) to E. coli (donor) was achieved [Notes9/10]. Although P. gingivalis is categorized as an obligate anaerobe it is able to survive without significant CFU loss (less than a log10) for up to 6 hours under aerobic conditions when incubated alone on BAPHK at 37°C [Note6].
The E. coli donor strain carrying the Mariner transposon on a suicide plasmid vector was conjugated with WT P. gingivalis using a bi-parental procedure where the E. coli donor strain and P. gingivalis recipient strain are cultured together on an agar plate to allow for plasmid transfer [Note6]. Conjugation was carried out aerobically at 37°C for 5 hr [Note6]. As P. gingivalis is naturally resistant to gentamicin, this antibiotic was used for selection against the donor E. coli following the conjugation [Note4]. The transposon contains an erythromycin resistance gene (ermG) used to select for P. gingivalis transposon insertion mutants [Note4].
Inoculate WT P. gingivalis from frozen stock onto BAPHK containing gentamicin (25 μg/ml) [Note8]
Incubate the BAPHK in GasPak at 37°C for 5–7 days [Note5]
Inoculate a single colony of the WT P. gingivalis from BAPHK into BHIHKSbcStgC without antibiotics and incubate anaerobically in GasPak at 37°C overnight
Back-dilute the P. gingivalis overnight into BHIHKSbcStgC without antibiotics in a 10:1 ratio [Note9]
Grow anaerobically in GasPak at 37°C to OD600 between 0.50 and 1.00
At the same time as step 3, plate E. coli strain S17-1 λpir containing the pSAM_Bt plasmid onto LB agar (carbenicillin 50 μg/ml); grow aerobically at 37°C
Inoculate LB broth (carbenicillin 50 μg/ml) with a single E. coli colony from the agar plate; grow to OD600 0.50 – 1.00 in 25 ml LB (carbenicillin 50 μg/ml); grow on a shaker, aerobically at 37°C
Remove fluid from the P. gingivalis and E. coli cultures in a 5:1 to 10:1 ratio of P. gingivalis (recipient) to E. coli (donor) and place into separate centrifuge tubes
Pellet each broth culture and resuspend in 2 ml BHIHKSbcStgC
Plate four 500 μL puddles onto BAPHK without antibiotics [Note6]
Incubate for 5–6 hours aerobically at 37°C
Resuspend conjugation reactions in a total of 25 mL PBS [Note7]
Plate 100–400 μL aliquots onto BAPHK supplemented with gentamicin and erythromycin [Note7]
-
Incubate anaerobically in Gaspaks at 37°C for at least 4 days, but up to 21 if desired [Note11]
[To determine transposition efficiency or for individual stocking]
Select individual colonies and transfer onto BAPHK containing erythromycin 5 μg/ml and gentamicin 25 μg/ml
-
Test individual P. gingivalis colonies using PCR to detect the presence of the transposon (ermG) as well as vector backbone components (bla and himar1C9a transposase) [Table 1] [Fig.1]
[For pooling mutant colonies instead of maintaining stocks of individual mutant strains]
Scrape and resuspend mutant strain colonies from BAPHK into BHIHKSbcStgC [Note12]
Table 1.
Primers necessary for checking for parts of the mutagenesis vector in individual mutant strains, nested semi-random PCR, and Tn-seq. The ermG and bla primers are for the genes of pGERM (EF155418.1) and the himar1c9a primers are for the gene of pBADC9 (PMCID: PMC18050).
| Primer | Sequence | Function |
|---|---|---|
| ermG + | TAGGTGCAGGGAAAGGTCAT | Vector |
| ermG − | CCATTTTTGCTGGCTTTCTT | Vector |
| bla + | TTGCCGGGAAGCTAGAGTAA | Vector |
| bla − | GCTATGTGGCGGTATTAT | Vector |
| himar1c9a + | GACGGAAAAACTCGGGTGTA | Vector |
| himar1c9a − | TTCAAGCGTGGTGAAATGAG | Vector |
| SAMseq1 | ACGTACTCATGGTTCATCCCGATA | Semi-random |
| SAMseq2 | GCGTATCGGTCTGTATATCAGCAA | Semi-random |
| SAMseq3 | TCTATTCTCATCTTTCTGAGTCCAC | Semi-random |
| ARB1 | GGCCACGCGTGCACTAGTACN10TACNG | Semi-random |
| ARB2 | GGCCACGCGTGCACTAGTAC | Semi-random |
| Olj376 | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGGGGGGGGGGGGGGG | Illumina |
| pSAM1 | CCTGACGGATGGCCTTTTTGCGTTTCTACC | Illumina |
| pSAM2 | AATGATACGGCGACCACCGAGATCTACACTCTTTGACCGGGGACTTATCATCCAACCTGTTA | Illumina |
| pSAM3 | ACACTCTTTGACCGGGGACTTATCATCCAACCTGTTA | Illumina |
Figure 1.
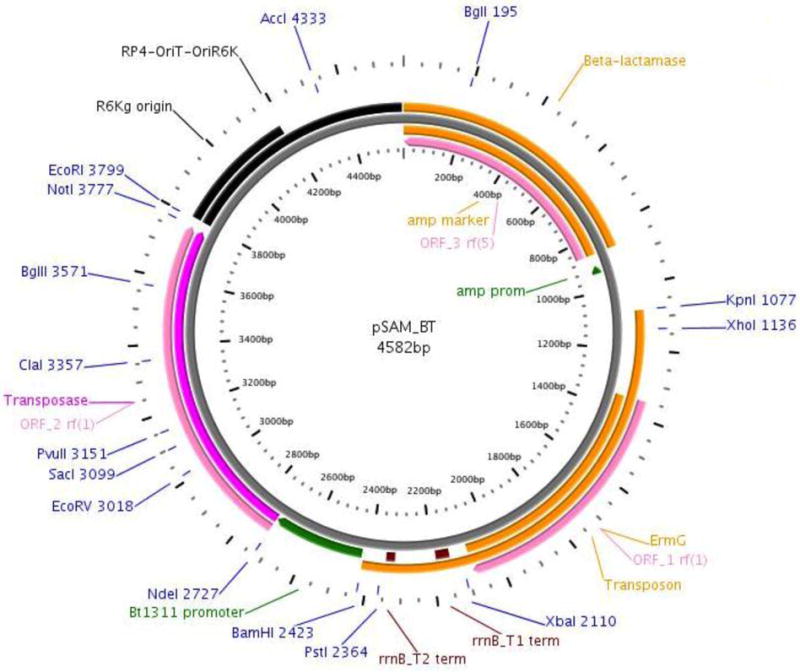
Map of the pSAM_Bt vector generated using Plasmapper. The three ORFs on the vector are denoted in pink. Antibiotic markers denoted in orange. The Amp and BT_1311 promoter regions are denoted in green. Unique restriction enzyme cut sites are labeled. (21)
Nested Semi-random PCR/Sequencing
While defining the library by Tn-seq can be done directly from the transformants, due to the time and expense of performing the sequencing repeatedly, we typically test the success of individual mutagenesis experiments by nested semi-random PCR. This allows us to quickly determine the success of an individual mutagenesis experiment in generating diverse insertions prior to adding the mutants to the library pool. This method can also be used when a mutant strain has been isolated from the pool and knowledge of the transposon insertion location is desired. Amplicons from the PCR reactions can be sequenced individually if desired, however, unless a mixture of strains has been prepared only one of the bands visible on the agarose gel following PCR will be recognized by the sequencing primer. In cases where a complete library has been tested by applying a selective pressure, nested semi-random PCR can be used to determine the complexity of the remaining mutants prior to subjecting to massively-parallel sequencing. If the nested semi-random PCR reveals limited remaining diversity of the surviving mutants, it may be more cost effective to sequence individual colonies.
-
1
Prepare genomic DNA from selected mutants/clones as well as wild-type progenitor strain; done by kit based (Qiagen) isolation.
- 2
-
3First round of ‘random’ PCR:
- 95°C for 2 min
- 95°C for 1 min
- 30°C for 1 min
- 72°C for 1 min
- Repeat steps 2–4 six cycles
- 95°C for 1 min
- 55°C for 1 min
- 72°C for 1 min
- Repeat steps 6–8 for 30 cycles
- 72°C for 5 min
- 4°C HOLD
[Do not electrophorese products on agarose gel at this step; you will either not see anything or will have a ladder of DNA. You can choose to or not to PCR-purify the products from reaction 1 prior to reaction 2 at this point, but this is not necessary.]
- 4
PCR Reaction 2
95°C for 2 min
95°C for 1 min
55°C for 1 min
72°C for 1 min
Repeat steps 2–4 for 40 cycles
72°C for 5 min
4°C HOLD
Test 5 μl of PCR reaction 2 product on agarose gel [multiple bands expected] [Fig.2]
PCR purify the remaining reaction round 2 PCR products using kit/kit protocol (Qiagen DNeasy)
Send PCR products for sequencing using SAMSeq3 primer [Table 1]
Use NCBI BLASTn [Note13] program to search sequence (minus the transposon sequence) and map for genomic location to identify the transposon-chromosomal junction
Figure 2.
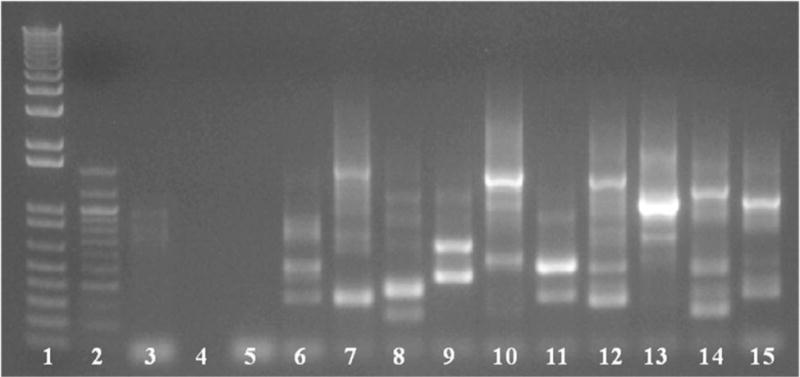
Nested semi-random PCR for sequencing/identification of individual mutants. PCR from individual colonies was carried out using primers specific to the transposon and random primers for the P. gingivalis genome. Agarose gel following second round of nested PCR shown above. Lanes 1 and 2 contain DNA ladder markers, lanes 3 through 5 are negative controls of WT P. gingivalis strain W83, template only and primer only, and individual mutants in the remaining lanes 6–15. Using SAMSeq3 primer for sequencing one will only receive a single product, even though there are multiple bands, unless the prepared gDNA contains two mutant strains. Of note, insertions within the same gene can give different banding patters, as similar banding patterns can be coincidental as well, so extrapolation of insertion location from PCR gel bands is not advisable.
Construction and sequencing of libraries
After confirming successful transposon mutagenesis, mutant library pools (or post-selection pools if experiments beyond the BAPHK mutagenesis have been performed) must be compiled and prepared for sequencing. Preparation for sequencing will include the elements necessary for bioinformatic analyses downstream of sequencing. Detailed methods specific for creation of DNA libraries for use with Tn-seq have been previously well described(18–20); below is a brief outline. For determining the in vitro blood agar based essential genes of P. gingivalis, a single-use mutant pool aliquot was processed using Tn-seq. First, genomic DNA from a mutant pool aliquot was sheared. Deoxycytidine triphosphate tails (C-tails) were then added to the sheared gDNA through PCR. Non-gDNA was removed during a gel filtration cleanup. Transposon containing fragments were amplified in a PCR reaction and adapters specific for sequencing and library identification were added. The Tn-seq-prepared mutant gDNA was run in a sequencing reaction using an Illumina sequencing platform.
We run between 8 and 12 samples per lane of the Illumina sequencer. Each sample is tagged with a unique barcode as part of the preparation for sequencing. Several dozen barcodes are available, which have been described by van Opijnen et al. and Gordon et al., as well as those available through Illumina and New England Biolabs. At the time of our initial publication in 2013, 8–12 samples in a lane resulted in an average of 5–10,000,000 reads per sample which was sufficient depth to provide reliable analysis for our purposies. Since then, Illumina reads per lane have increased dramatically, which would allow the inclusion of more bar codes per lane. However, this must be adjusted for the depth of sequencing required for the specific indication.
Prepare the mutant pool with a genomic DNA extraction kit/kit protocol (Qiagen DNeasy)
Elute genomic DNA with 100 μL elution buffer into a microfuge tube
Place eluent into a 2 mL round-bottom microfuge tube
Shear for 2 min in a Branson sonicator to generate fragments between 200–600 bp
Add poly C-tails to 1 μg of sheared DNA in a 20 μl reaction using TdT as per the manufacturer’s instructions (1hr incubation at 37°C)
Heat-inactivate at 75°C for 20 min
Remove ddCTP and other small molecules using a Performa gel filtration cartridge
Amplify transposon-containing fragments by PCR (primers pSAM1 and Olj376)
Remove PCR primers and other small molecules using a Performa gel filtration cartridge
Amplify the exact Tn-gDNA junction and add sequences needed for Illumina sequencing and indexing (primers: pSAM2 and indexing/barcoding)
Pool sequencing preparations if running more than one experiment per lane and run single-end sequencing reaction on Illumina GAII using pSAM3 and Illumina indexing primers
Data analysis
All read mapping and primary data analysis can be carried out on the Galaxy server in combination with Microsoft Office software (22). In-depth descriptions of specific scripts have been previously reported (and are either available on the open-access Galaxy server or can be generated from previous publications) (18–20). Sequencing output should be generated/received as either FASTQ or FASTA files. If received as FASTA, or potentially other formats, conversion to FASTQ prior to other downstream analyses will be required (and can be found on the Galaxy server under ‘NGS: QC and Manipulation’). Visualizations of Tn-seq data GenomeView allows for comparison of insertions and insertion frequency under multiple conditions (tracts) at once, can be customized manually, but also allows for overlay of NCBI (and other) documentation (23) [Fig.3].
Import FASTQ sequencing files (Galaxy→Get Data →Upload File)
Remove C-tails using adapter clipping script (Galaxy→NGS: QC and Manipulation →Clip)
Filter out low quality sequencing reads (Galaxy→NGS: QC and Manipulation→Filter By Quality)
Align clipped/filtered reads to reference genome (Galaxy→NGS: Mapping →Bowtie for Illumina)
Use bowtie output (SAM file) to tabulate insertion sites and instances an insertion site was sequenced
Generate Excel spreadsheet file indicating position in the genome, locus (if within a gene) that an insertion maps to, DNA strand associated with the insertion as well as the frequency of reads for each insertion site
Use Excel output to estimate complexity of a given transposon mutant library as well as to compare specific mutant strains in input and output samples
-
Convert Excel output file to BED file for visualization using GenomeView; alternatively, SAM file from step 5 can be converted to a BAM file (Galaxy→NGS: SAM Tools→Convert SAM-to-BAM) and then convert BAM file into a BED file (Galaxy→BED Tools→Convert BAM-to-BED)
[If visualization of insertions/reads is desired]
Download NCBI GenBank/FASTA files of reference genome for GenomeView visualizations
-
Use BED files from step 8 as input for GenomeView visualizations
[If attempting to determine or compare essentiality/conditional essentiality of a gene or intergenic region]
Retrieve nucleotide sequences of selected CDS using BROP [Note13]
Utilize BLASTn function in DEG (24) to determine putative essentially matches
Figure 3.

Using GenomeView for visualization of Tn-seq data. BED files (for Tn-seq data) and GenBank files (for NCBI) were uploaded to generate tracts, in which the insertion locus becomes an arrow and the numbers inside the arrow are the reads for that given insertions under the experimental condition. Top tract in blue are genes of the P. gingivalis ATCC 33277 genome from NCBI. Subsequent tracts are the BAPHK-based library followed by BHIHKSbcStgC- based condition. Under these conditions, insertions into an ISPg3 element and tnaA stay relatively constant (top), while insertions into ragA/ragB –operon show a BAPHK-essential gene, one with reduced fitness under BHIHKSbcStgC condition relative to BAPHK, as well as an unaffected gene (bottom).
Applications
There are many potential applications for transposon mutagenesis of a species followed by insertion site sequencing of the resultant mutant populations (14, 15, 25). Although for some species there have already been comprehensive, non-transposon mutagenesis-based essential gene studies (26–28), one can begin by attempting to define an in vitro essential gene list if no comprehensive clean deletion collection or saturated mutant library have been created in the species of interest. Beyond defining a set of genes that cannot be mutagenized under an in vitro condition, one can use a Tn-based mutant library to query genes and/or intergenic regions for relative ‘fitness’ under an in vitro or in vivo condition (16, 17, 29). In vitro conditions can include variations in growth medium nutrient compositions (carbohydrates, peptides, vitamins and minerals) [Fig.3], environmental stresses (temperature, salinity, oxygen levels and light) or host-derived immune stresses (complement components, antimicrobial peptides and phagocytosis). In vivo conditions can include colonization and survival within host-based niches as well as transit from host to host or reservoir to host.
Given that there had been no previous saturated mutant library or comprehensive deletion library created for P. gingivalis, we decided to use our transposon mutant library to define the in vitro (BAPHK-based) putatively essential genes of P. gingivalis (19). The genes identified during our study as essential have been deposited and curated in the DEG (24). An in silico bioinformatic analysis was performed using the P. gingivalis essential gene data as validation for the program GepTop, which is a tool for predicting gene essentiality in sequenced genomes (30).
Acknowledgments
We would like to thank Dr. Andrew Goodman (Yale University School of Medicine) for providing mutagenesis strains and plasmids. We are grateful to the Dr. Andrew Camilli, Dr. David Lazinski and the Tufts University Core Facility (Tufts University Sackler School of Sciences and Howard Hughes Medical Institute) for technical assistance with Illumina sequencing and analysis. We would also like to thank Drs. Michael Malamy (Tufts University Sackler School of Biomedical Sciences) and Pamela Baker (Bates College) for their insightful discussions pertaining to anaerobic bacteria, mutagenesis and genetics.
Funding: This project was supported by a Grant from the National Institute of Dental & Craniofacial Research, F31 DE022491 (BAK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Dental & Craniofacial Research or the National Institutes of Health.
Abbreviations
- Pg
Porphyromonas gingivalis
- PCR
Polymerase Chain Reaction
- BHIHKSbcStgC
supplemented Brain-Heart Infusion
- BAPHK
supplemented Blood Agar Plate
- BROP
Bioinformatics Resource Oral Pathogens
- BED
Browser Extensible Data format
- SAM
Sequence Alignment/Map format
- BAM
Binary Alignment/Map format
- BLAST
Basic Local Alignment Search Tool
- WT
Wild-Type
- DEG
Database of Essential Genes
Footnotes
- P. gingivalis ATCC 33277 (J. Slots→A.L. Coykendall→ATCC)
- P. gingivalis W83 (H. Werner→M. Sebald→ C. Mouton→ M.J. Duncan→ ATCC)
- P. gingivalis W50 (SUNY Buffalo [R. Genco])
- P. gingivalis A7A1-28/ATCC 53977 (SUNY Buffalo [R. Genco])
- P. gingivalis FDC-381 (A.C. Tanner/Forsyth Institute)
Each of the above strains proved to be a successful target/host for Mariner mutagenesis using E. coli S17-1 λpir/pSAM_Bt. Strain FDC-381 proved rather facile for mutant construction, much like strain ATCC 33277, while strain W50 proved difficult, much like strain W83. Strain A7A1-28 was intermediate in terms of ease of mutant construction. The differences for ease of mutant library construction probably come down to four main aspects: innate erm-resistance, innate aerotolerance, conjugation efficiency, and transformation efficiency.
E. coli
S17-1 λpir and plasmids pSAM, containing bla, and pSAM_Bt, containing bla, ermG and himar1c9a genes (Gift from Dr. Andrew Goodman, Yale University). E. coli S17-1 λpir contains the pir gene and has chromosomally integrated conjugational transfer functions (RP4/RK6) such that bi-parental mating can take place in lieu of tri-parental mating using helper strains. pSAM_Bt was used without modification, although it was originally designed for use in Bacteroides thetaiotaomicron. If desired, the Bt_1311 promoter could be swapped out for a potentially more active promoter from P. gingivalis.
Type and handling of blood
We use defibrinated sheep blood from Hemostat Laboratories. This blood is mechanically defibrinated and contains no anticoagulants or additives. Previously, studies with P. gingivalis have show its ability to grow on blood agar media made with blood from other animals such as horse and bovine, however, the blood and serum contents can vary drastically between species, as can the molecular structures of these contents that P. gingivalis is able to break down. Differing nutrient contents as well as molecular structures could lead to variability or differences from mutagenesis experiments done with sheep blood.
Antibiotic choices
Blood agar plates containing no antibiotics are necessary for the mixing/conjugation part of the mutagenesis, during which the donor E. coli will transfer the vector to the recipient P. gingivalis.
Gentamicin is added to media to maintain (in isolation) or select for P. gingivalis (counter-selecting E. coli). MICs of WT P. gingivalis strains range from ~100–300 μg/ml. However, during library construction, certain mutations to P. gingivalis will likely produce strains that have altered MICs to gentamicin. Mutations that cause an increase susceptibility to gentamicin would cause that strain to be lost prior to pooling into the library (unless one has lowered the gentamicin level significantly, which would in turn potentially allow for E. coli bleed-through into the mutant pools). Gentamicin concentrations ≥25 μg/ml were sufficient to eliminate E. coli growth on the selection plates.
Erythromycin is used to select for Mariner-mutagenized P. gingivalis strains and counter-select untransformed P. gingivalis parent cells. Erythromycin has no effect on E. coli because it carries the pSAM_Bt vector that has an ermG-cassette, therefore gentamicin is important to counter-select E. coli on the selection plates. The goal is to choose an erythromycin concentration that allows optimal recovery of Mariner-mutagenized P. gingivalis strains while keeping non-mutagenized (still WT) P. gingivalis from bleeding through onto the selection plates. We found that WT P. gingivalis strains varied slightly in their intrinsic resistance to erythromycin on the double-selective plates. No growth of WT P. gingivalis was observed when using erythromycin concentrations above 3 μg/ml. However, at concentrations between 5–10 μg/ml significantly more ATCC 33277-background mutants than W83-background mutants could be recovered in a single mutagenesis experiment. This suggests either a difference in basal erythromycin concentration tolerance or ability/efficiency to express the erythromycin resistance gene post-conjugation. The MIC of erythromycin for WT P. gingivalis was found to be between 1.5–1.75 μg/ml.
Gaspak/Anaerobic chamber
Gaspaks or anaerobic chambers can be used to generate the mutant libraries. In our hands, both yielded equal number of mutant colonies following transposition. However, steps in the mutagenesis protocol favor having access to both anaerobiosis-generating methods. Using an anaerobic chamber to pre-sparge (pre-reduce) the BHI broth can speed up growth and enhance survival. Pre-sparging the selective blood agar plates could also enhance survival of mutant strains that have become more susceptible to oxygen stress following transposition. Using Gaspaks for selection plates post-conjugation keeps plates from drying, which can happen quickly in anaerobic chambers that remove moisture from the agar. As some mutant strains will inherently grow more slowly than others, longer incubations 10–21 days, can improve recovery of slow growing or rare mutants. Blood agar plates maintain color longer in Gaspaks than in anaerobic chambers, which may be helpful in selection if morphological appearance is key.
‘Puddling’ and Conjugation
For conjugation, we found 5–6 hour incubations on blood agar plates to be sufficient. Older protocols used overnight aerobic incubations. Incubations shorter than 5 hours gave fewer total mutant colonies per conjugation; incubation times longer than 6 hours could cause loss of viability and can also lead to mutant strains dividing, giving forth sibling mutants which lead to ‘incorrect’ colony counts for a given pool. The mixture of bacteria should be ‘puddled’ centrally, not spread throughout the plate. This results in higher yields.
Plating for mutant selection
After the 5–6 hour incubation for conjugation, the bacterial mixture needs to be collected to be re-plated onto double-selective blood agar plates. BHIHKSbcStgC or PBS can be used for collection. In order to keep the mutants concentrated, which will reduce the number of plates needed, place 1–2ml of liquid onto the first plate to be collected. Repeatedly pipette the liquid over the plate to remove the cells. Transfer the collected liquid to the next plate and repeat the process. A small amount of liquid may be lost when wetting each plate so additional sterile liquid may be necessary every 5–10 plates. After collecting the bacterial mixture, spread 100–400 μl onto the double-selective media. Glass beads work very well for complete spreading, spreading via a glass rod seemed to give more concentrated patches, which made it harder to select individual colonies when necessary.
Reviving strains from −80°C vs. plate-to-plate
Strains, both WT P. gingivalis as well as the donor E. coli, should be revived from frozen stocks for each mutagenesis experiment. This will limit variation between experiments and avoid non-Mariner-based secondary mutations. Additionally, since most uses for the mutant library will require sequencing and matching back to previously-sequenced P. gingivalis genomes in lieu of also performing de novo sequencing, introducing variation through plate-to-plate passaging prior to sequencing should be avoided.
Dilution of overnight cultures
Many mutagenesis protocols call for dilution of overnight cultures started from a single colony prior to carrying out the mutagenesis. For P. gingivalis this is also helpful to better control the homogeneity of the culture on the whole. However, P. gingivalis broth culture and sub-culturing can be significantly less predictable than that of E. coli or B. subtilis. As such, we found that 1:10 or 1:100 back-dilutions worked best; instead of 1:1000 or 1:10000, which are often used with other bacterial species.
Centrifugation
For centrifugation, choosing the proper speed/force is important. Speed choice is a compromise between risking damage to pili/fimbriae, which may have an impact on mating with fast speeds and risking over exposure to aerobic conditions with slower speeds.
How long to grow post-conjugation before pooling/selection
Post-conjugation we recommend waiting at least three days prior to checking whether the mutagenesis was successful, as colonies are not easily visible even under 10–20× magnification before this time. Pigmentation of the colonies can help with identification of P. gingivalis versus a contaminant. Wild-type P. gingivalis pigments on supplemented blood agar plates between 5–7 days. Our libraries were made by scraping plates a minimum of 10 days and a maximum of 21 days post-conjugation. Of note, a few colonies that were not visible at day 10–14 were visible by day 21. These slow-growing mutants could be lost from the pool or only found at very low frequency if plates are scraped before day 14.
Pooling
Pooling of mutant colonies from plates requires sterile inoculating loops, 1–5 ml pipettes, 10–25 ml BHIHKSbcStgC, glycerol and cryogenic vials. Remove the double-selective plates from their anaerobic environment and into a sterile laminar flow hood. Set up a sterile conical of BHIHKSbcStgC inside the hood. Using a sterile inoculating loop (the plastic ones work best) scrape the mutant colonies from the plate and deposit them into the BHIHKSbcStgC (shake them off as a clump; they will be fully-resuspended later). After removing as much of the mutant colonies as possible with the inoculating loops, flood the plates with some BHIHKSbcStgC to collect any remaining cells. Carefully resuspend the collected mutant culture by pipetting. A 1–5 ml pipette works well and avoids the potential of lysing cells with a much larger electronic pipette or getting clumps stuck in a small pipette. Make 1 ml aliquots of the mutant library in cryovials for −80°C storage; add sterile glycerol to between 15–20% for cryopreservation.
Data analysis
Pfam (http://pfam.sanger.ac.uk/), Welcome Trust Sanger Institute
Prosite (http://prosite.expasy.org/), Swiss Institute of Bioinformatics
Interproscan (http://www.ebi.ac.uk/Tools/pfa/iprscan/), European Bioinformatics Institute
NCBI (http://www.ncbi.nlm.nih.gov/), National Center for Biotechnology Information
KEGG (http://www.genome.jp/kegg/), The Kyoto Encyclopedia of Genes and Genomes
BROP (http://www.brop.org/), Bioinformatics Resource Oral Pathogens, The Forsyth Institute
GenomeView (http://www.genomeview.org/), Thomas Abeel, Broad Institute
References
- 1.Holt SC, Ebersole J, Felton J, Brunsvold M, Kornman KS. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science. 1988;239:55–57. doi: 10.1126/science.3336774. [DOI] [PubMed] [Google Scholar]
- 2.Hajishengallis G, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host and Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nature Reviews Microbiology. 2012;10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holt SC, Kesavalu L, Walker S, Genco CA. Virulence factors of Porphyromonas gingivalis. Periodontol 2000. 1999;20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 5.Duncan MJ. Genomics of oral bacteria. Critical Reviews in Oral Biology and Medicine. 2003;14:175–187. doi: 10.1177/154411130301400303. [DOI] [PubMed] [Google Scholar]
- 6.Kuramitsu HK. Molecular genetic analysis of the virulence of oral bacterial pathogens: an historical perspective. Crit Rev Oral Biol Med. 2003;14:331–344. doi: 10.1177/154411130301400504. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama K. Molecular genetics of Porphyromonas gingivalis: Gingipains and other virulence factors. Current Protein and Peptide Science. 2003;4:389–395. doi: 10.2174/1389203033486983. [DOI] [PubMed] [Google Scholar]
- 8.Genco CA, Simpson W, Forng RY, Egal M, Odusanya BM. Characterization of a Tn4351-generated hemin uptake mutant of Porphyromonas gingivalis: evidence for the coordinate regulation of virulence factors by hemin. Infect Immun. 1995;63:2459–2466. doi: 10.1128/iai.63.7.2459-2466.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen T, et al. Identification and cloning of genes from Porphyromonas gingivalis after mutagenesis with a modified Tn4400 transposon from Bacteroides fragilis. Infect Immun. 2000;68:420–423. doi: 10.1128/iai.68.1.420-423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bélanger M, Rodrigues P, Progulske-Fox A. Genetic manipulation of Porphyromonas gingivalis. Current Protocols in Microbiology. 2007 doi: 10.1002/9780471729259.mc13c02s05. Chapter 13. [DOI] [PubMed] [Google Scholar]
- 11.Bryan G, Garza D, Hartl D. Insertion and excision of the transposable element mariner in Drosophila. Genetics. 1990;125:103–114. doi: 10.1093/genetics/125.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lampe DJ, Churchill MEA, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996;15:5470–5479. [PMC free article] [PubMed] [Google Scholar]
- 13.Lampe DJ, Akerley BJ, Rubin EJ, Mekalanos JJ, Robertson HM. Hyperactive transposase mutants of the Himar1 mariner transposon. Proc Natl Acad Sci U S A. 1999;96:11428–11433. doi: 10.1073/pnas.96.20.11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barquist L, Boinett CJ, Cain AK. Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biology. 2013;10:1161–1169. doi: 10.4161/rna.24765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Opijnen T, Camilli A. Transposon insertion sequencing: A new tool for systems-level analysis of microorganisms. Nature Reviews Microbiology. 2013;11:435–442. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodman AL, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci U S A. 2009;106:16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Opijnen T, Bodi KL, Camilli A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nature Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein BA, et al. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics. 2012;13 doi: 10.1186/1471-2164-13-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazinski DW, Camilli A. Homopolymer tail-mediated ligation PCR: A streamlined and highly efficient method for DNA cloning and library construction. BioTechniques. 2013;54:25–34. doi: 10.2144/000113981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong X, Stothard P, Forsythe IJ, Wishart DS. PlasMapper: A web server for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 2004;32:W660–W664. doi: 10.1093/nar/gkh410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goecks J, Nekrutenko A, Taylor J, Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11 doi: 10.1186/gb-2010-11-8-r86. R86-2010-11-8-r86. Epub 2010 Aug 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abeel T, Van Parys T, Saeys Y, Galagan J, Van De Peer Y. GenomeView: A next-generation genome browser. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gkr995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo H, Lin Y, Gao F, Zhang CT, Zhang R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42:D574–D580. doi: 10.1093/nar/gkt1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodman AL, Wu M, Gordon JI. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nature Protocols. 2011;6:1969–1980. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi K, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Molecular Systems Biology. 2006;2 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glass JI, et al. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A. 2006;103:425–430. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Opijnen T, Camilli A. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 2012;22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei W, Ning L-W, Ye Y-N, Guo F-B. Geptop: A Gene Essentiality Prediction Tool for Sequenced Bacterial Genomes Based on Orthology and Phylogeny. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0072343. [DOI] [PMC free article] [PubMed] [Google Scholar]