

Abstract
Tuberculosis (TB) remains one of the most threatening diseases in the world and the need for development of new therapies is dire. Herein we describe the rationale for the design and subsequent syntheses and studies of conjugates between pBTZ and both the imidazopyridine and cephalosporin scaffolds. Overall some compounds exhibited notable anti-TB activity in the range of 2 to 0.2 μM in the microplate alamar blue (MABA) assay.
Keywords: Tuberculosis, Benzothiazinones, Imidazopyridine, Cephalosporin
Graphical abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), rivals HIV/AIDS as the most notorious single infectious agent, posing a significant risk to global health. In 2013, it was estimated by the World Health Organization that one-third of the world population was infected with latent TB, approximately 9 million people fell ill with TB, and 1.5 million died of TB.1 The emergence of multi-drug resistant (MDR) and extensively drug resistant (XDR) TB have only put further pressure on the medicinal community, as these new strains pose great challenges to existing treatments.2 Further, the classic TB drug regimens have many issues associated with them, such as long treatment duration and adverse drug-drug interactions.3 In an effort to expand structure-activity-relationship studies of potential anti-TB agents, we were interested in designing hybrid scaffolds. Herein, we describe the synthetic coupling of piperazino-1,3-benzothiazin-4-ones (pBTZs)4 with 2,7-dimethylimidazo [1,2-a]pyridine-3-carboxylic acids and cephalosporins and anti-TB evaluations of the synthesized conjugates.
The current TB treatment regimen for drug sensitive forms of the disease is long and involves at least 6 months of therapy involving many of the front line orally active anti-TB agents (e.g. rifampin, isoniazid).5 This approach often suffers from poor patient compliance and can further lead to the progression of drug resistant TB forms. One potential way of slowing resistance has been the use of drug cocktails that keep the mycobacterial population in check by inhibiting not one, but multiple biochemical processes.6 Fortunately, a number of promising anti-TB agents in development have distinct targets. Nitroimidazoles (PA-824),7 benzothiazinones (BTZ043),8 and imidazopyridines carboxamides9 all have been reported to be potent anti-TB agents (Figure 1) and do not share target similarities.
Figure 1.

Promising anti-TB agents currently in development.
Work by Makarov et al. have shown that combination therapy of pBTZ169, bedaquiline, and pyrazinamide was more effective than the frontline TB regimen in murine models.10 Moreover, Lechartler et. al. recently demonstrated that the combination of clofazimine, a frontline anti-leprosy agent with pBTZ169 was found to be synergistic against both replicating and non-replicating Mtb.11 Intuitively, agents can be designed in a way that they might have multiple targets. Therefore, we envisioned chemical conjugation of 1,3-benzothiazin-4-ones, DprE1 inhibitors, with 7-acylamino cephalosporins and 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxylic acids. Cephalosporins and BTZs target the peptidoglycan and arabinogalactan component of the bacterial cell wall respectively, whereas the imidazo[1,2-a]pyridine-3-carboxamides have been shown to inhibit the cellular energy dependent process.12
The β-lactam family continues to be a hallmark of medicinal chemistry, having been discovered over 80 years ago, yet still making up the majority of the antibiotic market. Currently, the cephalosporin class of β-lactam antibiotics is the most widely used type of β-lactam, with sales estimated at 11.9 billion dollars, topping the list of antibacterial agents in 2009.13 As shown in Figure 2, one of the reasons the cephalosporins have endured the test of time is because a multitude of different functionalities have been successfully incorporated at the C-3’ position of the cephalosporin core (1). To date, a myriad of different agents, such as quinolone antibiotics (1)14 and pyridyl N-oxide toxins (2)15 have been conjugated to the cephalosporin core. With the above precedence in mind, we focused on the syntheses and anti-TB evaluations of conjugates of pBTZs and cephalosporins.
Figure 2.

Two examples of cephalosporin conjugates previously reported
The major scaffolds considered for this study are illustrated in Figure 3. Our synthetic strategy involved the synthesis of the 1,3-benzothiazin-4-one scaffold, wherein we opted to utilize the core of the recently reported analog of BTZ043, pBTZ,16 as one can introduce appropriate substituents at the terminal piperazinyl nitrogen potentially without affecting the target interaction (DprE1) with the nitro aromatic functionality. The next part involved syntheses of imidazopyridine-pBTZ conjugate without (Scheme 2) and with (Scheme 3) different linkers. Lastly, the syntheses of appropriately functionalized cephalosporins were performed (Schemes 4 and 5).
Figure 3.

Scaffolds of interest in this study
Scheme 2.

Syntheses of pBTZ-imidazopyridine conjugate 9.
Scheme 3.

Synthesis of pBTZ-Cephalosporin 13.
Scheme 4 .

Synthesis of pBTZ-cephalosporin 16.
The primary scaffold, pBTZ (Figure 3) for synthetic and biological activity studies, was synthesized as a TFA salt by removing the Boc group of its N-Boc-protected precursor (Boc-pBTZ-Boc, Figure 3), which in turn was synthesized according to the published procedure.17 For the syntheses of our first pBTZ-imidazopyridine conjugate, a previously published procedure was used to generate the necessary carboxylic acid.18 This intermediate was then reacted with pBTZ to give conjugate 4 (Scheme 1).
Scheme 1.

Synthesis of pBTZ-imidazopyridine conjugate (4) without any linker.
In order to explore the effect of a linker between pBTZ and the imidazopyridine scaffold, we chose to incorporate 4-aminomethyl benzoic acid as a representative amino acid linker. As shown in Scheme 2, the Boc group of benzyl 4-(((tert-butoxycarbonyl)amino)methyl)benzoate (5) was deprotected and the subsequent amine (6) was coupled with 3 to obtain 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide, 7. Compound 7 was then subjected to hydrogenolysis to give carboxylic acid 8 which was used without further purification for coupling with pBTZ in the presence of tetramethylfluoroformamidinium hexafluorophosphate (TFFH) and DIPEA to obtain the final conjugate 9 in 36% yield.
For our syntheses of pBTZ-cephalosporin conjugates, we began with the construction of two activated cephalosporins for eventual pBTZ conjugation. As shown in Scheme 3, starting with commercially available 7-aminocephalosporanic acid (7-ACA), tert-butyl esterification19 followed by acylation and enzymatic deprotection20 gave 10. The hydroxyl group was then converted to an activated carbonate by reaction with 1,2,2,2-tetrachloroethylchloroformate to give 11. Cephalosporin 11 was then coupled to pBTZ to give 12 and deprotection with TFA gave conjugate 13.
As with our pBTZ-imidazopyridine syntheses, we were also interested in exploring the effect of a linker on the anti-TB activities of the cephalosporin-pBTZ conjugates. As a result of the lipophilic cell wall (thick layer of mycolic acids) characteristic of Mtb, we tested various protected cephalosporin-pBTZ conjugates to see whether these more lipophilic protected cephalosporins would exhibit anti-TB activity. As shown in Scheme 4, we started with cephalosporin 10. Reaction with thionyl chloride gave 14a in good yield and subsequent reactions of 14a and 14b (commercially available) with NaI and pBTZ gave intermediates 15a–b. Deprotection of 15b with TFA gave conjugate 16.
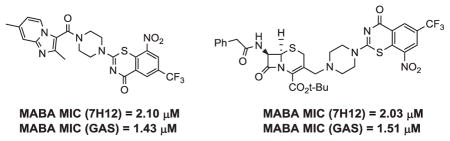
All compounds were then subjected to anti-TB evaluations against Mtb in the Microplate Alamar Blue assay (MABA). 21 As shown in Table 1, a number of compounds exhibited good activity against Mtb including the broad activity that conjugates 4 and 15a exhibited against the H37Rv strain of Mtb in two different mycobacterial growth media (7H12 and GAS). Although notable, the observed activities of all highlighted conjugates were still significantly lower than either the prototype imidazopyridine analogs or pBTZ 169, BTZ043 or the precursor pBTZ-Boc. The anti-TB evaluations of conjugate 9 indicated that the introduction of a linker between the imidazopyridine scaffold and pBTZ was detrimental for anti-TB activity. We additionally explored the syntheses and anti-TB evaluations of amino acid linkers such as β-alanine and γ-aminobutyric acid (see Supporting Information). Nevertheless, the anti-TB evaluations of these conjugates again indicated that the presence of a linker had a deleterious effect on the anti-TB activity. Interestingly, however, one of the imidazopyridine intermediates (7) for the syntheses of conjugate 9 was quite active with an MIC of 0.21 μM in 7H12 media and 4.1 μM in GAS media. Often, differences in activity in different media occur as the result of factors such as compound solubility, different carbon sources, and media age.22
Table 1.
MIC Determinations (μM) for Select Compounds Against Mycobacterium Tuberculosis (H37Rv)in the Microplate Alamar Blue Assay (MABA).
| Cmpd. | 7H12a | GASb | Cmpd. | 7H12a | GASb |
|---|---|---|---|---|---|
| 4 | 2.10 | 1.43 | 16 | 86.8 | 48.9 |
| 7 | 0.21 | 4.10 | Rifampin | 0.05 | 0.04 |
| 9 | >10 | >10 | BTZ-043 | <0.004 | <0.004 |
| 11 | >50 | 22.22 | pBTZ-Boc | 0.28 | <0.20 |
| 12 | 1.49 | 63.45 | |||
| 13 | 4.63 | 11.96 | |||
| 15a | 2.03 | 1.51 | |||
| 15b | >100 | >100 |
7H12 = 7H9 medium + casitone, palmitric acid, albumin, and catalase;
GAS = glycerol-alanine salts medium
SAR studies of the cephalosporin-pBTZ conjugates also revealed some media dependent activity against Mtb. Conjugate 15a was the most active, with MIC values of 2.03 and 1.51 μM in the 7H12 and GAS media, respectively. The difference in activity between conjugates 13 and 16; however, was somewhat surprising. Since both conjugates possessed the free carboxylate normally associated with potentiating β-lactams for activity, it was anticipated that activity might have been enhanced. Instead, the protected conjugates, (e.g. 12 and 15a) were more potent agents in general. This may be due to the greater lipophilicity of these intermediates relative to their free acid counterparts, thus allowing entry into Mtb. Interestingly, however, intermediate 15b was completely devoid of activity. Thus, it seemed that the choice of protecting group might significantly activate or deactivate these compounds.
Lastly, cephalosporin-pBTZ conjugates were also screened for antibacterial activity. As shown in Table 2, the conjugates targeted Gram-positive bacteria, exhibiting both potent zones of inhibition (see Supporting Information) and MIC values. Of outstanding interest was both cephalosporin-pBTZ conjugates 13 and 16, demonstrating notable inhibition against M. vaccae and B. subtilis, with MICs of 0.2 μM and <0.003 μM, respectively.
Table 2.
Minimum Inhibitory Determinations (μM) for Select Compounds.
| Cmpd. |
B. subtilis ATCC6633 |
S. aureus SG511 |
M. luteus ATCC 10240 |
M. vaccae IMET 10670 |
|---|---|---|---|---|
| 1, X = OAc | <0.1 | 0.2 | 3.13 | nt |
| BTZ043 | nt | >200 | >200 | 0.03 |
| 13 | 0.80–1.60 | 12.5 | nt | 0.20 |
| 15a | 1.56 | 12.5 | nt | 0.8 |
| 15b | 0.1–0.2 | 1.56 | nt | nt |
| 16 | <0.003 | 0.4 | 1.56 | 12.5 |
| Ciprofloxicin | 0.15–0.32 | nt | nt | nt |
Compounds were dissolved in MeOH/DMSO
KEY: B. subtilis = Bacillus subtilis, S. aureus. = Staphylococcus aureus, M. luteus = Micrococcus luteus, M. vaccae = Mycobacterium vaccae.
nt = not tested
To summarize, we have synthesized a focused set of conjugates between pBTZ and both imidazopyridines and 7-phenylacetamido cephalosporins and tested them for anti-TB activity in the MABA assay. The product of direct conjugation between pBTZ and imidazopyridine (compound 4) exhibited anti-TB activity albeit not as impressive as its precursors and the introduction of linkers between the two precursor scaffolds in 4 resulted in dramatic loss of activity. Anti-TB activity was observed only for the very lipophilic 15a, whereas similarly lipophilic analog 15b was completely inactive. Potent Gram-positive antibacterial activity was seen for cephalosporin-pBTZ conjugates 13 and 15–16.
Supplementary Material
Acknowledgments
We acknowledge the University of Notre Dame, and the NIH (NIH-2R01-AI054193-05A2) for support of this work. We acknowledge Nonka Sevova (Mass Spectrometry and Proteomics Facility, UND) for mass spectroscopic analyses and Viktor Krchňák for LC/MS and prep LC assistance. M.W.M acknowledges an ECK Global Health Fellowship (2014–2015, UND).
Footnotes
Synthetic procedures, compound characterization data, and MABA assay procedures are available online. This material is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Global Tuberculosis Report. 2014 http://www.who.int/tb/publications/global_report/en/
- 2.Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, Van Soolingen D, Jensen P, Bayona J. Lancet. 2010;375:1830–1843. doi: 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- 3.Zumla A, Nahid P, Cole ST. Nat Rev Drug Dis. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 4.Trefzer C, Skovierova H, Buroni S, Bobovska A, Nenci S, Molteni E, Pojer F, Pasca MR, Makarov V, Cole ST, Riccardi G, Mikusova K, Johnsson K. J Am Chem Soc. 2012;134:912–915. doi: 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- 5.Minarini PRR, de Souza AO, Soares EG, Barata LES, Silva CL, Bentley MV, Maria Vitoria LB. Drug Dev Ind Pharm. 2012;38:259–263. doi: 10.3109/03639045.2011.598535.b) http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5211a1.htm Zumla A, Raviglione M, Hafner R, von Reyn CF. N Engl J Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894.
- 6.Zumla A, Raviglione M, Hafner R, von Reyn CF. N Engl J Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 7.Singh R, Manjunatha U, Boshoff HIM, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., III Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makarov V, Manina G, Mikusova K, Mollmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Y, Moraski GC, Cramer J, Miller MJ, Schorey JS. PLoS One. 2014;9:e87483. doi: 10.1371/journal.pone.0087483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, Widmer N, Buclin T, Bitter W, Andries K, Pojer F, Dyson PJ, Cole ST. EMBO Mol Med. 2014;6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lechartier B, Hartkoorn RC, Cole ST. Antimicrob Agents Chemother. 2012;56:5790–5793. doi: 10.1128/AAC.01476-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moraski GC, Markley LD, Hipskind PA, Boshoff H, Cho S, Franzblau SG, Miller MJ. ACS Med Chem Lett. 2011;2:466–470. doi: 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamad B. Nat Rev Drug Dis. 2010;9:675–676. doi: 10.1038/nrd3267. [DOI] [PubMed] [Google Scholar]
- 14.a) Albrecht HA, Beskid G, Christenson JG, Deitcher KH, Georgopapadakou NH, Keith DD, Konzelmann FM, Pruess DL, Wei CC. J Med Chem. 1994;37:400–407. doi: 10.1021/jm00029a012. [DOI] [PubMed] [Google Scholar]; b) Albrecht HA, Christenson JG. Adv Med Chem. 1992;1:207–234. [Google Scholar]
- 15.O’Callaghan CH, Sykes RB, Staniworth SE. Antimicrob Agents Chemother. 1976;10:245–248. doi: 10.1128/aac.10.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao C, Ye TH, Wang NY, Zeng XX, Zhang LD, Xiong Y, You XY, Xia Y, Xu Y, Peng CT, Zuo WQ, Wei Y, Yu LT. Bioorg Med Chem Lett. 2013;23:4919–4922. doi: 10.1016/j.bmcl.2013.06.069. [DOI] [PubMed] [Google Scholar]
- 17.Peng CT, Gao C, Wang NY, You XY, Zhang LD, Zhu YX, Xv Y, Zuo WQ, Ran K, Deng HX, Lei Q, Xiao KJ, Yu LT. Bioorg Med Chem Lett. 2015;25:1373–1376. doi: 10.1016/j.bmcl.2015.02.061. [DOI] [PubMed] [Google Scholar]
- 18.Moraski GC, Markley LD, Cramer J, Hipskind PA, Boshoff H, Bailey MA, Alling T, Ollinger J, Parish T, Miller MJ. ACS MedChem Lett. 2013;4:675–679. doi: 10.1021/ml400088y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glinka T, Huie K, Cho A, Ludwikow M, Blais J, Griffith D, Hecker S, Dudley M. Bioorg Med Chem. 2003;11:591–600. doi: 10.1016/s0968-0896(02)00431-5. [DOI] [PubMed] [Google Scholar]
- 20.Patterson LD, Miller MJ. J Org Chem. 2010;75:1289–1292. doi: 10.1021/jo902406b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Collins L, Franzblau SG. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) DeVoss JJ, Rutter K, Schroeder B, Su H, Zhu Y, Barry CE., 3rd Proc Natl Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Antimicrob Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franzblau SG, DeGroote MA, Cho SH, Andries K, Nuermberger E, Orme IM, Mdluli K, Angelo-Barturen I, Dick T, Dartois V, Lenaerts AJ. Tuberculosis. 2012;92:453–488. doi: 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.