

Abstract
Background
De novo assembly of non-model organism’s transcriptomes has recently been on the rise in concert with the number of de novo transcriptome assembly software programs. There is a knowledge gap as to what assembler software or k-mer strategy is best for construction of an optimal de novo assembly. Additionally, there is a lack of consensus on which evaluation metrics should be used to assess the quality of de novo transcriptome assemblies.
Result
Six different assembly strategies were evaluated from four different assemblers. The Trinity assembly was used in its default 25 single k-mer value while Bridger, Oases, and SOAPdenovo-Trans were performed with multiple k-mer strategies. Bridger, Oases, and SOAPdenovo-Trans used a small multiple k-mer (SMK) strategy consisting of the k-mer lengths of 21, 25, 27, 29, 31, and 33. Additionally, Oases and SOAPdenovo-Trans were performed using a large multiple k-mer (LMK) strategy consisting of k-mer lengths of 25, 35, 45, 55, 65, 75, and 85. Eleven metrics were used to evaluate each assembly strategy including three genome related evaluation metrics (contig number, N50 length, Contigs >1 kb, reads) and eight transcriptome evaluation metrics (mapped back to transcripts (RMBT), number of full length transcripts, number of open reading frames, Detonate RSEM-EVAL score, and percent alignment to the southern platyfish, Amazon molly, BUSCO and CEGMA databases). The assembly strategy that performed the best, that is it was within the top three of each evaluation metric, was the Bridger assembly (10 of 11) followed by the Oases SMK assembly (8 of 11), the Oases LMK assembly (6 of 11), the Trinity assembly (4 of 11), the SOAP LMK assembly (4 of 11), and the SOAP SMK assembly (3 of 11).
Conclusion
This study provides an in-depth multi k-mer strategy investigation concluding that the assembler itself had a greater impact than k-mer size regardless of the strategy employed. Additionally, the comprehensive performance transcriptome evaluation metrics utilized in this study identified the need for choosing metrics centered on user defined research goals. Based on the evaluation metrics performed, the Bridger assembly was able to construct the best assembly of the testis transcriptome in Fundulus heteroclitus.
Introduction
Next generation sequencing (NGS) technologies have offered unprecedented opportunities to obtain genetic information for non-model organisms with little or no molecular information available [1]. This increasingly accessible technology provides an efficient and cost-effective approach for analyzing the transcriptome of non-model organisms that lack a fully-sequenced genome [2–6]. It has been employed to identify novel transcriptome sequences, single nucleotide polymorphisms (SNPs), simple sequence repeats (SSRs), splicing variants, transcript isoforms, new large intergenic noncoding RNAs, and relative levels of transcript expressions [1–2, 6–7]. With the arrival of NGS technologies, the number of publications characterizing de novo assemblies for non-model organisms have steadily been on the rise [8].
De novo transcriptome assembly is performed by taking the enormous amount of short read sequences produced by NGS and overlapping them to form contiguous sequences (contigs) [9]. The quality of the assembly output is reliant on the user designated k-mer value defined as the sequence overlap between two reads forming the contig [9–12]. Low k-mer values have a tendency to recover less abundant transcripts, while producing a large amount of contigs, with a number of them highly fragmented due to sequencing errors and lack of overlap [11, 13]. High k-mer values will produce a more contiguous assembly consisting of high coverage transcripts and splice variants. However, the assembly will contain fewer contigs leading to lower transcript representation [11, 13]. Therefore, utilizing a single k-mer approach when performing a de novo assembly can result in loss of relevant biological information due to the lack of transcript diversity [13]. A logical approach to resolve this dilemma is to cluster multiple single k-mer assembles together in order to take advantage of the characteristics of both the low and high k-mer values and thereby improve the accuracy of the assembly [9–11]. Investigations that compare the quality of transcriptome assembly using one or multiple k-mer values will contribute to standardizing NGS data processing.
The number of de novo transcriptome programs developed for assembly of short sequence reads has increased within the past few years. In 2010, Trans-AByss was reported to have the ability to merge multiple individual k-mer assemblies, allowing the transcriptome to be represented by wide levels of transcript expression [12]. In 2011, Trinity was reported to be able to fully reconstruct a large fraction of transcripts with low base error rates and have the ability to report alternative splice isoforms [14]. Trinity is currently regarded to be the best single k-mer assembler [15]. In 2012, Oases was reported to improve significantly on the Trans-ABySS and Trinity assemblers by merging the use of multiple k-mers presented in Trans-AByss with a topological analysis similar to that presented by Trinity [16]. In 2014, SOAPdenovo-Trans was reported to be able to perform multiple individual k-mer assemblies and provide higher contiguity, lower redundancy and faster execution when compared to Trinity and Oases [17]. All of these assemblers are founded on the de Bruijn graph-based assembly method to which programmers add their own algorithms [12, 14, 16–17]. In 2015, Bridger, which employs a new de novo assembly method that does not construct de Bruijn graphs, was created [18]. This assembler uses a rigorous mathematical model, called the minimum path cover, to construct splice graphs that are used to build compatibility graphs for transcriptome reconstruction from short RNA-seq reads [18]. This multiple k-mer assembler aims to build a bridge between the key concepts of two popular assemblers, the reference-based assembler, Cufflinks [19], and the de novo assembler, Trinity [18]. Given the number of programs available, there is a need for more definitive information on what assemblers and parameters work best for constructing a de novo transcriptome [10, 20].
Much effort has been dedicated towards improving the capabilities of de novo assembly software. However, methods to evaluate the assembler’s performance are still a few steps behind. For example, a common approach to assess genomes assemblies is to evaluate statistics such as the number of contigs, the amount of contigs over 1,000 bps, and the N50 value. The N50 value is defined as the length of the largest contig from all the contigs ranked smallest to largest that represents 50% of the assembly length [21–22]. However, these metrics are also routinely used to evaluate the quality of de novo transcriptome assemblies even though they may be misleading regarding their accuracy [21–23]. Evaluating mRNA characteristics such as the percentage of assembled full length transcripts and the number of long open reading frames (ORFs) are other common metrics for evaluating transcriptome assemblies [13, 15, 24]. A novel reference-free evaluation method to assess the quality of transcriptomes is Detonate RSEM-EVAL [21]. This program produces a statistically principled evaluation score using multiple factors, such as the compactness of the assembly and its support from the RNA-Seq reads used to create it [21].
Annotation-based metrics describe the percentage of sequences within an assembly that match protein sequences found in a related species or curated database [25]. An accurate assembly should contain a high percentage of these conserved proteins while a low percentage reflects mis-assemblies. These types of metrics can be challenging for non-model species that do not have an annotated phylogenetically related species to which it can be aligned. If the evolutionary distance between the two species is too great, then orthologs may have undergone nucleotide changes making alignments less likely to occur [23]. A database to gauge the performance of an assembly strategy in the absence of a well annotated phylogenetically related species is the CEGMA (Core Eukaryotic Genes Mapping Approach) database [26]. This is a manually curated database that contains 248 core proteins present in a wide range of taxa [26]. The same approach can be achieved by utilizing the manually curated BUSCO (Benchmarking Universal Single-Copy Orthologs) protein set for the quantitative assessment of transcriptome assembly and annotation completeness [27]. Using this database, assemblies are matched to 3,023 vertebrate genes and results return the percentage of unaligned sequences. Annotation metrics are useful for assessing both genome and transcriptome assemblies.
Evaluation metrics are important for assessing the quality of genome and transcriptome assemblies. Unfortunately, there is a lack of consensus as to which evaluation metrics work best let alone how many of them to use. For example, Chropra et al. performed a comparison study using peanut (Arachis spp.) RNA-Seq data. Assemblies were evaluated based on the N50 length, average contig length, number of contigs, the novelty of each assembly using the Mummer tool, the accuracy determined by RMBT, and the continuity by estimating the number of full length transcripts [13]. Moreton et al. relied on the RMBT and CEGMA percentages as well as the N50 length, the number of transcripts, and the number of transcripts >1kb when evaluating different assemblies of the duck (Amas platyrhynchos) [10]. He et al. assessed the assemblies of sweet potato fungus (Trametes gallica) and wild rice (Oryza meyeriana) based on N50 length, average contig length, number of contigs > 1,000 bps, as well as their ORFs and percent annotation using BLASTX results against phylogenetically related species [15]. More information on which evaluation metrics best predict the quality of de novo transcriptome assemblies would help establish “best practices” particularly for less experienced users.
The study presented compares assembly programs, k-mer strategies, and various metrics for determining de novo transcriptome assembly quality. Assembly performance is evaluated using the testis transcriptome of Fundulus heteroclitus, an estuary fish that is a sentinel teleost species commonly used in environmental toxicology studies [28]. To date, this is the first study to compare the performances of the commonly used de Bruijn graph-based de novo assemblers, Trinity, Oases, and SOAPdenovo-Trans, with a new de novo method employed by Bridger. This is also the first study to evaluate the effects of using a small and large multiple k-mer strategy for the Oases and SOAPdenovo-Trans assemblers within the same study. Based on the eleven evaluation metrics presented above, it was found that the product of those assemblies was more influenced by the assembler itself than the k-mer strategy. Overall, Bridger performed more often within the top three of each evaluation metric than the de Bruijn graph-based programs for the de novo transcriptome assembly of the killifish RNA-Seq reads.
Materials and Methods
Fish Collection and Nucleic Acid Isolation
Male fish identified as Atlantic killifish (Fundulus heteroclitus) were collected from Tuckerton, NJ (Little Sheepshead Creek) using baited minnow traps. The authority who issued the permission for capturing the killifish was The New Jersey Department of Environmental Protection; Division of Fish and Wildlife (Permit #1125). The vertebrate work done in this study was approved by The Rutgers University’s Institutional Animal Care and Use Committee (IACUC) (Protocol #08–025). In all instances the fish were alive when captured, and capture methods followed approved animal handling protocols. The fish were transported immediately back to Rutgers University in aerated containers containing water from the collection site to reduce stress. The fish were housed under laboratory conditions for one week before they were sacrificed with an overdose of MS-222 (tricaine methanesulphonate) followed by spinal cord dislocation. The testis were dissected and stored in RNAlater (Qiagen) at -20°C prior to RNA extraction.
Total RNA was extracted from the testis using TRIzol® Kit (Invitrogen™) following the manufacturer’s instructions. Potential genomic DNA contamination in the RNA sample was removed by DNase I digestion (Ambion, Inc.). Using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.), the RNA was quantified by measuring the absorbance at 260 nm. The purity of the RNA sample was assessed at an absorbance ratio of 260/280. The RNA integrity number (RIN) was measured with a BioAnalyzer 2100 (Agilent), for a RIN value > 7.0.
Illumina Short-Read Library Construction and Sequencing
Ribosomal depletion and mRNA selection was performed with MicroPolyA purist (Ambion). The mRNA was quantified and ribosomal RNA fractions under 2% were verified using a BioAnalyzer mRNA Nano Kit (Agilent). The dUTP-strand specific cDNA library was made using chemical hydrolysis and the Illumina Ultra Directional RNA-seq kit (NEB). Libraries were barcoded with Tru-Seq adaptors and amplified with 12–15 cycles of PCR (Illumina). Completed RNAseq libraries were quantified using Qubit DNA HS, BioAnalyzer High Sensitivity DNA (Invitrogen, Agilent, KAPA). The libraries were sequenced using the NextSeq 500 High Output 300 cycles kit, reading 155 x 155 bp Paired End Sequencing.
Sequence Data Processing
The quality of the raw Illumina sequence reads were initially assessed using FastQC v0.10.1 [29]. Based on the analysis report, Trimmomatic v0.32 was used to remove all the low quality reads with a Phred score below 20 as well as the Illumina adapters [30]. Contaminating sequences were removed from the reads by using Deconseq with the parameters set to 90% of the contig length with an identity of 94% [31]. FastQC was performed again to verify the integrity of the remaining raw Illumina sequence reads. Upon completion, the quality assessed reads were then ready to be used as the input for the various assembly strategies.
De novo Transcriptome Assembly
Six different assembly strategies were created from four different assemblers: Trinity (v2.0.6), Velvet (v1.2.07) and Oases (v0.2.08), SOAPdenovo-Trans (v1.03), and Bridger [14, 16–18, 32]. The Trinity assembly was the only single k-mer assembly. It was run with its default k-mer value of 25. The Bridger, Oases, and SOAPdenovo-Trans assemblies were performed with multiple k-mer strategies. Bridger, Oases, and SOAPdenovo-Trans used a small multiple k-mer (SMK) strategy consisting of the k-mer lengths of 21, 25, 27, 29, 31, and 33. The SMK strategy was based on the limitations of the Bridger assembler, which can only use k-mers values up to 33. Additionally, Oases and SOAPdenovo-Trans were performed using a large multiple k-mer (LMK) strategy consisting of k-mer lengths of 25, 35, 45, 55, 65, 75, and 85. All six assembly strategies incorporated the k-mer value of 25 to better compare their performances amongst each other. For the multiple k-mer strategies used in the Oases, Bridger, and SOAPdenovo-Trans assemblies, all seven individual k-mer assemblies from each group were concatenated followed by CD-HIT-EST (v 4.6.1) to further remove the redundancy and to cluster the contigs for annotation [33].
Statistics of Assemblies
The six different assembly strategies were assessed using typical statistics for the evaluation of de novo genome assemblies. These included the total number of contigs produced, each assemblies N50 length, and the amount of contigs over 1,000 bps long. These statistics were determined using Transrate (v1.0.0 beta3) http://hibberdlab.com/transrate/.
RMBT Analysis
The accuracy of each assembly was assessed by determining the percentage of raw reads that could be mapped back to transcripts (RMBT). First, indexes were generated using bowtie2-build [34]. Then Bowtie2 (v 2.2.5) was used to map the reads against each assembly and provide the metric of accuracy, which is the percentage of raw reads that align [34].
Phylogenetic Tree Alignments
Another common assessment tool to evaluate the quality of de novo transcriptome assemblies is to align the assembled contigs to a well annotated phylogenetically related species. Quality is based on the percentage of contigs that match the protein sequences of the related species. To determine which closely related fish genome to use as a reference, PhyloT (http://phylot.biobyte.de/contact.html) was used. This program generated a phylogenetic tree between killifish and the eleven publically available fish genomes on Ensembl: Amazon molly (Poecilia Formosa), Mexican tetra (Astyanax mexicanus), Atlantic cod (Gadus morhua), Japanese pufferfish (Takifugu rubripes), medaka (Oryzias latipes), southern platyfish (Xiphophorus maculatus), spotted gar (Lepisosteus oculatus), stickleback (Gasterosteus aculeatus), green spotted pufferfish (Tetraodon nigroviridis) Nile tilapia (Oreochromis niloticus), and zebrafish (Danio rerio). This program creates trees based on the NCBI taxonomy database, and it was visualized by the web based tool, Interactive Tree of Life (v2) [35]. Based on the created phylogenetic tree, southern platyfish (X. maculatus) and Amazon molly (P. formosa) were shown to be the closest relative to killifish. Therefore, the six different assemblies were aligned to the Ensembl proteins of southern platyfish and Amazon molly using BLASTX with an E-value cut-off of 1e-3 to quantify the percentage of previously annotated genes found in each assembly.
CEGMA and BUSCO Alignments
CEGMA (v.2.5) and BUSCO (v1.1) are other reference-based tools for assessing the degree of annotation. They were individually employed to quantitate assembly completion based on the percentage of contigs that do or do not (therefore mis-assembled) align to highly conserved proteins [28].
Full Length Transcript Analysis
The number of full length transcripts was quantified to further evaluate the performance of each assembly by following scripts provided by the Trinity software package (http://trinityrnaseq.sourceforge.net/). A modified ‘BLASTX’ script was used to calculate each assembly’s alignment coverage to the curator-evaluated database, SwissProt. Full length transcripts were defined in this study by having > 70% alignment coverage and > 90% alignment coverage to SwissProt proteins.
Open Reading Frames Analysis
The presence of long open reading frames (ORFs) was analyzed to determine the quality of each assembly by using scripts provided by TransDecoder (http://transdecoder.github.io/). BlastP (v 2.2.30+) was used to search the protein database SwissProt with an E-value cut-off of 1e-3. ORFs ranging from >799 bps, >999 bps, and >1,199 bps were determined using gawk to filter the Fasta files by length.
Detonate RSEM-EVAL Score
DETONATE’s RSEM-EVAL was used to evaluate the quality of the six different assemblies. This program offers a reference-free evaluation method that relies only on the assembly and the reads used to create it [22].
Results
Sequencing and De Novo Transcriptome Assembly
To globally profile the testis transcriptome of killifish, we employed Illumina NextSeq 500 technology to sequence the libraries generating 7,197,900 pair-end short reads encoding 1,033,683,143 bases (Table 1). All the raw sequencing reads were deposited into the Short Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) and can be accessed under the accession number SRX1058750. To perform quality control of the raw sequence reads, they were processed to remove Illumina adaptor sequences, low quality reads with a Phred score value less than 20, and contaminating sequences. The processing of the raw sequence reads resulted in 6,349,606 (88.21%) clean reads that were assembled by the following assembly strategies; SOAP LMK, SOAP SMK, Oases LMK, Oases SMK, Bridger, and Trinity (Table 1).
Table 1. Statistics of the raw reads after Illumina sequencing and processing.
| Killifish | |
|---|---|
| Number of nucleotide bases | 1,033,683,143 |
| Number of raw reads | 7,197,900 |
| Number of clean reads for assembly | 6,349,606 |
| Percent of used reads for assembly | 88.21% |
Statistics of Assembly
The statistics of each assembly was initially used to evaluate the performance of each assembly strategy.
Assemblies with the highest to lowest amount of contigs produced were: Bridger > SOAP LMK > SOAP SMK > Trinity > Oases SMK > Oases LMK (Table 2). Assemblies with the longest to shortest N50 length were: Oases SMK > Oases LMK > Bridger > Trinity > SOAP SMK > SOAP LMK (Table 2). Assemblies with the highest to lowest average contig length were: Oases SMK > Oases LMK > Bridger > Trinity > SOAP SMK> SOAP LMK (Table 2). Assemblies with the highest to lowest amount of contigs over 1kb were: Bridger > Oases SMK > SOAP SMK >Trinity > Oases LMK > SOAP LMK (Table 2). In summary, the Bridger assembly produced the most amount of contigs and contigs over 1kb. The Oases SMK assembly produced the longest N50 length and had the highest average contig length. The SOAP LMK assembly performed the worst for the N50 length, most amount of contigs over 1kb, and the average contig length. The Oases LMK assembly produced the least amount of contigs.
Table 2. Statistics of the Assemblies.
| SOAP LMK | SOAP SMK | Oases LMK | Oases SMK | Bridger | Trinity | |
|---|---|---|---|---|---|---|
| Contig Number | 198,085 | 187,104 | 99,567 | 135,312 | 303,906 | 180,658 |
| N50 Length | 917 | 1,042 | 1,676 | 1,743 | 1,668 | 1,189 |
| Minimum Contig Length | 200 | 200 | 200 | 200 | 201 | 224 |
| Largest Contig Length | 11,658 | 11,658 | 16,019 | 21,489 | 15,151 | 11,023 |
| Average contig length | 622 | 672 | 1,021 | 1,035 | 879 | 711 |
| Contigs Over 1k | 33,326 | 36,136 | 33,847 | 45,992 | 81,769 | 35,527 |
| RMBT | 83.78% | 80.77% | 85.28% | 84.18% | 87.71% | 82.96% |
RMBT Analysis
A common method to evaluate the accuracy of a de novo assembly without a reference genome is to determine the percentage of reads that can be mapped back to transcripts (RMBT) constructed by the assembler. Based on this metric, the Bridger assembly had the highest RMBT percentage (87.71%) and the SOAP SMK assembly had the lowest (80.77%). Results for Trinity, SOAP LMK, Oases SMK, and Oases LMK were similar with RMBT percentages ranging from 82.96% to 85.28% (Table 2).
Phylogenetic Tree Alignments
A strategy to gauge the credibility of a de novo assembly is to see how well its annotated sequences compare to those of a related species. Therefore, a phylogenetic tree of killifish and eleven publically available fish genomes was constructed using Ensembl. Based on the results, the southern platyfish and Amazon molly were determined to be the closest related genera to killifish (Fig 1). Therefore the contigs produced from each assembly strategy were aligned to the southern platyfish and Amazon molly genomes independently using BLASTX with an E-value of <1e-3. Results showed that the Oases LMK assembly had the best percentage of alignments to both the southern platyfish (50.14%) and Amazon molly (51.82%) databases (Table 3). The Trinity assembly had the lowest alignment percentage to both the southern platyfish (32.68%) and Amazon molly (34.15%) databases. Results for SOAP LMK, SOAP SMK, Bridger and Trinity were similar with alignment percentages ranging from 32.68 to 37.88% for southern platyfish and 34.15 to 39.35% for Amazon molly (Table 3).
Fig 1. Phylogenetic tree analysis.
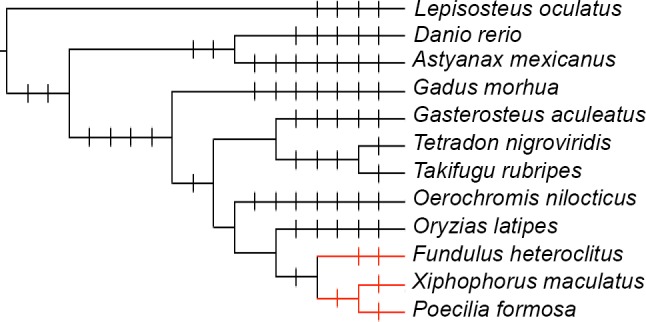
A phylogenetic tree analysis of the 11 publically available fish genomes and killifish testis. As highlighted in red, the results of Ensembl showed that southern platyfish (X. maculatus) and Amazon molly (P. Formosa) are the closest relatives to killifish (F. heteroclitus).
Table 3. BLASTX alignments from the six different assemblies against the southern platyfish and Amazon molly databases.
| Database | SOAP LMK | SOAP SMK | Oases LMK | Oases SMK | Bridger | Trinity |
|---|---|---|---|---|---|---|
| southern platyfish | 34.60% | 35.22% | 50.14% | 47.00% | 37.88% | 32.68% |
| Amazon molly | 36.46% | 37.19% | 51.82% | 48.71% | 39.35% | 34.15% |
CEGMA Alignments
A reference-based approach for assessing the quality of an assembly is to align the contigs to the 248 highly conserved proteins in the CEGMA dataset. All of the CEGMA proteins were present in the killifish testis transcriptome. However, none of the assembly strategies were able to incorporate all of them as seen in Table 4. Full length CEGMA proteins, defined as having at least 70% of the protein length found in the CEGMA dataset, ranged from 97.58 to 98.79%. Partial CEGMA proteins ranged from 99.60 to 100%. The Oases LMK and Oases SMK assemblies contained the highest percentage of full length (98.79%) and partial length (100%) CEGMA proteins. The Trinity assembly contained the lowest percentage (97.58%) of full length CEGMA proteins as well as the lowest percentage (98.79%) of partial CEGMA proteins (Table 4). Both of the SMK and LMK strategies of SOAP and Oases were unable to assemble the same CEGMA proteins at full or partial length (Table 4).
Table 4. BLASTX alignments of the six different assemblies to the CEGMA dataset.
| Assembly | CEGS | CEGs Missing | Partials | Partial Missing |
|---|---|---|---|---|
| SOAP LMK | 98.39% | KOG0261, KOG0209, KOG0462, KOG2311 | 99.60% | KOG2311 |
| SOAP SMK | 98.39% | KOG0261, KOG0209, KOG0462, KOG2311 | 99.60% | KOG2311 |
| Oases LMK | 98.79% | KOG0292, KOG2311, KOG4392 | 100% | |
| Oases SMK | 98.79% | KOG0292, KOG2311, KOG4392 | 100% | |
| Bridger | 98.39% | KOG0261, KOG0292, KOG0969, KOG2311 | 99.60% | KOG0969 |
| Trinity | 97.58% | KOG0292, KOG0209, KOG0434, KOG0469, KOG0481, KOG2623 | 98.79% | KOG0209, KOG0292, KOG0434 |
BUSCO Alignments
BUSCO is another reference-based program for assessing quality of de novo assemblies. The program determined the percentage of mis-assembled transcripts by trying to align all transcripts to highly conserved proteins within the BUSCO dataset. None of the assembly strategies were able to incorporate all of the 3,023 vertebrate BUSCOs genes as seen in Fig 2. Trinity performed best in terms of having the least amount of missing genes 675 (22.3%), followed by Bridger 762 (25.2%), Oases SMK 869 (28.7%), Oases LMK 940 (31%), SOAP SMK 980 (32.4%), and SOAP LMK 1,030 (34.1%).
Fig 2. BUSCO Analysis.

The Trinity assembly performed the best by having the least amount of missing BUSCOS.
Full Length Transcript Analysis
Another metric of assembler performance is quantifying the amount of transcripts that appear to be nearly full length. Based on the alignments with the manually annotated and reviewed SwissProt database, the Trinity assembly performed the best. This assembly produced 8,168 proteins that had greater than 70% alignment coverage and 5,664 proteins that had greater than 90% alignment coverage as seen in Fig 3A and 3B. Other assemblies with the highest to lowest amount of nearly full length proteins were: Oases SMK > Oases LMK > Bridger > SOAP LMK > SOAP SMK. This rank order was the same for 70% and 90% alignment analyses.
Fig 3. Full Length Transcript Analysis.

a) Trinity had the most proteins with greater than 70% alignment coverage. b) Trinity had the most proteins with greater than 90% alignment coverage.
Open Reading Frames Analysis
The contigs in this project were sequenced from mRNA; therefore, the best assembly strategy should produce a large number of open reading frames (ORFs). Overall, the assemblies performed in the same rank order for the presence of ORFs in sequence lengths ranging from >799 bps, >999 bps, and >1,199 bps. Assemblies with the highest to lowest amount of ORFs for each sequence length were: Bridger > Trinity > Oases SMK > Oases LMK > SOAP SMK > SOAP LMK. The Bridger assembly had the highest amount of ORFs for all three lengths. This assembly had 3,189 ORFs > 799 bps, 1,377 ORFs > 999 bps, and 593 ORFs > 1,999 bps (Fig 4). The SOAP LMK assembly had the lowest amount of ORFs for all three lengths. This assembly had 699, 187, and 62 ORFs for sequence lengths ranging from > 799 bps, > 999 bps, and > 1,999 bps, respectively (Fig 4)
Fig 4. Open Reading Frames Analysis.

The Bridger assembly produced the most amount of open reading frames for sequences with >799 bps (red), >999 bps (blue), and >1,199 bps (green).
Detonate RSEM-EVAL Score
A novel metric to evaluate the quality of each assembly is Detonate’s RSEM-EVAL score. This score is based on a probabilistic model that only requires the clean reads and the overall assembly for inputs [22]. Assemblies with higher RSEM-EVAL scores are considered better. The assemblies with highest to lowest RSEM-EVAL scores are as follows: Trinity > Bridger > SOAP LMK > Oases LMK > SOAP SMK > Oases SMK (Table 5). The scores for all six assemblies ranged from -5,426.0 x106 to -6,125.0 x106 indicating that all assemblies were very similar (Table 5).
Table 5. The Detonate’s RSEM-EVAL scores suggests that the Trinity assembly performed the best.
The higher the number value, the better the assembly.
| Assembly | SOAP LMK | SOAP SMK | Oases LMK | Oases SMK | Bridger | Trinity |
|---|---|---|---|---|---|---|
| Score | -5,488.0 x10 ˄6 | -5,715.0 x10˄6 | -5,602.0 x10 ˄6 | -6,125.0 x10 ˄6 | -5,448.0 x10 ˄6 | -5,426.0 x10 ˄6 |
Overview of Assembly Strategies
Based on the eleven evaluation metric categories used to assess the six different assembly strategies, it was determined that no one particular assembly strategy performed the best in all categories tested (Table 6). The assembly strategies that performed within the top three of most metrics was the Bridger assembly (10 of 11), followed by the Oases SMK assembly (9 of 11), and then the Oases LMK assembly (6 of 11). The assembly strategies that occurred within the top three the least were the Trinity assembly (5 of 11), followed by the SOAP LMK assembly (4 of 11) and then the SOAP SMK assembly (3 of 11).
Table 6. Summary of the top three performers for each evaluation metric category.
| First | Second | Third | |
|---|---|---|---|
| Contig Number | Bridger | SOAP LMK | SOAP SMK |
| N50 Length | Oases SMK | Oases LMK | Bridger |
| Contigs >1kb | Bridger | Oases SMK | SOAP SMK |
| RMBT | Bridger | Oases LMK | Oases SMK |
| southern platyfish DB | Oases LMK | Oases SMK | Bridger |
| Amazon molly DB | Oases LMK | Oases SMK | Bridger |
| CEGMA | Oases LMK and Oases SMK | Bridger, SOAP LMK, and SOAP SMK | |
| BUSCO | Trinity | Bridger | Oases SMK |
| Full Length Transcripts | Trinity | Oases SMK | Oases LMK |
| ORFs | Bridger | Trinity | Oases SMK |
| Detonate | Trinity | Bridger | SOAP LMK |
Discussion
Currently, there is not a gold standard for a de novo transcriptome assembler. It is known that different de novo assemblers using the same transcripts with similar user defined parameters have produced assemblies that vary amongst each other [10, 13, 15, 25]. One of the goals of this work was to compare four recently published de novo assemblers and one user defined parameter, the k-mer value, to determine if one strategy was better than another for de novo assembly of a non-model organism transcriptome. The assemblers included Trinity, Oases, and SOAPdenovo-Trans (commonly used de Bruijn graph-based de novo assemblers) as well as Bridger (uses the minimum path cover model to construct splice graphs that are used to build compatibility graphs). Additionally, eleven metrics of assembly quality were compared in order to better form a consensus as to which ones should be used to assess de novo transcriptome assemblies. Overall, eleven metrics were used to evaluate six assembly strategies.
Commonly used evaluation statistics, such as the number of contigs, the N50 value, and contig length, were developed for genome assemblies but have also been used to evaluate transcriptome assemblies [22]. Better assemblies should theoretically have more reads assembled into longer contigs and thereby higher N50 values. However, importance of the N50 metric has been questioned [21, 36]. Research indicated that N50 values could be artificially increased based on k-mer strategy and or the user defined minimum contig length. Short contigs occurred when high k-mer values did not assemble short reads of low abundance transcripts or low k-mer values assembled short fragmented transcripts due to lack of overlap [11, 13]. In both cases, if the resulting transcripts were shorter than the minimum contig length parameter established by the user, they were eliminated by the program ultimately generating artificially high N50 values. In the study presented, the influence of high (LMK) and low (SMK) k-mer values on evaluation statistics was determined for SOAP and Oases assemblies. Both showed somewhat higher N50 values for low k-mer assemblies, but greater differences were found for the assembler. For example, the N50 lengths for SOAP LMK and SMK were 917 and 1,042, respectively, while those for Oases LMK and SMK were 1,676 and 1,743, respectively (Table 2). Bridger also used a low k-mer strategy, and results showed that contig number as well as contigs over 1k were twice those of the other assemblies. Taken together, the assembler program had more influence on these traditional, genomic metrics than the k-mer values, and the Bridger assembler performed the best in two out of the three metrics.
The quality of an assembly can be assessed by its ability to construct transcripts that align to genes in publically available databases. In the study presented, the six assemblies were evaluated using two well annotated phylogenetic tree relatives, the CEGMA database, and the BUSCO database. For the phylogenetic tree analyses, killifish transcriptome assemblies were aligned to the reference databases of southern platyfish (X. maculatus) and Amazon molly (P. formosa) (Fig 1). Oases assemblies had the best percentages of alignments ranging from 47.00 to 51.82%. Results for the other assemblies were similar but lower with alignments ranging from 32.68 to 39.35%. The low percentage of killifish matches in general could be attributed to the evolutionary distance between killifish and the other two species as other de novo transcriptome assembly studies using non-model fish had similar findings [37–39]. Additionally, the BLASTX results were based on aligning the testis transcriptome of killifish with the entire genomes of the southern platyfish and Amazon molly. This would have inherently caused a lower percentage of matches. Furthermore, some of the unannotated contigs may be short and consist of sequences lacking well characterized protein domains, comprised of 3’ or 5’ untranslated regions, or be non-coding RNAs [40]. Results also showed that the general performance of each assembly’s BLASTX alignment was more dependent on the assembler software than k-mer value. For example, assemblies using Oases SMK and LMK had percent alignments of 47.00 and 50.14% in southern platyfish while SOAP had percent alignments of 35.22% and 34.60% for the same species (Table 3).
CEGMA and BUSCO evaluated assembly quality by aligning contigs to core proteins. For CEGMA, most of the 248 core proteins were found with each assembler; therefore, this metric did not distinguish between them (Table 4). Interestingly, some assemblers left out CEGMA proteins found by others. This indicated that particular assemblers had different proficiencies in reassembling clean reads into transcripts as previously reported by Naksugi et al. [25]. For example, KOG0292 (Vesicle coat complex COPI, alpha subunit) was only present in the SOAP SMK an LMK strategies, and KOG2311 (NAD/FAD-utilizing protein possibly involved in translation) was only present in Trinity. In addition, both SOAP and Oases were unable to properly assemble the same transcripts regardless of the k-mer strategy. For example, both SOAP SMK and LMK did not assemble at full length KOG0261, KOG0209, KOG0462 or KOG2311. Both Oases SMK and LMK did not assemble at full length KOG0292, KOG2311 or KOG4392. This indicated that the assembler’s algorithms had a more influential role than the k-mer selection when assembling transcripts. BUSCO showed greater differences between assemblers than CEGMA most likely due to the larger database of 3,023 vertebrate genes (Fig 2). Missing BUSCO genes for the six different assemblies ranged from 675 (22.3%) in Trinity to 1,030 (34.1%) in SOAP LMK. As with CEGMA and BLASTX alignments, this metric showed that the assembler rather than the k-mer strategy had more influential over the outcome. Both the SMK (28.7%) and LMK (31%) strategies of Oases outperformed the SMK (32.4%) and LMK (34.1%) strategies of SOAP. Overall, results indicated that for either genomes or transcriptomes, if a user’s goal was to annotate the most genes possible for a non-model organism, Oases was the assembler of choice. If the goal was to assemble the most accurate transcriptome, Trinity or Bridger would be the best choice. As of 2015, CEGMA is no longer actively supported and has been essentially replaced by BUSCO.
This study incorporated several evaluation metrics specifically for transcriptomes including number of full length transcripts, ORFs, Detonate’s RSEM-EVAL and RMBT. Shown here as well as other studies, the Trinity assembler proved able to reconstruct the most full length transcripts [13–14, 41]. Alignment coverages were greater than 70% (8,168) and 90% (5,664) even though it utilized a single k-mer value (Fig 3A and 3B). The Bridger assembler was able to reconstruct the most ORFs (Fig 4). Results showed that transcripts of >799 bps, >999 bps, and > 1199 bps had 3,189, 1,377 and 593 ORFs, respectively. This number of ORFs was 23.9% (>799 bs), 19.3% (>999 bps), and 16.3% (>1199 bps) higher than those for Trinity, the next closest assembler. SMK and LMK assemblies of Oases and SOAP were similar for both full length transcripts and ORFs; therefore, the overall quality of each assembly's performance was not reliant on the k-mer strategy. Detonate’s RSEM-EVAL provided a reference-free evaluation score where a high value indicated an accurate transcriptome assembly. Results showed that the Trinity assembly appeared slightly better than the Bridger and SOAP LMK (Table 5). However, all scores were similar (ranging from 5,426.0 x10 ˄6 to 6,125.0 x10 ˄6); and therefore, this metric did not distinguish well between the assemblers. The RMBT statistic determined assembly accuracy based on the philosophy that the higher the amount of processed reads that can be mapped back to an assembly the fewer the errors (i.e. mis-assembly) introduced by the assembler program. Results for the six assembly strategies evaluated showed RMBT percentages ranging from 87.71% for the Bridger assembly to 80.77% for the SOAP SMK assembly (Table 2). Other studies comparing assembly strategies also reported differences in RMBT percentages [9–10, 13, 20, 25]. In the study presented, the assembler software and not the k-mer strategy appeared to have the greater impact. RMBT results showed that the SMK and LMK assemblies for both SOAP (80.77 and 83.78%, respectively) and Oases (84.18 and 85.28%, respectively) performed similarly. Overall, both Trinity and Bridger performed well according to transcriptome-specific metrics and k-mer sizes were not a factor.
Conclusion
This research presents a comprehensive multi k-mer strategy assessment with a thorough multiple assembler performance evaluation analysis. To date, this is the first study to compare the performances of the commonly used de Bruijn graph-based de novo assemblers- Trinity, Oases, and SOAPdenovo-Trans with a new de novo method employed by Bridger. Additionally, this is the first study to analyze a small and large multiple k-mer strategy for the same assembler software. Based on eleven evaluation metrics, it was found that the assembler had more influence than the k-mer strategy. This contradicted Chopra et al., 2014, Chang et al., 2015, and Surget-Groba and Montoya-Burgos, 2010, who highlight the advantages and disadvantages between low and high k-mer values (11, 13, 18). For assemblers, killifish data indicated that Bridger performed more often within the top three of each evaluation metric performed better in more of the matrices tested (10 of 11) than the other programs. Oases generated the most alignments with phylogenetically related species. Trinity generated the most full length transcripts, while Bridger generated the most ORFs. Bridger and Trinity also both performed well with BUSCO and Detonate’s RSEM-EVAL. Something that differentiated Trinity and Bridger was time for transcriptome assembly. Bridger was faster, which the authors found to be an important consideration. Other researchers have shown that Trinity takes longer to assemble transcriptomes than other programs including SOAPdenovo-Trans and Oases [17]. Overall, Bridger was the preferred assembler program for producing a quality de novo assembly of the killifish testis transcriptome based on its performance within the top three of each evaluation metric along with its time for completion.
Acknowledgments
The authors wish to thank Dr. Keith Cooper and Dan Milemann at Rutgers University, for their technical support in capturing and housing the killfish.
Data Availability
The data is available at the SRA of NCBI. The accession number is SRX1058750 (http://www.ncbi.nlm.nih.gov/sra/SRX1058750) [accn].
Funding Statement
This project is sponsored by funding provided by the Louisiana Department of Wildlife and Fisheries (LDWF)-LSU Contract #69670.
References
- 1.Gordo SM, Pinheiro DG, Moreira EC, Rodrigues SM, Poltronieri MC, de Lemos OF, et al. High-throughput sequencing of black pepper root transcriptome. BMC Plant Biology 2012; 12:168 10.1186/1471-2229-12-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan H, Xiao Y, Yang Y, Xia W, Mason AS, Xia Z, et al. (2013) RNA-Seq Analysis of Cocos nucifera: Transcriptome Sequencing and De Novo Assembly for Subsequent Functional Genomics Approaches. PLoS ONE 8(3): e59997 10.1371/journal.pone.0059997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng V, Villanueva KE, Ewen-Campen BS, Alwes F, Browne WE, Extavour CG. De novo assembly and characterization of a maternal and developmental transcriptome for the emerging model crustacean Parhyale hawaiensis. BMC Genomics. 2011; 12:581 10.1186/1471-2164-12-581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg R, Patel RK, Tyagi AK, Jain M. De Novo Assembly of Chickpea Transcriptome Using Short Reads for Gene Discovery and Marker Identification. DNA Research. 2011; 18 (1): 53–63. 10.1093/dnares/dsq028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar A, Congiu L, Lindström L, Piiroinen S, Vidotto M, Grapputo A. Sequencing, De Novo Assembly and Annotation of the Colorado Potato Beetle, Leptinotarsa decemlineata, Transcriptome. PLoS ONE. 2014; 9(1): e86012 10.1371/journal.pone.0086012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma N, Jung C-H, Bhalla PL, Singh MB. RNA Sequencing Analysis of the Gametophyte Transcriptome from the Liverwort, Marchantia polymorpha. PLoS ONE. 2014; 9(5): e97497 10.1371/journal.pone.0097497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nandety RS, Kamita SG, Hammock BD, Falk BW. Sequencing and De Novo Assembly of the Transcriptome of the Glassy-Winged Sharpshooter (Homalodisca vitripennis). PLoS ONE. 2013; 8(12): e81681 10.1371/journal.pone.0081681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashrafi H, Hill T, Stoffel K, Kozik A, Yao J, Chin-Wo SR, et al. De novo assembly of the pepper transcriptome (Capsicum annuum): a benchmark for in silico discovery of SNPs, SSRs and candidate genes. BMC Genomics. 2012; 13:571 10.1186/1471-2164-13-571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haznedaroglu BZ, Reeves D, Rismani-Yazdi H, Peccia J. Optimization of de novo transcriptome assembly from high-throughput short read sequencing data improves functional annotation for non-model organisms. BMC Bioinformatics 2012, 13:170 10.1186/1471-2105-13-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreton J, Dunham S. Emes R. (2014) A consensus approach to vertebrate de novo transcriptome assembly from RNA-seq data: assembly of the duck (Anas platyrhynchos) transcriptome. Front Genet. 2014; 5:190 10.3389/fgene.2014.00190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surget-Groba Y and Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010; 20(10):1432–40. 10.1101/gr.103846.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, et al. (2010) De novo assembly and analysis of RNA-seq data. Nat Methods 7: 909–912. 10.1038/nmeth.1517 [DOI] [PubMed] [Google Scholar]
- 13.Chopra R, Burow G, Farmer A, Mudge J, Simpson CE, Burow MD. Comparisons of De Novo Transcriptome Assemblers in Diploid and Polyploid Species Using Peanut (Arachis spp.) RNA-Seq Data. PLoS ONE.2014; 9(12): e115055 10.1371/journal.pone.0115055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011; 29: 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He B, Zhao S, Chen Y, Cao Q, Wei C, Cheng X, et al. Optimal assembly strategies of transcriptome related to ploidies of eukaryotic organisms. BMC Genomics. 2015; 16:65 10.1186/s12864-014-1192-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012; 28: 1086–1092. 10.1093/bioinformatics/bts094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014; 30: 1660–1666. 10.1093/bioinformatics/btu077 [DOI] [PubMed] [Google Scholar]
- 18.Chang Z, Li G, Liu J, Zhang Y, Ashby C, Liu D, et al. Bridger: a new framework for de novo transcriptome assembly using RNA-seq data. Genome Biology 2015; 16:30 10.1186/s13059-015-0596-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010; 28:511–5. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics 2011; 12(Suppl 14): S2 10.1186/1471-2105-12-S14-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, Dewey C. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biology 2014; 15:553 10.1186/s13059-014-0553-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker M. De novo genome assembly: what every biologist should know. Nat Meth. 2012; 9(4):333–7. 10.1038/nmeth.1935 [DOI] [Google Scholar]
- 23.O'Neil ST, Emrich SJ. Assessing De Novo transcriptome assembly metrics for consistency and utility. BMC Genomics 2013; 14: 465 10.1186/1471-2164-14-465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakasugi K, Crowhurst R, Bally J, Waterhouse P. Combining Transcriptome Assemblies from Multiple De Novo Assemblers in the Allo-Tetraploid Plant Nicotiana benthamiana. PLoS ONE. 2014; 9(3): e91776 10.1371/journal.pone.0091776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schliesky S, Gowik U, Weber A, Braeutigam A: RNA-Seq assembly–Are we there yet?. Front Plant Sci. 2012; 3:220 10.3389/fpls.2012.00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007; 23: 1061–1067. [DOI] [PubMed] [Google Scholar]
- 27.Simão FA, Waterhouse RM, Loannidis P, Kriventseva EV, Zdobnov EV. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- 28.Burnett KG, Bain LJ, Baldwin WS, Callard GV, Cohen S, Di Giulio RT, et al. Fundulus as the premier teleost model in environmental biology: opportunities for new insights using genomics. Comparative Biochemistry and Physiology Part D, Genomics & Proteomics. 2007; 2: 257–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 30.Bolger AM, Lohse M., & Usadel B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics. 2014; 30(15): 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmieder R, Edwards R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE. 2011. 6(3): e17288 10.1371/journal.pone.0017288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zerbino DR, Birney E. (2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008; 18(5): 821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Godzik A Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- 34.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Letunic I, Bork P. Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucl Acids Res. 2011; 39: 475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, et al. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Research 2012; 22: 557–567. 10.1101/gr.131383.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji P, Liu G, Xu J, Wang X, Li J, Zhao Z, et al. Characterization of Common Carp Transcriptome: Sequencing, De Novo Assembly, Annotation and Comparative Genomics. PLoS ONE. 2012; 7(4): e35152 10.1371/journal.pone.0035152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huth TJ, Place SP. De novo assembly and characterization of tissue specific transcriptomes in the emerald notothen, Trematomus bernacchii. BMC Genomics. 2013; 14:805 10.1186/1471-2164-14-805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salisbury JP, Sirbulescu RF, Moran BM, Auclair JR, Zupanc GKH, Agar J The central nervous system transcriptome of the weakly electric brown ghost knifefish (Apteronotus leptorhynchus): de novo assembly, annotation, and proteomics validation. BMC Genomics 2015; 16:166 10.1186/s12864-015-1354-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Wang X, Zou Z, Jia X, Wang Y, Zhang Z. Transcriptome analysis of the differences in gene expression between testis and ovary in green mud crab (Scylla paramamosain). BMC Genomics. 2014; 15:585 10.1186/1471-2164-15-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duan J, Xia C, Zhao G, Jia J, Kong X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics. 2012; 13: 392 10.1186/1471-2164-13-392 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data is available at the SRA of NCBI. The accession number is SRX1058750 (http://www.ncbi.nlm.nih.gov/sra/SRX1058750) [accn].