

Abstract
Development of therapeutic vaccines/strategies to control chronic hepatitis B virus (HBV) infection (CHB) has been challenging due to HBV-induced tolerance. In this study, we explored strategies for breaking tolerance and restoring the immune response to the HBV surface antigen in tolerant mice. We demonstrated that immune tolerance status is attributed to the level and duration of circulating HBsAg in HBV carrier models. Removal of circulating HBsAg by a monoclonal anti-HBsAg antibody in tolerant mice could gradually reduce tolerance and reestablish B cell and CD4+ T cell responses to subsequent Engerix-B vaccination, producing protective IgG. Furthermore, HBsAg-specific CD8+ T cells induced by the addition of a TLR agonist, resulted in clearance of HBV in both serum and liver. Thus, generation of protective immunity can be achieved by clearing extracellular viral antigen with neutralizing antibodies followed by vaccination.
Keywords: antibody, HBs, anti-HBs, tolerance, vaccine
Introduction
An estimated two billion people worldwide have been infected with the hepatitis B virus (HBV), with 350 million of them living with chronic HBV infection. Approximately one quarter of adult HBV carrier and chronic hepatitis B (CHB) patients later die from HBV-related complications of liver cirrhosis and hepatocellular carcinoma (1, 2). Despite enormous efforts to develop antiviral agents, no successful treatment has been reported to eradicate HBV or produce protective anti-hepatitis B surface antigen antibodies (anti-HBs), the clinically desired goal of anti-HBV therapy (3, 4). Anti-HBs antibody production and virus-specific CD8+ T cell response are impaired in CHB patients (5, 6), as well as in the reported HBV-carrier mouse model (7), which has been attributed to viral antigen-induced immune tolerance (8). A study using AAV/HBV mouse model also found that DNA vaccine could induce T/B cell response but was much weaker than in WT mice and failed to reduce HBsAg, suggesting that tolerance was not broken (9). It has been challenging but essential to develop translational strategies that can break tolerance in HBV carriers and CHB patients and induce neutralizing antibody response and/or HBV-specific CTL response to eradicate HBV infection.
A widely used preventive vaccine, Engerix-B (EnxB), containing HBV surface antigen (HBs) in aluminum (alum), induces protective anti-HBs in 90% of non-infected individuals but fails to produce protective anti-HBs in either chronic carriers or CHB patients. In recent large-scale clinical trials, there was no increase of HBV carriers who developed neutralizing anti-HBs against infecting virus despite repeated vaccination, compared to the placebo control group (10, 11). Those data have elucidated the difficulties in breaking tolerance and inducing anti-HBs. HBeAg is another HBV-secreted protein which usually serves as a marker of a high level of HBV-replication. It is also thought to act as a major T cell tolerogen for both HBe (precore) and core-antigens, and regulates the immune response to HBV infection (12). However, the HBeAg-negative variant, a potential CTL-escape mutant, has shown a selective advantage over wild-type HBV within the livers of CHB patients, indicating the complex roles of this HBV protein (13).
For patients with seroconversion from HBs+/anti-HBs− to HBs−/anti-HBs+, liver inflammation and fibrosis reduced gradually over time (14, 15). HBV DNA in the serum of these patients always decreases to an undetectable level, suggesting a correlation between serum HBs depletion and HBV control (16). This raises the possibility that existing levels of circulating HBs induce a specific tolerance that prevents the host from responding to HBs vaccination. Therefore, we hypothesize that clearance of circulating HBs might sufficiently reduce the tolerance enough for the host to reestablish a protective immune response to the clinical HBs vaccine. Firstly, using HBV carrier mouse models that have defined low and high levels of serum HBs (9, 17), we have observed a close correlation between HBs level and tolerance status. By pre-treating the AAV/HBV infected tolerant mice with HBs neutralizing antibodies (NAb), we cleared circulating HBs efficiently and restored protective anti-HBs responses to the prophylactic HBV vaccine currently used in clinic.
Materials and Methods
Mice
C57BL/6j (B6) mice were provided by Beijing Vital River Co., Ltd. Rag1-deficient C57BL/6j strain (Rag1−/−) producing no mature T cells and B cells were obtained from the Model Animal Research Center (Nanjing, China). Mice were maintained under specific pathogen-free condition in BSL-2+ animal facility and animal experiments were followed with protocol number DWSWAQ (ABSL-2) 2012205 at the Institute of Biophysics, Chinese Academy of Sciences. Animal experiments performed at the University of Chicago were approved by UC Institutional Animal Care Committee protocol (#71866 for UC).
Reagents
AAV/HBV virus, an adeno-associated-virus-serotype-8 gene transfer vector expressing 1.3-fold whole HBV genome (genotype D, serotype ayw), was provided by Beijing Five Plus Molecular Medicine Institute (Beijing, China). Peptides were purchased from China Peptide Co., Ltd (Shanghai, China). HBs and HBeAg ELISA kits were purchased from Kehua Bioengineering Co., Ltd (Shanghai, China). EngerixB (EnxB), containing HBsAg genotype A/serotype adw2, was purchased from GlaxoSmithKline Biological (Shanghai, China). Antibody-H (Ab-H), a mouse IgG1 monoclonal antibody to HBsAg, and antibodies TIB210 (ATCC#: TIB-210, anti-CD8) and GK1.5 (ATCC#: TIB-207, anti-CD4) were prepared and purified in house. HBs (ayw) were purified by CsCl density gradient centrifugation from the supernatant of HEK293 transient transfected by HBs (ayw)-expressing plasmid.
HBV carrier mouse model
Generation of the AAV/HBV carrier mouse model was described previously (9, 17). Briefly, C57BL/6 (male, 5–7 weeks of age) were intravenously injected with 5 × 109, 5 × 1010, or 1 × 1011 viral particles through the tail vein (200 µl/injection, diluted in PBS). The model was used throughout this study and is abbreviated as “carrier” in the text or figures. The HDI/HBV carrier mouse model was generated by hydrodynamic injection of plasmid pAAV/HBV1.2 into male B6 mice (7). In brief, 5 µg or 0.25 µg plasmid DNA were diluted to the equivalent of 8% of the individual mouse’s body weight; the total injection volume was delivered within 5–8 seconds.
HBV antigen, antibody, and serum alanine aminotransferase (ALT) analysis
Sera were prepared from blood collected from the retro-orbital sinus of the mouse at the indicated time points. Serum levels of HBs, HBeAg, and anti-HBs were measured by standard ELISA after a series of dilutions with sterile phosphate-buffered saline containing 2% fetal bovine serum, according to the manufacturer’s instructions. Diluted commercial HBV antigens (Beijing Controls & Standards Biotechnology Co., Ltd.) were used as the standards. Endogenous HBs-antibodies were monitored by a pre-coated plate with the recombinant ayw-specific HBs proteins or peptides with the same encoding sequence in AAV/HBV1.3. Peptides used for measurement of subtype-specific antibodies by ELISA: HBs ayw 113–137 (SSTTSTGPCRTCMTTAQGTSMYPSC), and HBs-adw-113–137 (STTTSTGPCKTCTTPAQGNSMFPSC). Serum alanine aminotransferase (ALT) activity was determined using a fully automatic biochemical analyzer (BioSino Bio-technology and Science Inc.) after an appropriate dilution with sterile 0.9% NaCl solution.
Measurement of HBV genomic DNA
Serum HBV genomic DNA was extracted from 100 µl of serum following the manufacturer’s manual (Qiagen, Hilden, Germany). Capsid-associated HBV DNA in the liver was extracted as described previously (18). Briefly, liver tissues were homogenized in a lysis buffer, and viral cores were then precipitated by adding polyethylene glycol, after removing host genomic DNA by DNase I. Viral DNA was extracted by treatment of core particle with proteinase K and SDS, and then was precipitated with isopropanol. Quantitative real-time PCR (Q-RT-PCR) was performed to detect HBV DNA levels with HBV-specific primers, HBV-Real-F (5’-CACATCAGGATTCCTAGGACC-3’) and HBV-Real-R (5’-GGTGAGTGATTGGAGGTTG-3’). The HBV surface antigen containing plasmid was used to create standard curves.
Lymphocyte depletion, sorting and adoptive transfer
In vivo depletion of CD4+ T cells or CD8+ T cells was performed by using anti-CD4 (GK1.5) or anti-CD8 (TIB210) antibody before transfer of splenocytes from the indicated groups of treated or control mice (200 µg of individual antibody per mouse). Lymphocyte sorting and purification was performed by using LS columns (Miltenyi Biotec) and monoclonal antibodies of anti-B220 (RA3-6B2), anti-CD4 (RM4-5), anti-CD38 (90), and by anti-APC microbeads (Cat.No.130-090-855, Miltenyi Biotec), following manufacturer’s instructions. 5 × 107 splenocytes from indicated donor mice were transferred intravenously to Rag−/− mice via the tail vein.
Antigen specific T cell ELISPOT Assay
An ELISPOT assay was performed according to the manufacturer’s instructions (BD, San Diego, CA, USA. catalog: 551083) using lymphocytes isolated from the liver and spleen. An enzymatic digestion method was utilized for the isolation of intrahepatic lymphocytes. In brief, liver tissues were digested by collagenase IV (Roche, Basel, Switzerland) at 37°C for 15 minutes. The suspension was centrifuged at 30 g for 1 minutes to remove hepatocytes. Lymphocytes were then pelleted by centrifugation at 400 g for 10 minutes and further purified with 40% and 70% Percoll solutions by centrifuging at 800 g for 20 minutes at room temperature. Cells were collected from the interface, and red blood cells were removed with ACK buffer to make a single-cell suspension. To detect an antigen-specific immune response, liver lymphocytes were incubated for 48 hours at 37°C in a complete medium containing 10 µg/ml H2-KbENV190–197 peptide (VWLSVIWM) or OVA257–264 peptide (SIINFEKL) in an IFNγ ELISPOT plate.
Statistical Analysis
Statistical analyses were performed on GraphPad Prism Software (California, USA). The unpaired two-tailed student’s t test was used to compare variables between the two groups. Differences among multiple groups were analyzed by the student’s t test following one-way ANONA. All experiments were repeated at least twice.
Results
Humoral immune tolerance is dependent on the level of circulating HBs
Persistence of HBV was developed by introducing the HBV genome using a liver tropic type 8 adeno-associated virus vector (AAV/HBV) (17). AAV/HBV carries the entire HBV genome that will express HBV proteins, finish HBV replication, and release both pseudoviruses and complete HBV virions. HBV-specific immune tolerance was also observed in this mouse model, with no HBs to anti-HBs seroconversion, even after repeated vaccination (9, 17). Thus the AAV/HBV mouse, as an animal model, could provide critical information for CHB immunotherapy studies. To investigate the role of the level of circulating HBs on the regulation of HBV-induced humoral tolerance, we infected two groups of male B6 mice with either a high dose (1 × 1011 vg per mouse) or a low dose (5 × 109 per mouse) of AAV/HBV. Serum levels of HBs reached to 1761.3 ± 165.2 ng/ml in the high antigenemia (>1000ng/ml) group and 41.1 ± 7.2 ng/ml in the low antigenemia (<50ng/ml) group, at week 4 post infection. Then, these mice were subcutaneously vaccinated with a commercially available prophylactic HBs vaccine, EnxB, which is a potent anti-HBs inducer. We monitored the serum level of HBs (serotype ayw) as well as ayw-specific anti-HBs antibodies at weeks 1, 2, 4 post vaccination, and found that no antibodies to HBs (ayw) were detected and serum HBs was not significantly reduced in the high antigenemia group. In contrast, the low antigenemia mice exhibited rapid depletion of serum HBs, which became undetectable at week 2 post vaccination, while generating a considerable level of HBs (ayw)-specific antibodies (Fig. 1A, B). In the untreated control group, serum HBs persisted stably at high levels for at least 6 months (Supplementary Fig. 1). Another HBV mouse model made by hydrodynamic tail vein injection (HDI) of a plasmid containing 1.2-fold of the entire HBV genome was also used in this study, which has been used before to investigate persistent HBV expression and HBV-induced immune tolerance (7). Similar results of immune tolerance were also observed in the HDI/HBV mouse model (Fig. 1C, D). Taken together, our data indicate that HBV-induced humoral immune tolerance is strongly associated with the level of circulating HBV antigens, such as HBs.
Fig. 1. Antibody responses to EnxB-vaccine are inversely correlated to the level of circulating HBs.
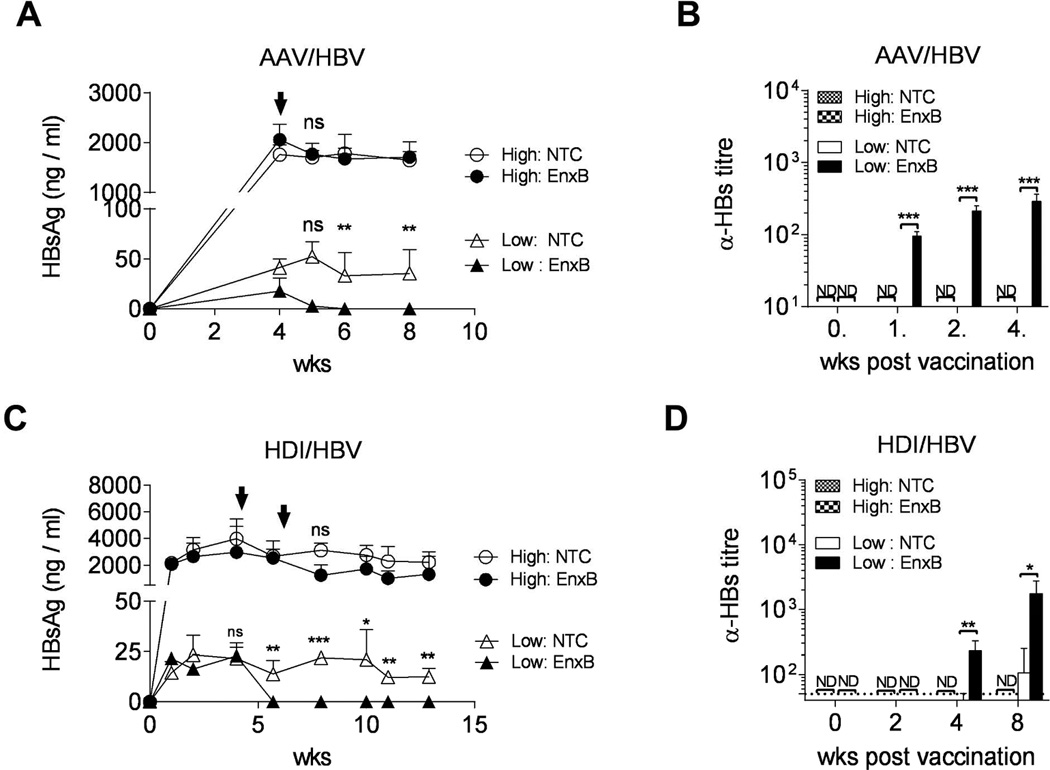
(A) Serum levels of HBs in AAV/HBV carrier mice were monitored by ELISA after vaccination with EnxB. HBs values are presented as a nanogram per milliliter and are expressed as means ± SD of three mice in each time point. (B) Ayw subtype-specific anti-HBs antibody responses in AAV/HBV carrier mice were monitored by ELISA at indicated time points (n=3). (C) Serum HBs in HDI/HBV mice after EnxB- vaccination was monitored by ELISA (n=4). (D) Anti-HBs antibody responses in HDI/HBV mice were monitored by ELISA at indicated time points post vaccination (n=4). The arrows indicate the time points of EnxB vaccination. High, high antigenemia; Low, low antigenemia; NTC, no treatment control; EnxB, s.c. vaccinated with EngerixB; ND, not detected; NS, not significant; *P < 0.05, **P < 0.01, and ***P < 0.001versus corresponding control mice (throughout all figures). The data presented are representative of three independent experiments.
HBs is a major humoral immune tolerogen in the CHB model
To determine whether unresponsiveness to EnxB in high antigenemia HBV carrier mice is due to immune tolerance, we vaccinated the high antigenemia HBV carrier mice with CpG-adjuvanted EnxB to enhance the efficacy of vaccination. Type B CpG ODN1826 is a strong TLR9 adjuvant in mice (19, 20). Compared to EnxB vaccination alone in naive mice, which induced a strong antibody response but with no CTL, EnxB/CpG could promote not only a much stronger humoral immune responses, but also a robust cytotoxicity responses (Supplementary Fig. 2). Similar to EnxB (Fig. 1A, B), EnxB/CpG vaccination did not result in serum HBs decline (Fig. 2A) or induction of corresponding ayw-specific anti-HBs antibodies in carrier mice, while a strong immune response was induced in uninfected control mice (Fig. 2B). This result indicates that there is a severe tolerance to HBs in carrier mice. However, spontaneous anti-HBV-core antibodies can be easily detected in most (7/10) unvaccinated carrier mice (Fig. 2C), indicating that HBcAg might not be a humoral tolerogen in this model. As expected, the serum level of HBeAg, which is a well-known major viral T-cell tolerogen, was not influenced by either EnxB or EnxB/CpG vaccination (data not shown). Anti-HBs conversion is the clinical term for the cure of HBV infection. Thus, we focused on mechanistic studies of HBsAg-induced humoral tolerance in the rest of our study, and chose HBeAg seroconversion as one of the indicators for the breaking of T-cell tolerance and HBV clearance via HBs-based strategies. To test whether the nonresponsive is systemic or unique to HBs, we vaccinated carrier and uninfected control mice with EnxB and HSV-1 at the same time. We observed a strong antibody responses against HSV-1 gD in both groups of mice. Again, antibody response to HBs was detected only in the uninfected mice but not in the HBV carrier mice (Fig. 2D). Thus, these results suggest that HBs is a major humoral immune tolerogen in the chronic HBV mice model and this tolerance is antigen-specific with no effect on either HBc or HSV-1 gD responses.
Fig. 2. HBs is the major humoral immune tolerogen in the CHB model.
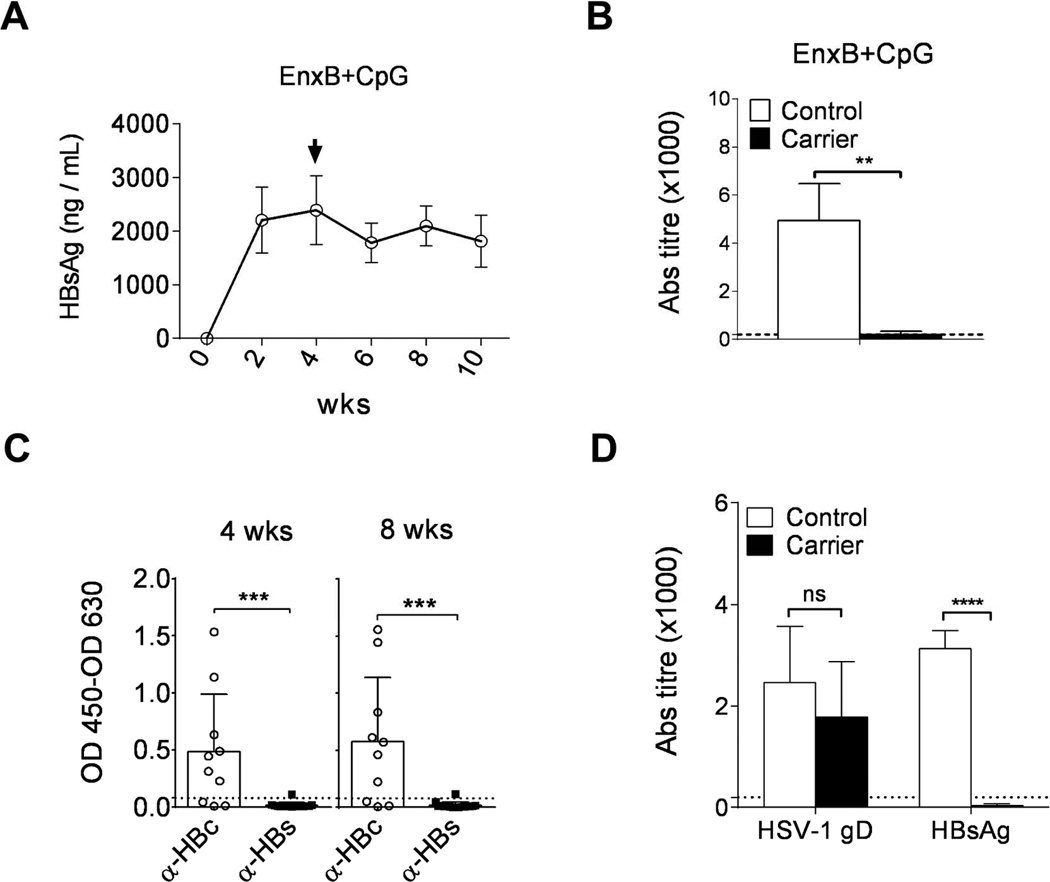
Serum levels of HBs (A) and ayw subtype-specific anti-HBs antibody (B) in AAV/HBV carrier mice (n=3) were monitored by ELISA after vaccination with EnxB (2 µg) plus CpG (30 µg). (C) Spontaneous antibody responses to viral core antigen (n=10) and surface antigen (n=16) were monitored by ELISA at 4 and 8 weeks post infection. (D) Antibody responses to HBV surface antigen as well as HSV-1 gD antigen (n=3) were tested in AAV/HBV carrier mice and control mice infected with HSV-1 (5 × 107 pfu) and vaccinated with EnxB (2 µg). Control, C57BL/6j mice that were not infected with AAV/HBV. The data presented are representative of at least two independent experiments.
The duration of HBs existence plays an important role in the induction and maintenance of HBs tolerance
High levels of circulating HBs could induce tolerance in carrier mice, but the induction process was unknown. To clarify how long the presence of HBs would be required to induce humoral tolerance, we vaccinated carrier mice with EnxB at a series of time points post AAV/HBV infection while monitoring serum levels of HBs and anti-HBs. We observed that vaccination on day 1, week 1, and week 2 post infection resulted in rapid reduction of serum HBs, which became undetectable on the week 4 after primary vaccination. Anti-HBs antibodies could be detected immediately after disappearance of HBs. In contrast, mice under prolonged exposure to HBs (4 weeks) could not respond to EnxB. Serum HBs could be detected on the 4th week after post primary vaccination, and even an additional EnxB-immunization could not stimulate ayw-specific anti-HBs antibody to clear HBs (Fig. 3A). These results indicate that aside from HBs levels, the duration of HBs presence is another important factor for inducing and maintaining HBs-specific immune tolerance in carrier mice.
Fig. 3. The duration of HBs existence contributes to inducing and breaking tolerance to HBs.
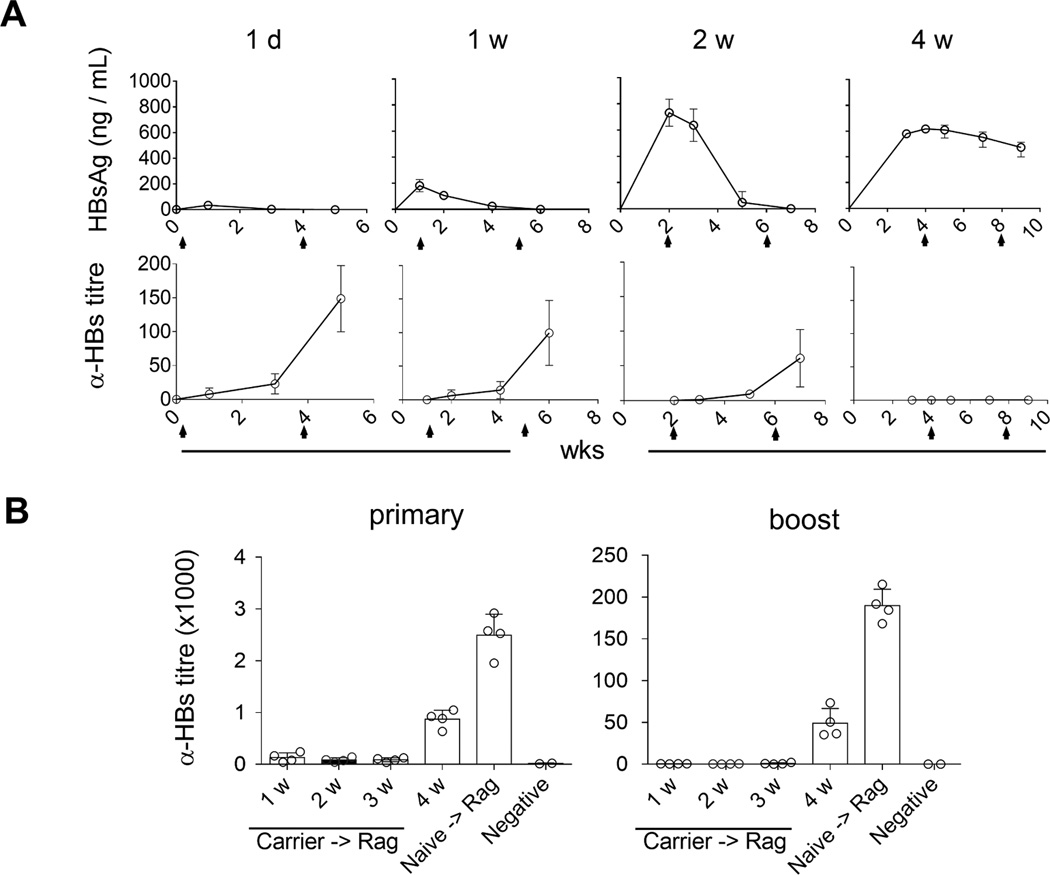
At one day (1d), one week (1w), two weeks (2w) and four weeks (4w) post AAV/HBV (2.0 × 1010 vg) infection, mice were vaccinated with EnxB, then boosted with the same dose of EnxB in four-week intervals as indicated by arrows. Serum levels of HBs (A) and anti-HBs (B) were monitored over time (n=3). (B) Splenocytes from tolerant AAV/HBV carrier mice were adoptively transferred to Rag1−/− mice, which were vaccinated and boosted with EnxB on 1, 2, 3, or 4 weeks post transfer respectively. Ayw subtype-specific humoral responses were estimated by ELISA. Mice that were transferred with splenocytes from naïve mice were used as the positive control (n=4). The data presented are representative of at least two independent experiments.
The roles of level and persistence of circulating HBs in the induction of HBs-specific humoral tolerance raises the possibility that the clearance of circulating HBs for a specific length of time could gradually diminish HBs-induced tolerance. To test this hypothesis, splenocytes from tolerant HBV carrier mice were transferred to Rag1−/− mice to create an antigen-free environment, followed by subcutaneous vaccination with EnxB at specific time points after adoptive transfer. We observed that the antibody response to HBs was undetectable in mice with splenocytes transferred within three weeks, while reconstituted Rag1−/− mice that were vaccinated on the 4th week post transfer had a strong antibody response to HBs (Fig. 3B). This result indicated that humoral immune tolerance to HBs still existed within the first few weeks even in a tolerogen free environment, but could be diminished gradually starting at week 4 as hosts became responsive to vaccination.
Reversal of immune tolerance by neutralizing HBs
To develop a clinically relevant strategy that could decrease serum HBs to a minimum level efficient for therapeutic vaccination in HBV carriers, we treated tolerant carrier mice with Ab-H, a NAb against HBs (ayw), to remove circulating HBs, mimicking the HBs-free status (Fig. 4A). Serum HBs rapidly decreased to an undetectable level after NAb administration and never rebounded after EnxB immunization and cessation of NAb treatment for at least 84 days (Fig. 4B, triangular black line). In contrast, without EnxB vaccination, HBs rebounded immediately once administration of NAb was halted, even after four weeks of constant NAb treatment (Fig. 4B, circular blue line). EnxB alone cannot significantly reduce serum HBs in tolerant carrier mice (Fig. 4B, square red line). Consistent with depletion of HBs, serological conversion to anti-HBs was observed in the combined treatment group, but not in the monotherapy group with either NAb or vaccine (Fig. 4C). The current commercial HBV-vaccine (HBs in alum) is a potent inducer of anti-HBs production but a weak inducer of CTL proliferation, which could result in CTL-mediated severe liver pathogenesis. Indeed, significant liver injury was not observed in mice that were treated with either monotherapy or combined therapy of EnxB with NAb as determined by the ALT test (Fig. 4D) and pathologic analysis of liver tissue (Fig. 4E). Our data support that the neutralization of circulating HBs using NAb can reduce humoral immune tolerance, assist the host response to the HBs-based vaccine, and produce long-term protective anti-HBs antibody without inducing detectable liver injury, a clinically desirable end point.
Fig. 4. Reversal of immune tolerance by neutralizing HBs: a new strategy.
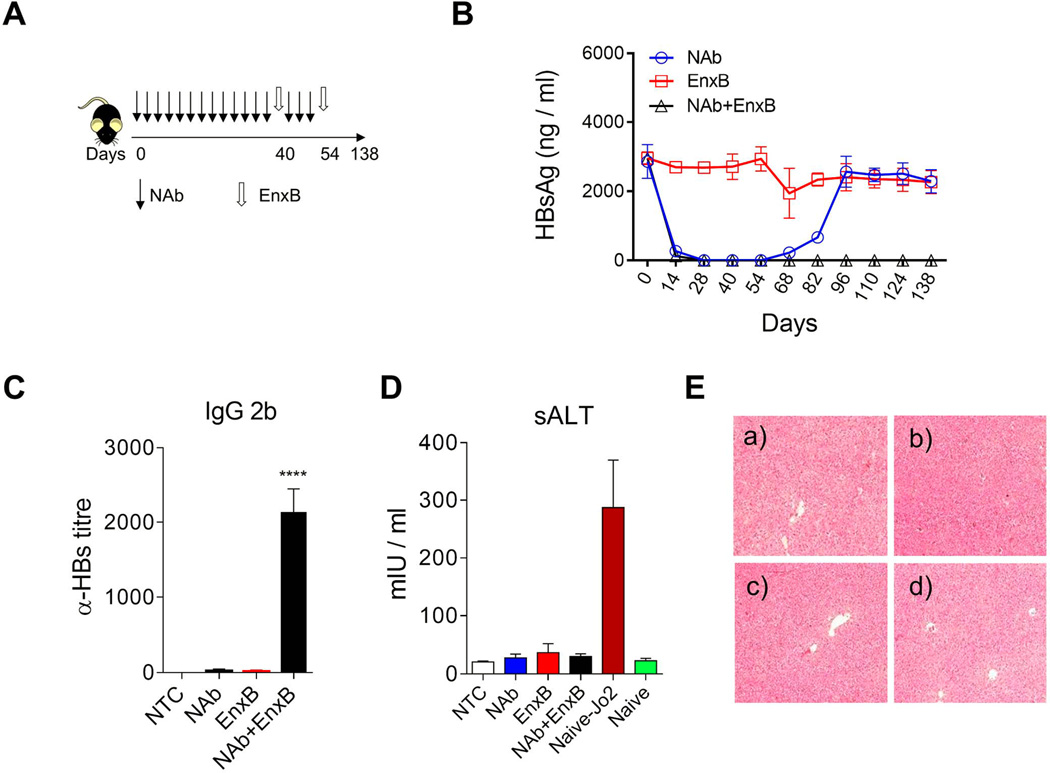
(A) Schematic treatment schedule of tolerant HBV carrier mice with a combination of HBs-neutralizing antibody (Ab-H, 1 mg/mouse with 3-days intervals) and vaccine (EnxB, 2 µg). (B) Serum levels of HBs in different treatment groups were monitored by ELISA (n=3). (C) De novo anti-HBs generation was evaluated on day 21 after the 2nd EnxB vaccination, by monitoring IgG subtype-2b antibodies via ELISA using ayw-HBs antigen identical to the HBV serotype in animal model (n=3). (D) Serum level of ALT was monitored on day 21 after the previous vaccination. Sera from Jo2 (a Fas agonistic antibody) treated B6 mice were used as the positive control (n=3). (E) HE staining for pathologic analysis of liver injury or inflammation. a) No treatment control, b) NAb treated group, c) EnxB treated group, d) combination treatment with NAb plus EnxB group. Experiments were repeated at least three times. Data shown here is one representative of three independent experiments.
Reconstituted immunity to EnxB in HBV carrier mice depends on both B cells and CD4+ T cells
To further study the mechanism of this immune reconstitution, we investigated which subsets of the cell population participate in controlling HBV antigenemia and inducing immune memory after combined therapy. Rag1−/− mice were adoptively transferred with splenocytes from naïve mice, EnxB vaccinated WT mice, EnxB vaccinated carrier mice, or carrier mice resolved from NAb/EnxB treatment, respectively. These recipient mice were then challenged with AAV/HBV on the day following splenocytes transfer. Serum levels of HBs were measured on days 3, 14, and 28 post-transfer. We observed that persistent circulating HBsAg was detectable in Rag1−/− mice transferred with splenocytes from naïve mice as well as from carrier mice that were vaccinated with EnxB, but not in those transferred with splenocytes from EnxB-vaccinated WT or resolved carrier mice after combination treatment. Although HBs appeared in the serum on day 3 post infection (dpi 3), it disappeared on dpi 14 and the serum remained HBs-negative as long as dpi 28 (Fig. 5B), indicating that HBV-specific memory response may play a role in protecting the host from setting up persistent infection by AAV/HBV. Our data support that resolved HBV carrier mice have gained immune memory to HBs, and that this memory is transferrable and protective.
Fig. 5. Reconstituted immunity to EnxB in CHB depends on both B cells and T cells.
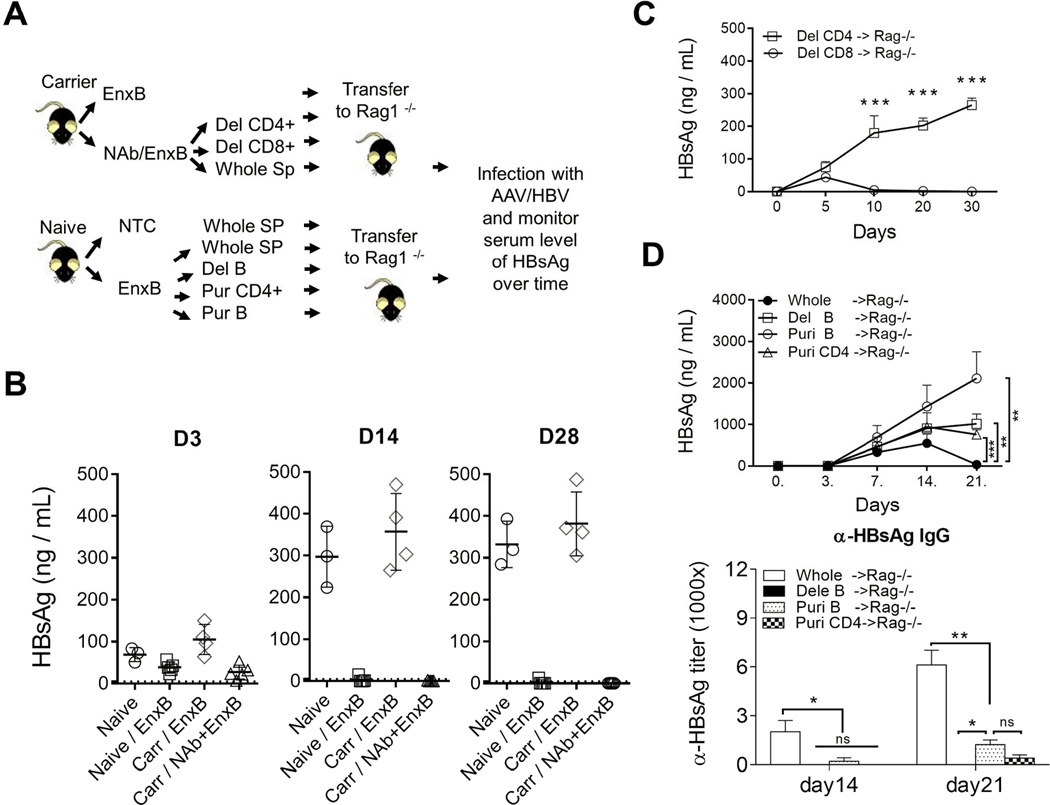
(A) Schematic diagram of adoptive transfer assay. (B) Rag1−/− mice were adoptively transferred with splenocytes from naïve mice (circle), mice vaccinated with EnxB (square), HBV carrier mice vaccinated with EnxB (diamond), or recovered carrier mice treated with NAb plus EnxB regimen (triangle), followed by AAV/HBV infection on the next day after adoptive transfer. Serum levels of HBs were monitored on days 3, 14 and 28 after infection (n=3). (C) Adoptive transfer of splenocytes with CD8+ or CD4+ T cell depletion. These splenocytes were from the resolved mice treated with NAb plus EnxB and transferred to Rag1−/− mice. Then rag−/− mice were challenged with AAV/HBV. Serum levels of HBs were monitored by ELISA (n=3). (D) Rag −/− mice were adoptively transferred respectively with either whole splenocytes, or B cells depleted, or purified B cells, or purified CD4+ T cells from the EnxB vaccinated WT mice. Then mice were challenged with AAV/HBV. Serum levels of HBs (D upper) and anti-HBs antibody (D lower) were monitored by ELISA (n=3). Experiments were repeated at least two times The data shown here is a representative of three independent experiments.
We expected CD4+ T helper cells to be the dominant functional T cells stimulated by an alum-based vaccination. To test this, depletion assays were performed using anti-CD8 (TIB210) or anti-CD4 (GK1.5) monoclonal antibody before transferring splenocytes to Rag1−/−mice. Depletion of CD8+ T cells did not impair the transferred immunity from resolved carrier mice. However, loss of antibody-production was observed when CD4+ T cells were depleted (Fig. 5C). This is consistent with the notion that vaccination with an alum adjuvant can mainly induce CD4+ T helpers and antibody responses but not CTL responses (21, 22). To clarify whether the memory response resulted from a direct antiviral neutralizing effect or an indirect mechanism that promotes antibody generation by CD4+ T cells, Rag1−/− mice were infected with AAV/HBV1.3 after adoptive transfer with whole splenocytes, B cells depleted, purified B cells, or purified CD4+ T cells or from EnxB vaccinated WT mice respectively. We observed a loss of Ag-neutralization in all groups except those receiving a whole splenocyte transfer, which suggests that both CD4+ T cells and B cells are required for optimal protection. Anti-HBs was generated in mice with whole splenocyte transfer, tested on day 14 and day 21 post transfer, but was significantly reduced or undetectable for all other groups in which serum HBs could not be cleared and kept at a high level (Fig. 5D). Taken together, our data suggest that both B cells and CD4+ T cells play essential roles in reestablishing memory immunity to HBs after administration of our vaccine.
Reconstitution of HBs-specific cytotoxic T cell for viral eradication by CpG-combination therapy
Although combination therapy with NAb and EnxB could reverse humoral tolerance and generate anti-HBs antibodies in the AAV/HBV1.3 mouse model, it did not decrease serum HBeAg or eradicate intracellular HBV (Supplementary Fig. 3A). Furthermore, splenocytes from EnxB primed WT mice could not directly control the production of either HBs or HBeAg when adoptively transferred to carrier mice (data not shown). It is commonly believed that viral-specific CTL response plays a key role in eradication of virus-infected hepatocytes. EnxB-primed hosts can generate protective antibodies but fail to generate sufficient CTLs. In WT B6 mice, EnxB/CpG could induce vigorous CD8+ T cell response (Supplementary Fig. 2A) as well as a strong humoral response (Supplementary Fig. 2B). To further improve our combination treatment to induce HBV-specific CTL response, we immunized carrier mice with EnxB mixed with CpG. We tested the potential of EnxB/CpG on therapeutic effects and the reconstitution of HBV-specific CTLs in tolerant AAV/HBV carrier mice with pretreatment of NAb infusion to provide an “antigen-free” time window (Fig. 6A). Compared to those treated with NAb alone, or EnxB/CpG alone, mice treated with NAb plus EnxB/CpG exhibited a significant increase in HBV-specific T cell activity, as shown by ELISPOT analysis (Fig. 6B). In accordance with the enhanced T cell response, a slight increase of an alanine aminotransferase (ALT) was observed in the combination-treated group (Fig. 6C), indicating that functional CTLs targeting HBV infected hepatocytes might be induced inside the liver. Most importantly, serum HBV proteins and DNA were reduced to an undetectable level six weeks after terminating combination therapy with NAb plus EnxB/CpG (Fig. 6D), while mice receiving either NAb or EnxB/CpG monotherapy did not experience such a reduction. To further evaluate the therapeutic efficacy for clearance of hepatic HBV, HBV DNA from hepatocyte extracts was measured by real time PCR. We found that HBV DNA in the liver was reduced to an undetectable level in the carrier mice treated with NAb/CpG-EnxB but not in other groups (Fig 6E left). Meanwhile, HBsAg, HBeAg, and HBcAg in the liver were diminished in mice receiving the NAb/CpG-EnxB treatment, compared to the control and other treatment groups (Fig. 6E middle, right, and Supplementary Fig. 4). These results suggest that HBV-induced CTL tolerance can be broken and even hepatic HBV DNA can be cleared with a combination of NAb pre-treatment and subsequent CpG-adjuvant vaccination even in the HBV carrier model.
Fig. 6. Reconstitution of HBs-specific cytotoxic T cells by combination treatment with NAb and EnxB plus TLR9 agonistics.
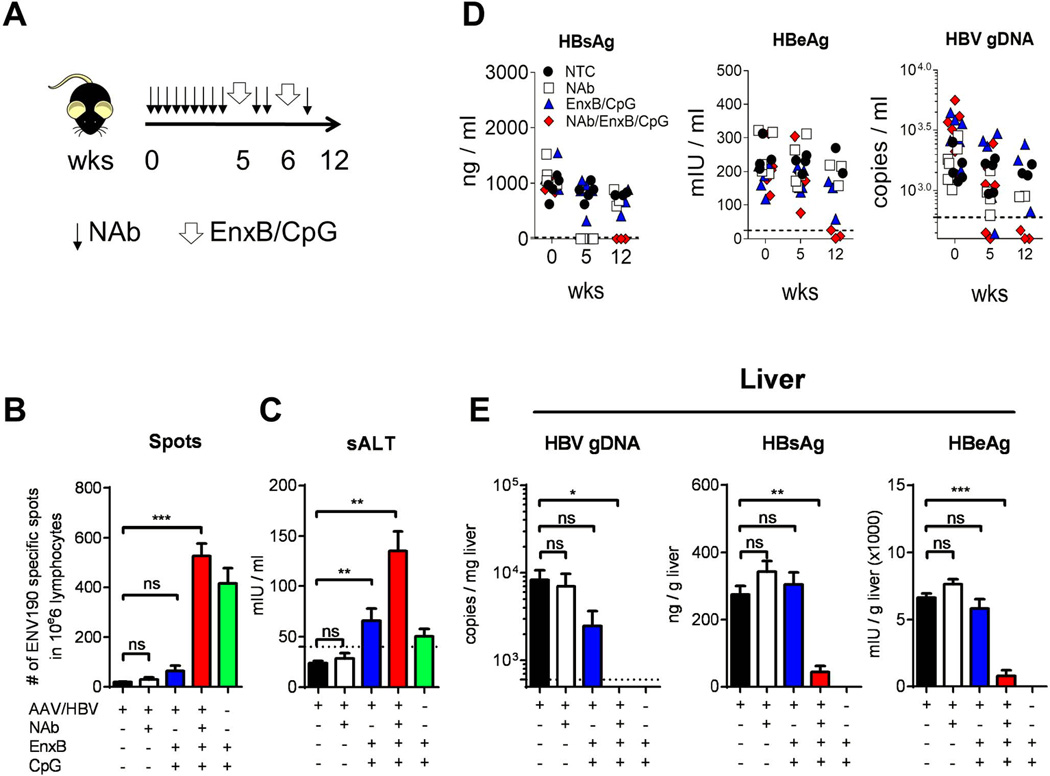
(A) The time course of combination treatment with NAb and EnxB (1 µg), plus CpG (50 µg) administered to HBV carrier mice. (B) Mice were sacrificed on day 16 post first immunization. Intra-hepatic HBs-specific CTLs were monitored by ELISPOT assays after stimulation with ENV190–197 or control peptide OVA257–264. (C) Serum level of ALT (sALT) on 7 days post the last vaccination from (n=5). (D) Serum levels of HBs (left), HBeAg (middle), and HBV genomic DNA (right) were evaluated on day 0 (starting NAb treatment), week 5 (starting vaccine treatment), and week 12 (6 weeks after 2nd vaccination) respectively. (E) Intra-hepatic capsid-associated HBV genomic DNA (left), HBs (middle), and HBeAg (right) were monitored on the day when the experiment was terminated and mice were sacrificed (n=3). The data presented are representative of three independent experiments (mean± SD).
Discussion
Therapeutic vaccines against chronic HBV have demonstrated the extreme difficulty in restoring HBV-specific immunity. In recent large-scale clinical trials including prolonged anti-virus drugs and repeated vaccination, some HBV carriers indeed developed anti-HBs T and B cell responses against epitopes contained in the vaccine, but could not clear serum HBs and the infecting HBV (10, 11, 23). This suggests that such treatments may respond mainly to the non-shared epitopes of recombinant HBs different from the sub-serotype or spontaneous mutations of the infecting HBV in the host, so fail to produce protective antibodies to the shared epitopes with infecting HBV and likely fail to reverse tolerance. Using AAV/HBV and HDI/HBV models, we have observed that immune tolerance is attributed to the level and duration of HBs. Furthermore, clearing HBs from the host might reverse HBV-induced tolerance over time and gradually allow the host to respond to vaccination, which finally become responsive to CpG-adjuvanted HBs-vaccines after 4 weeks of antibody treatment, clearing serum or hepatic HBV by generating a strong protective immune response. This simple but translational strategy may also be applied to the treatment of other chronic infections.
Breaking HBV-induced immune tolerance, especially inducing protective IgG responses to HBs from the infecting HBV strain, has been a difficult goal to achieve in the clinic for decades (2, 24, 25). Many experimental HBV vaccines, such as DNA and new-adjuvant vaccines, have been also investigated for improving therapeutic vaccines to break HBV immune tolerance (26, 27). These studies demonstrate that both antibody response and proper CTL response are required for partially breaking HBV tolerance. However, it was difficult to evaluate the function of vaccines in clearing virus due to the integration of the HBV genome in the transgenic mouse. DC and CTL transfer treatments for CHB have been tested in HBV-transgenic mice, but neither was successful, due to a weak T/B cell response (28) or toxic nonspecific inflammation (29). Current anti-HBV drugs or IFNs can inhibit viral replication and reduce DNA load, but cannot clear HBs and induce protective antibodies. If long-term use of HBV suppressor drugs can greatly reduce antigen load, reactivity of HBV-specific T cells could be partially restored (25). In our study with both the AAV/HBV and HDI carrier models, we observed that clearance of HBs with NAb over time may help to restore the host response to vaccination, with no detectable side effects (Supplementary Fig. 3B and 3C). When circulating HBs had been cleared for several weeks, tolerant HBV carrier mice indeed regained the responsiveness to vaccination and reconstituted anti-viral immunity that can clear circulating HBV particles. Endogenous anti-HBs may coexist with HBsAg in CHB patients and has no influence on HBsAg quantification (30). Our results suggest that HBV-induced tolerance can be broken by a combination of pre-treatment with NAb and subsequent vaccination in HBV carrier models.
Viral-specific adaptive response is weak or undetectable in CHB patients (5, 6), and it has not been defined how to restore anti-HBs responses or functional virus-specific CTLs by vaccination. A recent study using AAV/OVA-induced tolerance showed that antigen expression level threshold tunes the fate of CD8+ T cells during the primary hepatic immune response, which may explain why AAV/HBV infection resulted in a profound tolerance in adult mice (31). The T-cell exhaustion mechanism in HBV animal models have been reported by several studies recently. Xu et al. demonstrated that Tr1-like cells can migrate to the DLN and participate in inducing systemic tolerance by inhibiting GC formation upon HBsAg vaccination in an HBV 1.2 model. The role of Tregs in this HBV1.2 HBV mouse model had been shown not to be the dominant mechanism for tolerance by Xu et al (32). But clinical studies demonstrated that in vitro depletion of Tregs from PBMC taken from HBV-infected patients led to an increase of IFN-γ production following HBV antigen stimulation. The PD1-PDL1 pathway is reported to be involved in HBV induced T cell tolerance, and anti-PD-1 blockage could reverse the exhausted phenotype in intrahepatic T lymphocytes and lead to virus clearance in vivo (33). Some clinical studies also demonstrated that T cell apoptosis or deletion were involved in HBV induced immune tolerance (34). In conclusion, the mechanisms of HBV induced immune tolerance are complicated, and the precise mechanism of immune tolerance will be carefully studied in our subsequent works.
Without TCR and BCR transgenic mice, it is difficult to show the reverse of tolerance at the single-cell level in HBV animal models. However, using adaptive transfer or combined treatment, we have shown, at the lymphocyte subpopulation level, that host response to HBsAg from CHB could be restored with our therapeutic strategy. Dependency of CD4+ T and B cells is consistent with the knowledge that alum-adjuvant antigen generates stronger helper T cells for antibody than CTLs response (35, 36). We have shown that HBV reactive immune cell clones were not completely deleted and could be recovered in circumstances with proper immune stimuli (Fig. 5). These long-term benefits to patients are potentially valuable considerations for future clinical trials. A lack of CTL in response to the alum-based vaccination could limit liver injury, while still providing protective antibodies to prevent viral spread to healthy hepatocytes, including newly generated hepatocytes. However, remaining viral DNA might allow HBV relapse when immune responses are compromised. Proper CTL proliferation is required for intracellular HBV clearance in the liver, but the possibility of severe liver injury should be neglected. Effective CTL response and protective antibodies are achievable, but which strategy is the best for patients remains to be determined in large scale clinical trials. Although B cell tolerance has been the major clinical challenge, it is likely that generation of both CTL and antibody might be induced simultaneously and help each other in clearing HBV while limiting the spread of virus and liver injury via our newly developed strategy.
Therapeutic vaccines against CHB infection are still under extensive exploration (24, 37). Most recently, at least three versions of HBsAg + HBcAg vaccines have been carried by several pharmaceutical companies for animal and even clinical studies, with promising immune response in naïve mice or healthy patients, but with no report yet for significant therapeutic effects, such as HBsAg clearance, HBeAg seroconversion or HBV clearance (38). In this study, we designed a simple and translational approach that successfully achieved two goals: 1) NAb treatment effectively clearing the circulating HBs to provide a relatively antigen-free window in lymphoid tissues allowing immune cells to recover from tolerant status; and 2) the currently used HBV vaccine prompting HBV-carrier hosts to generate protective IgG. Furthermore, advanced vaccination with CpG could generate additional CTL to clear intracellular HBV once HBs tolerance was weakened by HBs-depletion. Thus, reducing extracellular antigens in chronically infected hosts either by treatment with neutralizing antibodies or other means, such as prolonged anti-viral drugs, is a pre-requisite for effective therapeutic vaccination in order to induce protective IgG or CTL immunity. This new therapeutic strategy could provide a practical solution for treatment of chronic infections.
Supplementary Material
Acknowledgments
We thank Daryl Harmon for editorial assistance.
Grant support:
This work was supported by the National Key Basic Research Program of China (No. 2012CB910203, No. 2012CB519000, and No. 2009CB522502) and the National Grand Program on Key Infectious Disease (No. 2012ZX10002006 and No. 2013ZX10002001-001-003) to Y.X.F. and H.P.; National Nature and Science Foundation of China (No. 81172814) to H.P.; Ministry of Science and Technology (2009CB522507 & 2010-Biols-CAS-0201 & KSCX20YW-R-50) to L.Z.; the Ministry of Health (2011ZX10004503-007) to L.Z., L.S.; and NIH funding (R01AI095097) to L.S and Y.X.F.
Abbreviations
- HBV
Hepatitis B virus
- CHB
chronic hepatitis B
- EnxB
EngerixB
- NAb
neutralizing antibody
- AAV
adeno-associated virus
- IFNγ
interferon gamma
- NTC
no treatment control
Footnotes
Disclosures:
The authors have no financial conflicts of interest.
Reference
- 1.Brechot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004;127:S56–S61. doi: 10.1053/j.gastro.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 2.Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 3.Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 4.Lok AS, McMahon BJ. AASLD Practice Guidelines. Chronic hepatitis B: update of therapeutic guidelines. Rom J Gastroenterol. 2004;13:150–154. [PubMed] [Google Scholar]
- 5.Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, Zerbini A, Cavalli A, Missale G, Bertoletti A, Ferrari C. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. Journal of virology. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster GJ, Reignat S, Brown D, Ogg GS, Jones L, Seneviratne SL, Williams R, Dusheiko G, Bertoletti A. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. Journal of virology. 2004;78:5707–5719. doi: 10.1128/JVI.78.11.5707-5719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang LR, Wu HL, Chen PJ, Chen DS. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc Natl Acad Sci U S A. 2006;103:17862–17867. doi: 10.1073/pnas.0608578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boni C, Penna A, Ogg GS, Bertoletti A, Pilli M, Cavallo C, Cavalli A, Urbani S, Boehme R, Panebianco R, Fiaccadori F, Ferrari C. Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology. 2001;33:963–971. doi: 10.1053/jhep.2001.23045. [DOI] [PubMed] [Google Scholar]
- 9.Dion S, Bourgine M, Godon O, Levillayer F, Michel ML. Adeno-associated virus-mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA-A2 and HLA-DR1 molecules. Journal of virology. 2013;87:5554–5563. doi: 10.1128/JVI.03134-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu B, Wen X, Huang C, Wei Y. Unraveling the complexity of hepatitis B virus: from molecular understanding to therapeutic strategy in 50 years. The international journal of biochemistry & cell biology. 2013;45:1987–1996. doi: 10.1016/j.biocel.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 11.Xu DZ, Wang XY, Shen XL, Gong GZ, Ren H, Guo LM, Sun AM, Xu M, Li LJ, Guo XH, Zhen Z, Wang HF, Gong HY, Xu C, Jiang N, Pan C, Gong ZJ, Zhang JM, Shang J, Xu J, Xie Q, Wu TF, Huang WX, Li YG, Xu J, Yuan ZH, Wang B, Zhao K, Wen YM Y. I. C. E. T. S. Team. Results of a phase III clinical trial with an HBsAg-HBIG immunogenic complex therapeutic vaccine for chronic hepatitis B patients: experiences and findings. Journal of hepatology. 2013;59:450–456. doi: 10.1016/j.jhep.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Chen M, Sallberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud JN, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. Journal of virology. 2005;79:3016–3027. doi: 10.1128/JVI.79.5.3016-3027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frelin L, Wahlstrom T, Tucker AE, Jones J, Hughes J, Lee BO, Billaud JN, Peters C, Whitacre D, Peterson D, Milich DR. A mechanism to explain the selection of the hepatitis e antigen-negative mutant during chronic hepatitis B virus infection. Journal of virology. 2009;83:1379–1392. doi: 10.1128/JVI.01902-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu CM, Liaw YF. HBsAg seroclearance in asymptomatic carriers of high endemic areas: appreciably high rates during a long-term follow-up. Hepatology. 2007;45:1187–1192. doi: 10.1002/hep.21612. [DOI] [PubMed] [Google Scholar]
- 15.McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes of 1536 Alaska Natives chronically infected with hepatitis B virus. Annals of internal medicine. 2001;135:759–768. doi: 10.7326/0003-4819-135-9-200111060-00006. [DOI] [PubMed] [Google Scholar]
- 16.McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology. 2009;49:S45–S55. doi: 10.1002/hep.22898. [DOI] [PubMed] [Google Scholar]
- 17.Yang D, Liu L, Zhu D, Peng H, Su L, Fu YX, Zhang L. A mouse model for HBV immunotolerance and immunotherapy. Cellular & molecular immunology. 2014;11:71–78. doi: 10.1038/cmi.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keasler VV, Hodgson AJ, Madden CR, Slagle BL. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. Journal of virology. 2007;81:2656–2662. doi: 10.1128/JVI.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ballas ZK, Krieg AM, Warren T, Rasmussen W, Davis HL, Waldschmidt M, Weiner GJ. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. Journal of immunology. 2001;167:4878–4886. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]
- 20.Beloeil L, Tomkowiak M, Angelov G, Walzer T, Dubois P, Marvel J. In vivo impact of CpG1826 oligodeoxynucleotide on CD8 T cell primary responses and survival. Journal of immunology. 2003;171:2995–3002. doi: 10.4049/jimmunol.171.6.2995. [DOI] [PubMed] [Google Scholar]
- 21.Jordan MB, Mills DM, Kappler J, Marrack P, Cambier JC. Promotion of B cell immune responses via an alum-induced myeloid cell population. Science. 2004;304:1808–1810. doi: 10.1126/science.1089926. [DOI] [PubMed] [Google Scholar]
- 22.Marrack P, McKee AS, Munks MW. Towards an understanding of the adjuvant action of aluminium. Nature reviews. Immunology. 2009;9:287–293. doi: 10.1038/nri2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandepapeliere P, Lau GK, Leroux-Roels G, Horsmans Y, Gane E, Tawandee T, Merican MI, Win KM, Trepo C, Cooksley G, Wettendorff M, Ferrari C H. B. V. V. G. o. I. Therapeutic. Therapeutic vaccination of chronic hepatitis B patients with virus suppression by antiviral therapy: a randomized, controlled study of co-administration of HBsAg/AS02 candidate vaccine and lamivudine. Vaccine. 2007;25:8585–8597. doi: 10.1016/j.vaccine.2007.09.072. [DOI] [PubMed] [Google Scholar]
- 24.Bertoletti A, Gehring A. Therapeutic vaccination and novel strategies to treat chronic HBV infection. Expert review of gastroenterology & hepatology. 2009;3:561–569. doi: 10.1586/egh.09.48. [DOI] [PubMed] [Google Scholar]
- 25.Boni C, Laccabue D, Lampertico P, Giuberti T, Vigano M, Schivazappa S, Alfieri A, Pesci M, Gaeta GB, Brancaccio G, Colombo M, Missale G, Ferrari C. Restored function of HBV-specific T cells after long-term effective therapy with nucleos(t)ide analogues. Gastroenterology. 2012;143:963–973. e969. doi: 10.1053/j.gastro.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Buchmann P, Dembek C, Kuklick L, Jager C, Tedjokusumo R, von Freyend MJ, Drebber U, Janowicz Z, Melber K, Protzer U. A novel therapeutic hepatitis B vaccine induces cellular and humoral immune responses and breaks tolerance in hepatitis B virus (HBV) transgenic mice. Vaccine. 2013;31:1197–1203. doi: 10.1016/j.vaccine.2012.12.074. [DOI] [PubMed] [Google Scholar]
- 27.Isogawa M, Kakimi K, Kamamoto H, Protzer U, Chisari FV. Differential dynamics of the peripheral and intrahepatic cytotoxic T lymphocyte response to hepatitis B surface antigen. Virology. 2005;333:293–300. doi: 10.1016/j.virol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Farag MM, Tedjokusumo R, Flechtenmacher C, Asen T, Stremmel W, Muller M, Protzer U, Weigand K. Immune tolerance against HBV can be overcome in HBV transgenic mice by immunization with dendritic cells pulsed by HBVsvp. Vaccine. 2012;30:6034–6039. doi: 10.1016/j.vaccine.2012.07.057. [DOI] [PubMed] [Google Scholar]
- 29.Guidotti LG. The role of cytotoxic T cells and cytokines in the control of hepatitis B virus infection. Vaccine. 2002;20(Suppl 4):A80–A82. doi: 10.1016/s0264-410x(02)00392-4. [DOI] [PubMed] [Google Scholar]
- 30.Pancher M, Desire N, Ngo Y, Akhavan S, Pallier C, Poynard T, Thibault V. Coexistence of circulating HBsAg and anti-HBs antibodies in chronic hepatitis B carriers is not a simple analytical artifact and does not influence HBsAg quantification. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2015;62:32–37. doi: 10.1016/j.jcv.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 31.Tay SS, Wong YC, McDonald DM, Wood NA, Roediger B, Sierro F, McGuffog C, Alexander IE, Bishop GA, Gamble JR, Weninger W, McCaughan GW, Bertolino P, Bowen DG. Antigen expression level threshold tunes the fate of CD8 T cells during primary hepatic immune responses. Proc Natl Acad Sci U S A. 2014;111:E2540–E2549. doi: 10.1073/pnas.1406674111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu L, Yin W, Sun R, Wei H, Tian Z. Liver type I regulatory T cells suppress germinal center formation in HBV-tolerant mice. Proc Natl Acad Sci U S A. 2013;110:16993–16998. doi: 10.1073/pnas.1306437110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tzeng HT, Tsai HF, Liao HJ, Lin YJ, Chen L, Chen PJ, Hsu PN. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PloS one. 2012;7:e39179. doi: 10.1371/journal.pone.0039179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes AR, Kellam P, Das A, Dunn C, Kwan A, Turner J, Peppa D, Gilson RJ, Gehring A, Bertoletti A, Maini MK. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. The Journal of clinical investigation. 2008;118:1835–1845. doi: 10.1172/JCI33402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sokolovska A, Hem SL, HogenEsch H. Activation of dendritic cells and induction of CD4(+) T cell differentiation by aluminum-containing adjuvants. Vaccine. 2007;25:4575–4585. doi: 10.1016/j.vaccine.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 36.Yip HC, Karulin AY, Tary-Lehmann M, Hesse MD, Radeke H, Heeger PS, Trezza RP, Heinzel FP, Forsthuber T, Lehmann PV. Adjuvant-guided type-1 and type-2 immunity: infectious/noninfectious dichotomy defines the class of response. Journal of immunology. 1999;162:3942–3949. [PubMed] [Google Scholar]
- 37.Michel ML, Deng Q, Mancini-Bourgine M. Therapeutic vaccines and immune-based therapies for the treatment of chronic hepatitis B: perspectives and challenges. Journal of hepatology. 2011;54:1286–1296. doi: 10.1016/j.jhep.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 38.Elvidge S. Blockbuster expectations for hepatitis B therapeutic vaccine. Nature biotechnology. 2015;33:789. doi: 10.1038/nbt0815-789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.