

Abstract
To target oncolytic measles viruses (MV) to tumors, we exploit the binding specificity of designed ankyrin repeat proteins (DARPins). These DARPin-MVs have high tumor selectivity while maintaining excellent oncolytic potency. Stability, small size, and efficacy of DARPins allowed the generation of MVs simultaneously targeted to tumor marker HER2/neu and cancer stem cell (CSC) marker EpCAM. For optimization, the linker connecting both DARPins was varied in flexibility and length. Flexibility had no impact on fusion helper activity whereas length had. MVs with bispecific MV-H are genetically stable and revealed the desired double-target specificity. In vitro, the cytolytic activity of bispecific MVs was superior or comparable to mono-targeted viruses depending on the target cells. In vivo, therapeutic efficacy of the bispecific viruses was validated in an orthotopic ovarian carcinoma model revealing an effective reduction of tumor mass. Finally, the power of bispecific targeting was demonstrated on cocultures of different tumor cells thereby mimicking tumor heterogeneity in vitro, more closely reflecting real tumors. Here, bispecific excelled monospecific viruses in efficacy. DARPin-based targeting domains thus allow the generation of efficacious oncolytic viruses with double specificity, with the potential to handle intratumoral variation of antigen expression and to simultaneously target CSCs and the bulk tumor mass.
Introduction
In 2012, breast and ovarian carcinoma accounted for 25.2 and 3.6% of worldwide 3.5 million cancer mortalities in women, respectively.1 Especially aggressive forms of both tumor entities are characterized by some shared markers. Among these are the epithelial cell adhesion molecule (EpCAM) and the human epidermal growth factor receptor 2 (HER2/neu, or short HER2). Targeting of the tumor-associated antigen HER2 by monoclonal antibodies has been a showcase for the development of targeted cancer medicine, which has first been authorized for the treatment of metastatic breast cancer.2 HER2, a receptor tyrosine kinase, is amplified in 15–30% of all breast3,4 and 9–32% of ovarian cancer patients.5 This cellular surface glycoprotein is an active “driver” oncogene, which has no known ligand, and its expression is clinically associated with poor prognosis. HER2-targeted therapies radically changed the way HER2-overexpressing tumors are treated, but a significant number of patients with recurrent, HER2-positive breast cancer do not respond to antibody therapy with or without chemotherapy (only 25–50% response6). For ovarian carcinoma, numbers are even worse.7 Moreover, even responsive tumors eventually become resistant. Reasons for the disappointing durability of response to HER2-targeted therapies in advanced breast and ovarian carcinoma are complex, but can be partially explained by acquired resistance or a priori heterogeneity in tumor cell populations.8
High mutation rates and heterogeneity of tumor cells are a general hallmark of cancer. Consequently, cell populations of advanced tumors are inherently diverse, also with regard to expressed markers, and can acquire escape mutations. Moreover, the stem cell theory of cancer proposes that among all cancerous cells within a tumor, a few act as stem cells that reproduce themselves and sustain the tumor. In this view, it is especially these cancer stem cells (CSCs) that need to be targeted by a successful therapy in order to prevent the tumor from recurrence and becoming therapy resistant. Among the markers that identify potential CSCs9,10 is EpCAM.11–13 It is often upregulated in epithelial tumors and is in general better accessible for therapeutics here, since in the course of transformation the polarity of EpCAM expression at tight junctions is lost and EpCAM molecules are homogenously distributed on the cancer cell surface.11 Interestingly, EpCAM is present at low levels in 48%, and overexpressed in approximately 35–42% of all breast tumor patients samples.14–16 In ovarian cancer, EpCAM-expression is upregulated even in 69% of all patients.17 Coexpression of HER2 and EpCAM occurs in a significant number of breast cancer patients (13.2%) and further worsens prognosis.15 Thus, the treatment of this particular subtype of carcinomas could be improved by combination of HER2- and EpCAM-targeted therapies. The potential for tumor escape might be reduced when therapy is combined in one setting or even a single drug, an approach that can be realized by an oncolytic virus (OV) therapy.
OVs are novel cancer therapeutics and are intensively studied in preclinical and clinical studies. Most recently, an oncolytic herpesvirus named talimogene laherparepvec (Imlygic®) has received a positive recommendation for marketing authorization by the European Medicines Agency and the Federal Drug Agency for the treatment of melanoma.18 Notably, being replication competent viruses, OVs have a completely different mode of action than conventional drugs.19,20 Due to the strongly lytic nature of its replication, measles virus (MV) appears as an ideal virus for use as OV with currently ongoing phase 1 trials for the treatment of six different tumor entities and a phase 2 study for multiple myeloma, which is recruiting patients.21 In general, MV is well tolerated and one patient with advanced multiple myeloma went into full remission after high-dose MV treatment, with flu-like symptoms during infusion reported as most striking side effects.22 MV is the prototypic Morbillivirus and belongs to the Paramyxoviridae family. It is the causative agent of the measles, but live attenuated vaccine strains have been developed, which are among the most efficient and safest vaccines known.23 MV vaccine strains use CD46 as entry receptor24,25 in addition to the receptors used by pathogenic wildtype MV strains: SLAM on activated lymphocytes,26 or nectin-4 on epithelial cells lining the upper airways.27,28 CD46 is expressed on all human nucleated cells, but regularly found upregulated in certain tumors.29 Accordingly, the tumor tropism of unmodified oncolytic MV derived from the Edmonston B vaccine strain has been correlated to CD46 upregulation.30 However, this tropism is only relative, since the CD46 is ubiquitously expressed on human cells.
On the virus surface, the viral glycoprotein hemagglutinin (H) is responsible for receptor attachment followed by triggering cell-entry.31 MVs’ receptor usage can be altered by changing the binding specificity of H. This can be achieved by introducing four specific point mutations to ablate recognition of the native receptors32 and the genetic fusion of binding domains specific for the desired target receptor to the C-terminus of H. Usually, these have been single-chain antibody fragments (scFv’s),33 natural receptor ligands34 or peptides.35 Thereby, MV tropism can be redirected to virtually any surface-exposed structure of choice.33 As an alternative binding domain, we recently developed a strategy36 using designed ankyrin repeat proteins (DARPins)37 to target tumor markers HER2 (ref. 38), EpCAM,39 or EGFR.40 The unique structural properties of DARPins enabled us to generate a bispecific MV using HER2 and/or EpCAM, as entry receptors.36 This virus was generated to address resistance development to mono-targeted therapies, but its qualities have only been partially characterized.36
Here, we aim to demonstrate the advantage of bispecific compared to monospecific MVs and to optimize bispecific targeting by variation of the linker connecting both DARPins. All bispecific binding cassettes were demonstrated to bind to cells expressing HER2/neu or EpCAM, albeit to different extent. Linkers composed of nine proline residues or constituting two α-helical turns were identified to convey best fusion helper function in vitro. Viruses generated with these linkers as well as the prototype virus MV-Ec4-G3 (ref. 36) specifically infected cells via EpCAM or HER2. The viruses were found to be cytotoxic for two human carcinoma cell lines, while only one of these replicated the virus efficiently. Their oncolytic potential was shown in a disseminated ovarian cancer xenograft model in mice. Furthermore, the superior efficacy of bispecific over monospecific viruses was demonstrated in a cell culture model mimicking tumor heterogeneity, thus providing evidence for the versatility and efficacy of such viruses for the therapy of advanced tumors.
Results
Generation and analysis of bispecific targeting cassettes with optimized linkers
With the final aim to further enhance the oncolytic efficacy of MV by targeting two receptors of choice simultaneously, new bispecific binding cassettes were constructed by exchanging the linker peptide connecting both DARPin domains (Figure 1a). The prototype virus encoded a glycine-serine linker ((G4S)2) between DARPins Ec4 (binding EpCAM with a KD of 1.7 nmol/l) and G3 (binding HER2 with a KD of 0.09 nmol/l), genetically fused in tandem to the carboxy-terminus of the mutated H protein.36 The same orientation of DARPins was used for testing different linkers, since for initial experiments using H-DARPin-linker-DARPin proteins with the prototypic ((G4S)2-linker, the order of the DARPins did not matter with respect to fusion helper-activity (data not shown). Three different types of linkers were used: highly flexible glycine-serine linkers, more rigid poly-proline linkers, and α-helical peptide linkers, which however can unfold. The linkers were varied in length covering between 5 (G4S) and 20 amino acids (aa) ((G4S)4), 9 aa (two full helical turns; αHelix2) and 17 aa (4 turns; αHelix4), or 9 and 18 aa of consecutive prolines. Furthermore, simple glycine-serine hinges (hi) were added to both ends of the rigid linkers to increase flexibility (Figure 1a).
Figure 1.

Construction and function of different bispecific receptor-attachment proteins. (a) Schematic representation of the engineered H gene cassette. Restriction sites used for cloning are indicated. (b) Protein expression was analyzed in lysates of 293T cells transfected with plasmids encoding the indicated constructs 48 hours post-transfection with an antiserum (α-Hcyt) detecting the cytoplasmic tail of H, i.e. potentially also degradation products of H having (partially) lost the DARPin binding domain. β-actin served as loading control. (c) Fusion helper activity of all recombinant bispecific H variants was assayed by cotransfection of the respective pCG-H variants and pCG-F into single receptor-positive CHO-HER2-K6 (black) or CHO-EpCAM #6 (gray) cells and counting of nuclei from 25 individual syncytia. Relative fusion helper activities normalized by the amount of nuclei triggered by respective monospecific H proteins are depicted. Target receptor-independent background fusion activity of each monospecific construct is indicated by a black (Ec4) or gray (G3) horizontal line. Dashed black line indicates 100% fusion-helper activity of the respective monospecific H-DARPin proteins. (d) Binding of recombinant mono- and bispecific DARPin-Fc proteins (10 μmol/l) to CHO-HER2 and CHO-EpCAM61 cells analyzed by flow cytometry.
The respective genes encoding the different H-DARPin-linker-DARPin constructs were first cloned into pCG-1-derived expression plasmids. After transfection of these plasmids into 293T cells, all recombinant H-proteins were detected by immunoblot analysis (Figure 1b). All proteins revealed the expected shift in molecular weight, compared to unmodified H, of approximately 30 kDa, consistent with the size of two connected DARPin units. No proteins with lower molecular weight became evident suggesting that there was no loss of the distal DARPin due to linker proteolysis. Furthermore, all proteins were successfully transported to the cell surface of transfected 293T cells, as demonstrated by flow cytometry detecting the C-terminal surface-exposed His6-tag (Supplementary Figure S1).
To analyze functionality, i.e., fusion helper function, the H protein variants were transiently coexpressed with the MV fusion protein (F) in CHO-HER2-K6 or CHO-EpCAM #6 cells with defined receptor density.36 After binding its receptor, H initiates fusion of neighboring receptor-positive cells by triggering F to fuse the cell membranes, resulting in syncytia, i.e., multi-nucleated giant cells. Compared to the monospecific H-Ec4 or H-G3, the best fusion activity of the evaluated bispecific formats, indicated by formation of syncytia, was observed with medium sized linkers and regardless of their flexibility: (G4S)2 (10 aa), αHelix2 (9 aa), or Pro9 (9 aa). After binding to HER2, these proteins revealed almost completely conserved fusion helper function of between 80% (αHelix2 & Pro9) and 90% ((G4S)2) as compared to monospecific H-G3 (Figure 1c). After binding to EpCAM, fusion helper function of bispecific constructs ranged from 40% ((G4S)2) to 60% (αHelix2 & Pro9) compared to H-Ec4. It should be noted that the anti-EpCAM DARPin is placed between the H-protein and the anti-HER2 DARPin, and may thus be more sterically constrained than the latter, shown in lower percentages compared to Ec4 itself. Longer linker peptides or additional hinge regions impaired function, as well as shorter linker peptides such as Gly-Ser (X/B; introduced XmaI/BstBI restriction sites) or a Gly4-Ser sequence. Hence, the linker variants αHelix2 and Pro9 were chosen as alternatives to the prototype (G4S)2-linker for the generation of bispecific MVs.
For additional characterization, soluble versions of the bispecific binding domains were generated, which were fused to the N-terminus of human IgG1-Fc-portion. The Fc-part induces homo-dimerization and thus potentially results in bivalent binding of the DARPin-Fc complexes. Recombinant DARPin-Fc proteins were expressed in 293T cells, purified (Supplementary Figure S2), and tested for binding to cell lines differing in HER2/neu or EpCAM surface expression (Table 1) by flow cytometry (Figure 1d, Supplementary Figure S3). Most interestingly, Fc-dimerized (G4S)2- and αHelix2-linked proteins bound either HER2- or EpCAM-positive target cells with comparable efficacy also to monospecific DARPin-Fc-proteins. These comparably constructed proteins demonstrated unimpaired binding capacity of the DARPin units when fused to the Fc-portion. The Pro9-connected soluble bispecific cassette bound both HER2 and EpCAM-expressing cells significantly less efficiently indicating a negative impact of the Pro9 linker in the binding efficiencies of both DARPins, as discussed below.
Table 1. Surface density of targeted receptors as determined previously.
| Cell line | HER2/cella | EpCAM/cella | CD46/cella |
|---|---|---|---|
| SK-OV-3 | 5.72 × 105 | 4.59 × 103 | 9.10 × 104 |
| Caco-2 | 1.81 × 104 | 1.01 × 105 | 1.77 × 105 |
| LN-308 | 4.76 × 103 | -b | 6.30 × 104 |
| MCF-7 | 1.24 × 104 | 9.57 × 104 | 7.82 × 104 |
Data taken from ref. 36.
Number of respective HER2/neu, EpCAM, or CD46 molecules per cell.
Background level as determined by isotype control.
Rescue and characterization of bispecific DARPin-MV with alternative linker peptides
After extension of the carboxy-terminal His6-tag by one extra histidine to coincide with the rule-of-six,41 the targeted H-ORFs were cloned into the genome of attenuated Edmonston MV strain NSe (MVNSe) encoding eGFP as reporter protein (Figure 2a). All respective viruses were successfully rescued. These recombinant MVs were amplified on Vero-αHis cells and subsequently analyzed by immunoblot for H protein expression in infected cells (Figure 2b). Both MV-Ec4-Pro9-G3 and MV-Ec4-αHelix2-G3 showed expression of the recombinant H proteins with an approximately 30 kDa increase in molecular weight, as expected (Figure 2b).
Figure 2.
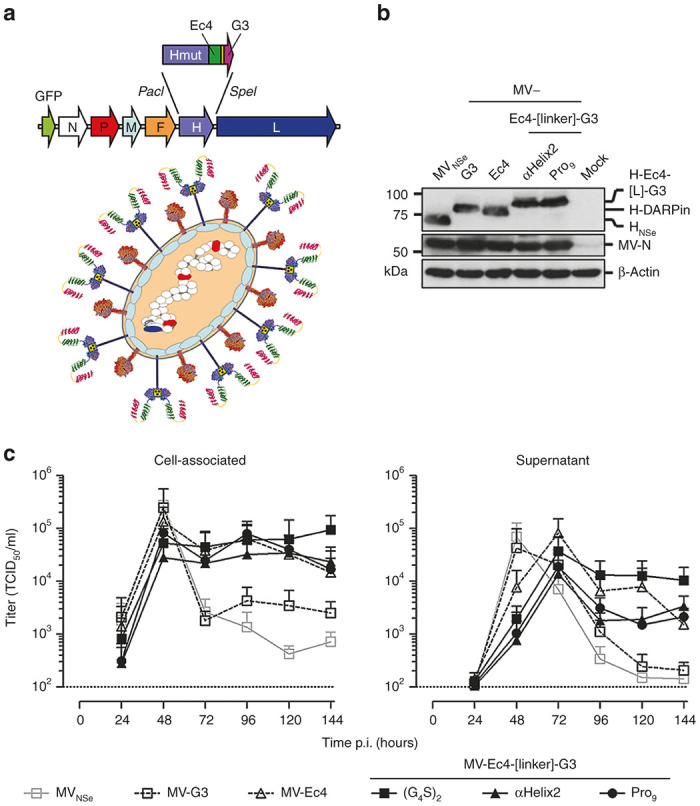
Generation of bispecific viruses. (a) Schematic representation of bispecifically retargeted measles virus (MV) and its genome with cloning sites used for exchange of the H gene cassette. The same color code for proteins and their corresponding genes is used. Scheme73 modified with crystal structures from H, F74, and DARPins.75 (b) Immunoblot for detection of virus proteins. Lysates of infected Vero-αHis cells harvested 48 hours after infection (MOI = 0.1) were analyzed for indicated proteins. Protein content was normalized to MV-N and β-actin. (c) Multi-step growth analysis of parental MVNSe, monospecific-, bispecific prototype- (MV-Ec4-G3) and generated bispecific MV (MV-Ec4-αHelix2-G3 & MV-Ec4-Pro9-G3) on the producer cell line Vero-αHis after infection (MOI = 0.03) by analyzing cell associated (left panel) or released (right panel) virus titers.
In multi-step growth curves on Vero-αHis cells, similar maximal cell-associated titers around 1 × 105 TCID50/ml were reached 48 hours after infection by all MVs (Figure 2c). As expected, the replication kinetics of the bispecific constructs closely reflected those seen for monospecific MV-Ec4: Virus titers did not drop after reaching their maximum, but stayed at a plateau until 6 days post infection (Figure 2c). Titers of all recombinant MV in the supernatant of infected Vero-αHis revealed a similar pattern, but were reduced by half an order of magnitude and delayed by 24 hours (Figure 2d).
The receptor tropism of retargeted bispecific MVs was assessed by infecting a panel of transgenic CHO cell lines expressing either one of the native MV vaccine strain receptors (CD46, hSLAM, or nectin 4), or the targeted receptors (HER2 or EpCAM). As expected, neither CHO-K1 cells (not expressing any putative receptor) nor the CHO lines expressing one of the native MV receptors were infected by any retargeted virus, demonstrating successful detargeting from natural MV receptors. While monospecific MVs infected either CHO-HER2 or CHO-EpCAM cells according to their respective specificity, bispecific MVs were able to infect both (Figure 3). Thus, target specificity was retained irrespective of the peptide linkers connecting both targeting domains of the bispecific MVs.
Figure 3.
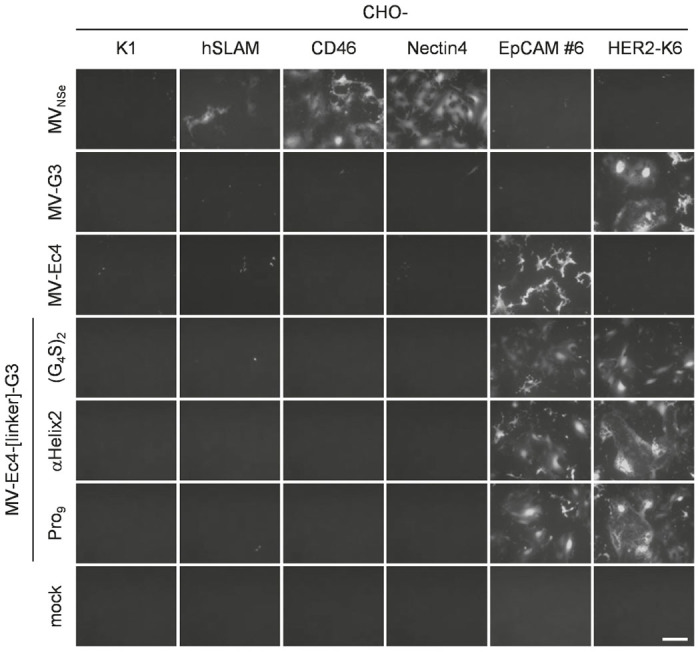
Receptor tropism of bispecific measles viruses (MVs). A panel of receptor-transgenic CHO cell lines (as indicated) including the parental CHO-K1 cells was infected with parental MVNSe, monospecific, or bispecific MVs (MOI = 0.3). Infected cultures were analyzed 48 hours after infection by fluorescence microscopy. Representative pictures are shown. Scale bar, 400 μm.
Cytotoxicity and replication of the recombinant MVs in vitro
Next, the oncolytic properties of the bispecific MVs were characterized using the human carcinoma cell lines SK-OV-3 (ovarian) and Caco-2 (colorectal), expressing different levels of HER2 or EpCAM (Table 1, Figure 4). First, cytotoxicity of the bispecific MVs was assessed by monitoring cell viability in MTT-assays in comparison to nontargeted or monospecific MV. Both tumor cell lines revealed a rapid decline in viability within the first 72 hours after infection with any of the viruses, except Caco-2 infected with MV-G3. This virus exhibited only moderate cytotoxicity in this setting (71.7% viability 72 hours after infection, which declined to 28.7% 168 hours after infection). However, MV-G3 was more cytotoxic for SK-OV-3 cells than MV-Ec4 (27.7 versus 44.7% viability 72 hours after infection), which is likely explained by the 100-fold higher density of HER2 versus EpCAM in these cells. Interestingly, all bispecific viruses were at least as cytotoxic as MV-G3 on either cell line. In comparison to the nontargeted parental virus MVNSe, whose efficiency can be explained by the robust overexpression of its receptor CD46 on both cell lines (Table 1), MV-Ec4-Pro9-G3 even reached comparable cytotoxicity (15.3% versus 9.5% residual viability of infected SK-OV-3 72 after infection) (Figure 4a). In Caco-2 cells, residual viability dropped to almost undetectable levels 168 hours after infection with any bispecific virus (Figure 4b). Thus, bispecific MVs demonstrated high oncolytic efficacy in vitro irrespective of the targeted receptor.
Figure 4.

Cytolytic efficacy of bispecific measles viruses (MV) in vitro. (a,b) The human ovarian adenocarcinoma cell line SK-OV-3 (a) or the human colorectal adenocarcinoma cell line Caco-2 (b) were infected with parental MVNSe, monospecific, or bispecific MVs (MOI = 1) and their viability was determined at the indicated time points by MTT assay. Depicted is metabolic activity indicating viability relative to the mock infected control culture. Mean of three independent experiments each consisting of four replicates. Error bars indicate SD. (c–f) Multi-step growth analysis of recombinant MV on SK-OV-3 (c,e) or Caco-2 (d,f) cells after infection at an MOI of 0.03. Cell-associated (c,d) or released viruses (e,f) were titrated on Vero-αHis cells. Mean of three independent experiments, error bars indicate standard deviation. (g) Impact of DARPins on MV infection. SK-OV-3 cells were incubated with recombinant DARPin-Fc proteins and infected with nontargeted MVNSe at an MOI of 0.03. Cell-associated titers were determined 48 hours after infection. (h) Immunoblot analysis of purified virus particles from supernatants of infected Vero-αHis cells. Proteins separated on 10% SDS-PA-gel were detected by antibodies directed against the indicated viral proteins. H, hemagglutinin; H-D, hemagglutinin fused with designed ankyrin repeat protein(s); MV, measles virus; N, nucleocapsid protein.
Surprisingly, analysis of viral replication on both target cell lines revealed a different picture. On SK-OV-3 cells, cell-associated virus titers peaked 48 hours (MVNSe) or 72 hours after infection (all targeted MVs). MVNSe and monospecific MV-Ec4 reached maximal cell-associated titers of 8.2 ± 2.7 × 103 TCID50/ml and 0.7 ± 1.0 × 104 TCID50/ml, respectively. Ninety-six hours after infection, titers of both viruses stabilized at around 1 × 103 TCID50/ml. Peak titers of MV-G3 were reduced (2.7 ± 2.1 × 103 TCID50/ml) and dropped below the limit of detection (1 × 102 TCID50/ml) 96 hours after infection. Titers of the bispecific viruses were only slightly above the detection limit on SKOV-3 cells (Figure 4c), but within the range of the monospecific and parental viruses on Caco-2 cells (Figure 4d). The maximal cell-associated titers were between 3.0 ± 4.8 × 104 TCID50/ml (MV-Ec4) and 5.5 ± 8.2 × 103 TCID50/ml (MV-G3). MVNSe and MV-Ec4 stabilized at about 10-fold lower levels during later time points, whereas MV-G3 and the bispecific MVs stayed at peak levels until 120 hours after infection (Figure 4d).
Interestingly, the time point for maximal titers varied between 72 and 120 hours after infection. Thus, while the cytotoxicity of bispecific MVs was comparable with or even exceeded that of nontargeted or monospecific viruses, maximal titers were significantly reduced in SK-OV-3, but not in Caco-2 or Vero-αHis cells. Titers of virus released into the supernatant revealed a similar pattern as those of cell-associated virus, but were roughly one order of magnitude lower, as expected for MV (Figure 4e,f).
To exclude any effect on virus replication caused by sole bispecific binding to HER2 and EpCAM, we monitored virus titers after infection with nontargeted MVNse in the presence of bispecific DARPins potentially triggering inhibitory signaling via the targeted receptors. For this purpose, DARPins were added to SK-OV-3 cells during infection with nontargeted MVNSe. No differences were found in peak virus titers 72 hours after infection irrespective of the binding proteins used for pretreatment of SK-OV-3 cells (Figure 4g).
Compared to the other cancer cell lines, SK-OV-3 cells express especially HER2/neu in very high amounts, and also EpCAM is detectable on this cell line.36 This high receptor expression may block virus replication, if receptor abundance and high-affinity interaction with recombinant H proteins would interfere, e.g., with protein processing in the endoplasmic reticulum. Although expression of recombinant H-DARPin-linker-DARPin proteins was comparable to that of monospecific H-DARPin (Figure 2b) and MV encoding the latter replicated well, we analyzed the abundance of recombinant H proteins on MV particles to reveal, if differential incorporation into virus particles during assembly may render bispecific MV vulnerable to slowed down replication in tumor cells with extraordinary receptor density. Indeed, when analyzing recombinant MVs purified from the supernatant of infected Vero-αHis cells, we detected a significant reduction of H-Ec4-linker-G3 proteins in virus particles as compared to unmodified H, or monospecific H-G3 and H-Ec4 (Figure 4h).
Taken together, bispecific MV variants with DARPin-based targeting domains containing different linkers were rescued and demonstrated proper replication in two out of three analyzed cell lines. The infection by these viruses was highly toxic for both cancer cell lines tested, mandating the evaluation of this virus panel in an advanced in vivo setting.
Efficacy of bispecific MVs against disseminated orthotopic ovarian cancer
Oncolytic efficacy of the targeted MVs was tested in a model of disseminated orthotopic ovarian carcinoma.42 For this purpose, luciferase-expressing SK-OV-3 cells (SK-OV-3-luc) were intraperitoneally (i.p.) injected into immuno-deficient nude mice. Since luciferase signals in the peritoneum were readily detectable 2 dpi by in vivo imaging, the virotherapy treatment was started at this point. Subsequently, luciferase expression was monitored over time to determine tumor growth (Figure 5a).
Figure 5.

Therapeutic efficacy of bispecific measles viruses (MV) in disseminated orthotopic ovarian cancer. (a) Schematic depiction of the treatment schedule in a human xenograft tumor model in athymic mice implanted i.p. with SK-OV-3-luc cells. Two days thereafter, mice were injected i.p. four times as indicated with viruses (2 × 106 TCID50/injection) or control. n = 7–8. To monitor tumor burden, mice were imaged for luciferase activity starting on day 2, twice a week from day 9 to 27, then once a week until day 34 and finally once every 2 weeks, thereafter. (b) Representative IVIS images of mock-treated or bispecific MV-treated mice at the start of the treatment cycle (d2) and at the point of minimal residual disease (d34). (c) Mean luciferase activities over the course of the experiment in the differently treated groups (n = 7–8). Error bars, SD. (d) Tumor progression as defined by doubling of the initial luciferase signal, or when animals had to be sacrificed due to critical tumor location. Logrank test, **P < 0.01 (mock versus (G4S)2-treated).
Mock-treated control animals revealed a steady progression of the tumor burden. In a state of minimal residual disease on day 34, shown for representative mice treated with bispecific MV in comparison to untreated mock mice (Figure 5b), the tumor burden of all virus-treated animals was reduced by 76% (MV-Ec4-Pro9-G3) to 95% (MV-Ec4) as compared to the control group (Figure 5b,c). Interestingly, at least two MV, monospecific EpCAM-targeted MV-Ec4 and bispecific MV-Ec4-(G4S)2-G3, revealed a tendency for a higher oncolytic efficacy in vivo than MVNSe. This is remarkable since unmodified MVNSe is de facto also “targeted” to the tumor cells, which are the only cells expressing human CD46 in this in vivo model, since athymic nude mice are not susceptible to MV and do not express human CD46. MVs’ therapeutic efficacy became even more apparent when analyzing the time to progression. Tumor growth progressed rapidly in mock-treated animals while it was significantly delayed in all MV-treated animals (Figure 5d).
However, most tumors relapsed and the mice had to be sacrificed. The sacrificed control animals had disseminated disease throughout the peritoneal cavity and showed signs of ascites. In contrast, the virus-treated animals did not develop ascites and there were fewer but larger tumors detectable (Supplementary Figure S4, arrows). Moreover, no alterations in the expression levels of the targeted receptors were detectable in dissociated tumor cells derived from representative explanted tumors (data not shown). Additionally, the cells could readily be reinfected with the respective viruses used for treatment (data not shown) indicating no treatment resistance in postentry steps. Thus, all recombinant MVs exhibited a significant oncolytic efficacy in a disseminated tumor model.
Bispecific MVs are superior in targeting heterogeneous tumor cell populations
Having provided proof of principle for the oncolytic efficacy of bispecific MVs in vitro and in vivo, we aimed next at analyzing their potential for treating heterogeneous tumor cell populations. Two tumor cell lines (LN-308 and MCF-7) with distinct HER2 and EpCAM36 expression patterns (Table 1, Figure 6), were cocultivated and infected with the mono- or bispecific MVs. For better discrimination between the cell lines, LN-308 cells were genetically labeled with TurboFP635. Infection of these red-fluorescent LN-308red cells with GFP-expressing MVs resulted in yellow syncytia (due to mixed red and green fluorescence), in contrast to unmodified MCF-7 giving rise to green syncytia after infection (Supplementary Figure S5). Of note, the shape of the MV-induced syncytia is cell-specific, too. Infection of a mixed culture of LN-308red and MCF-7 with the various viruses induced syncytia of different shapes and shades of yellow (red and green fluorescence), as both cell populations are fused (Figure 6a). The only exception was MV-Ec4 which induced only green syncytia (Figure 6a) matching the shape of syncytia formed by MCF-7 cells in the infected mono-culture (Supplementary Figure S5). This is in accordance with the inability of MV-Ec4 to infect isolated LN-308red cells (Supplementary Figure S5).
Figure 6.

Cytolytic efficacy of bispecific measles viruses (MVs) for heterogeneous tumor populations in vitro. (a) LN-308red and MCF-7 were cocultivated at approximately 1:1 ratio and infected by parental MVNSe, monospecific, or bispecific MVs at an MOI of 0.1, respectively. Representative pictures after fluorescence microscopy 40 hours after infection are depicted. Scale bar, 400 μm. (b–d) LN-308 (b), MCF-7 (c) or both cell lines in coculture (d) were infected with parental MVNSe, monospecific, or bispecific MVs (MOI = 1) and their viability was determined at indicated time points by MTT assay. Depicted is metabolic activity indicating viability relative to the mock infected control culture. Mean of three independent experiments each consisting of four replicates. Error bars, SD. n-way analysis of variance with factors virus group (mono versus bispecific), virus (nested in virus group), and time (hours postinfection); ***P < 0.001.
Cytotoxicity of infection was subsequently determined with MTT assays on infected, isolated (Figure 6b,c), or mixed cultures (Figure 6d) of unlabeled LN-308 and MCF-7 cells. These assays demonstrated that both monospecific MVs were cytotoxic for the respective cell line (Figure 6b,c), which expressed the targeted receptor at high levels, but not vice versa (Table 1). In either cell line, the most efficient bispecific MVs were at least as effective in cell killing as the respective monospecific MV targeting the appropriate receptor (Figure 6b,c). On MCF-7, the bispecific viruses were even superior to MV-Ec4 in tumor cell killing (Figure 6c). Interestingly, linker variation again enhanced the oncolytic efficacy of the bispecific candidates. On LN-308, MV-Ec4-αHelix2-G3 and MV-Ec4-Pro9-G3 were more cytotoxic than the prototypic MV-Ec4-G3 virus (Figure 6b). Seventy-two hours after infection, the cytotoxicity of MV-Ec4-Pro9-G3 was enhanced, whereas that of MV-Ec4-αHelix2-G3 was comparable to that of MV-Ec4-G3 on MCF-7 cells (Figure 6c).
The full potential of the bispecific viruses became evident when infecting the coculture of both cell lines. All bispecific viruses revealed superior oncolytic efficacy (MV-Ec4-G3: 58%, MV-Ec4-αHelix2-G3: 34%, MV-Ec4-Pro9-G3: 30% residual viability 72 hours after infection) compared to either monospecific virus (MV-G3: 78%, MV-Ec4: 76% residual viability 72 hours after infection) (Figure 6d). Moreover, the optimized constructs MV-Ec4-αHelix2-G3 and MV-Ec4-Pro9-G3 exhibited an almost twofold enhanced cytotoxic effect compared to the prototype virus MV-Ec4-G3 at this time point. Noteworthy, the viability was further declining in infected cultures at the end of the observation period, indicating the potential of the bispecific MVs for eradication of the mixed culture over prolonged infection periods.
Discussion
The general suitability of DARPins as targeting domains for oncolytic MVs and a prototypic MV targeted against two different antigens have been presented in our previous studies.36 Here, our data reveal the advantages of optimized oncolytic viruses, which are targeted to two different tumor markers, simultaneously. Thereby, enhancement of the bispecific targeted viruses’ fusion activity was one of the aims of the presented studies. For this purpose, the linker peptide connecting both targeting entities was varied to study its impact on glycoprotein-induced fusion, a major determinant of recombinant MVs’ cytotoxicity.43 Fusion helper function of bispecific MV-H was best, when medium-sized linkers were used, irrespective of their flexibility. Bispecific oncolytic MVs replicated to identical titers as the monospecific viruses or parental MVNSe in two out of three tested cell lines, and revealed the expected conserved specificity. All viruses showed oncolytic activity comparable to monospecific MVs and parental MVNSe in in vitro cultures and in vivo tumor models. Thereby, no hint for attenuation by the presence of two linked DARPins in series on the virus coat was found. Finally, the advantage of bispecific over monospecific viruses became evident in a mixed tumor cell model, representative for tumor heterogeneity found in vivo.
The connection of two DARPins turned out as an important parameter for their function as targeting domain for tumor-targeted oncolytic viruses. When used for oncolytic MVs, the linker determines the spatial orientation and flexibility of the two DARPins with respect to each other. In our study, accessibility of the individual DARPins for receptor binding most likely depends on the linker that may or may not cause steric hindrance on the individual DARPin domains. For example, optimal efficacy for delivering siRNA coupled to EpCAM-specific DARPins was reached if they were arranged such that they could simultaneously bind to two EpCAM epitopes at the tumor cell surface.44 In our study, linkers of intermediate length revealed best biological function in vitro regardless of flexibility. Thus, the distance of both binding domains seems to be a critical parameter for this application.
As DARPins have a highly stable fold,45 distance effects likely influence their spatial orientation, or the transmission of the molecular force exerted by ligand binding to the globular head of H. If the linker is too short, the DARPins might mutually block binding to their designated targets. If the linker is too long, the distance between cellular and virus particle membranes might be too long for efficient induction of fusion. This distance has been demonstrated to be a critical determinant for MV glycoprotein-mediated fusion.46 Thus, the intermediate linker size most likely reflects the best suited compromise. At least receptor binding was found to be conserved when analyzing the binding of Fc-tagged recombinant binding domains to the respective target cells, even though the Fc part was fused at the C-terminus of the DARPins and is thus not directly comparable to the fusion of H at the N-terminus of the DARPins.
The only exception found was the cassette with the Pro9-linker, but one could speculate about interference of the stiff linker with protein folding when expressed as Fc fusion. This recombinant protein was expressed at by far lowest levels as N-terminal fusion proteins to the Fc-tag after transfection of 293T cells. Thus, degradation seems to be induced under these conditions, which can be most easily explained by co- or post-translationally misfolded protein. In contrast, expression of the targeting cassette with the Pro9-linker when C-terminally fused to H seemed not to be impaired. Therefore, it is consistent with this observation that all viruses had high oncolytic efficacy.
For cell fusion and the fusion helper function of H, linker flexibility seems to play just a secondary role for. Only the extent of receptor usage changed from a HER2-dominated targeting for (G4S)x-linkers to a more balanced pattern for more rigid linkers such as αHelix2 and Pro9, while hinge-encompassing linkers did worse. Fusion is triggered by H, whose globular head binds to a receptor, leading to subsequent conformational changes that are transmitting the activation signal to the fusion protein.47 Since the EpCAM-binding DARPin is directly connected to H in our constructs, transmission of molecular forces caused by the DARPin-mediated binding of the globular head of H to EpCAM should not be modulated by the linker peptide sequence. Our data indicate an enhanced EpCAM-dependent fusion activity when more rigid linkers are used, likely due to an enhanced spatial separation of both DARPin units. In contrast, binding of the outer DARPin unit is transmitted to the H head irrespective of the linker flexibility.
When these more rigid linkers of intermediate length were introduced into recombinant MVs, the resulting viruses revealed a conserved replication pattern in Vero-αHis cells as compared to the EpCAM-targeted MV, while their receptor tropism was solely determined by the binding specificity of the displayed DARPins. Thereby, the phenotype of the bispecific targeting units, as expected, reflects the behavior of each of the monospecifically targeted MVs using monospecific targeting domains,33–36 or the prototypic bispecific MV.36 The oncolytic potency of the newly generated bispecific MVs correlated well with the fusion helper activity of the respective recombinant H proteins. Such correlation has been described before for MV strains “naturally” differing in their fusion activity43,48 as well as for monospecific targeted oncolytic MVs.36 Fusion-helper activity and cytotoxicity are dependent on receptor density, for both nontargeted30,49 and monospecifically targeted viruses.36,50 Thus, it was not surprising to observe such a correlation for the bispecific MVs as well.
Surprising differences became, however, evident for the replication of bispecific MVs on a certain tumor cell line. In SK-OV-3 cells, cell-associated titers of bispecific MVs were reduced compared to other MVs. SK-OV-3 cells express both receptors in high density at the cell surface36 and are killed by the bispecific viruses more efficiently than by either monospecific virus. Impaired productive replication of bispecific MVs seems to be a specific feature of SK-OV-3 cells, since the pattern of replication of bispecific viruses in Caco-2 and Vero-αHis cells followed that of monospecific or nontargeted viruses. Enhanced cytotoxicity as potential cause of impaired replication in SK-OV-3 might curb virus titers due to premature cell killing, as observed before,43 but nontargeted MVNSe was at least as cytotoxic, and replicated better. In addition, toxicity of bispecific MVs for Caco-2 cells was found to be enhanced compared to solely HER2-targeted MV, but replication was at least as good. Thus, cytotoxicity seems not to be the differentiating factor, here.
A self-contained effect of the bispecific DARPin units on cell physiology through targeted receptor signaling, like recently described for other bispecific HER2-binding DARPins,51 could be excluded by incubation of SK-OV-3 cells with bispecific DARPin-linker-DARPin-Fc proteins and subsequent infection with MVNSe. Cell-specific impairment of virus assembly in SK-OV-3 cells and reduced virus release due to the high receptor expression levels in SK-OV-3 cells may be alternative explanations for reduced infectivity, as some cell-specific features of MV replication affecting titers have been previously described also for clinical grade vector production.52
The only difference between bi- and monospecific MV was the reduced incorporation of H-Ec4-linker-G3 proteins into recombinant MV particles. Obviously, the amount of recombinant H on the respective virus particles was sufficient for replication on two other cell lines and for significant cytotoxicity in all five cell lines tested. Nevertheless, high-affinity interaction between the displayed DARPin domain and the HER2 receptor in the endoplasmic reticulum of the virus-infected cell may interfere with virus spread especially on the SKOV cells, which have a very high density of the HER2 receptor. However, elucidating this issue may be a matter of future experiments.
In the model of peritoneal carcinomatosis, all viruses, including MVNSe, controlled tumor burden to a similar extent. If at all, monospecific MV-Ec4 and bispecific MV-Ec4-(G4S)2-G3 performed best, even doing slightly better than nontargeted virus. Interestingly, in vitro they had been least cytotoxic. In a comparable experiment, treatment with nontargeted recombinant MV (closely reflecting MVNSe used in this study) resulted in similar level of residual tumor burden as evaluated by tumor cells’ luciferase activity in the state of minimal residual disease.53 Remarkably, in our experiment, virus treatment resulted in remission of the tumors below the starting volume, whereas tumors in the parallel study only went into stasis.53 Thus, therapeutic efficacy in our experiments was clearly within the expected range for an oncolytic virus. Interestingly, oncolytic MVs seem to be superior to other experimental treatment modalities previously tested in this tumor model. When HER2-affibodies coupled to a bacterial toxin were administered, the exponential tumor growth rate was attenuated, but no remission of the tumor burden was observed.54
Moreover, our oncolytic MVs changed the tumor phenotype in this in vivo model from a poly-focal to an oligo-focal pattern going along with absence of ascites. The latter had also been previously observed with nontargeted MV.42 Also, in these experiments, only few and small residual tumors were isolated at day 80. Most likely, only a small fraction of the initially injected tumor cells survived virus treatment, which then relapsed to few large tumors. It is tempting to speculate that these remaining tumor nests can be better controlled by using an extended treatment schedule. Indeed, a significantly prolonged schedule (16 injections over 6 weeks of treatment) and 14 times higher total dose of virus (1.6 × 108 pfu) can result in smaller tumor loads on day 80 post-treatment.42 However, with (targeted) therapies being present over a longer period of time, selective pressure for tumor cells escaping the therapy increases, as already documented in vitro for oncolytic MV.36 For trastuzumab targeting HER2, compensatory activation of different pathways, altered intracellular signaling6,55,56 or receptor downregulation57 have been described.
To assess the potential for resistance against our viruses described here, we isolated representative tumors 17–32 weeks after treatment. No signs for resistant tumor cells explaining the relapse could be identified including target receptor expression und MV-susceptibility. However, the postulated advantage of bispecific targeted MVs with respect to resistance development cannot be properly documented in this setting given (i) the absence of virus in isolated tumors that might have induced resistance and (ii) lack of heterogeneity in the orthotopic tumor model used here. Nevertheless, the picture might change, when the selective pressure is increased by using more treatment cycles or heterogenic tumors are established in vivo, thus enforcing resistance development to mono-targeted therapies.
Indeed, in an in vitro model mimicking tumor heterogeneity, all bispecific MVs were superior to monospecific viruses in eradicating mixed tumor cells. Again, higher rigidity of the linker improved the efficacy (MV-Ec4-αHelix2-G3 and MV-Ec4-Pro9-G3). These in vitro data indicate the power of simultaneous targeting of two different receptors, especially when treating heterogeneous cell populations. Combination of two monospecific MV differs from simultaneous targeting by bispecific MV, since the latter have an inherent bispecificity of each single particle. Bispecificity of the targeted agents is thus continuously present in any infected cell, and thereby, a single virus should be able to spread throughout heterogeneous tumor tissue, whereas two simultaneously applied viruses may encounter regions that cannot be crossed by one of the viruses preventing effective spread and therapy.
Moreover, two completely independent oncogenic pathways can simultaneously be targeted using bispecific MVs. Both, HER2 and EpCAM are associated with bad prognosis for the patient.3,16 Combined targeting of cells expressing either one or two markers should therefore extend the potential of MV to kill critical tumor cell populations with different roles in uncontrolled tumor growth such as tumor evolution and metastasis. Whether the high in vivo efficacy of EpCAM-targeted MV-Ec4 can be attributed to the targeting of cancer stem cells, or not, remains to be elucidated. By careful choice of the targeted pathways, development of resistance might potentially also be suppressed.
Taken together, bispecific MVs targeted by two combined DARPins are a novel and unique type of virotherapeutic antitumoral agent. They harbor an intrinsic and robust bispecificity that allows them to replicate in an extended proportion of heterogeneous tumor cell populations. In addition, they are independent of a singular target structure; thereby the chance for easy resistance development should be suppressed. Furthermore, using the highly versatile DARPin technology,37 specificity of the viruses, in principle, can be directed to almost any target of choice, including critical antigens on cancer stem cells and pathways critical for tumor cell survival and metastasis.
Materials and Methods
Cells
293T (CRL-11268), CHO-K1 (CCL-61), SK-OV-3 (HTB-77), Caco-2 (HTB-37), and MCF-7 (HTB-22) cells were purchased from ATCC (Manassas, VA) and grown in media recommended by ATCC at 37 °C in a humidified atmosphere containing 6% CO2 for no longer than 6 months of culture after thawing of the original stock. Vero-αHis,33 CHO-hSLAM,26 CHO-CD46 (ref. 58), CHO-nectin4 (ref. 59), CHO-HER2 (ref. 60), CHO-EpCAM,36,61 and LN-308 (ref. 62) cells and their maintenance have been described. SK-OV-3-luc and LN-308red were generated by transduction of SK-OV-3 (MOI 0.5) or LN-308 (MOI 50) cells, respectively, with an HIV-1 derived lentiviral vector system pseudotyped with VSV-G63 encoding firefly luciferase or the TurboFP635 gene (pS-TFP635-W64). SK-OV-3-luc single cell clones were subsequently analyzed for luciferase and HER2/neu expression. One clone with high luciferase expression and unaltered HER2/neu expression was used for further experiments. LN-308red cells were used as bulk culture.
Plasmids
For cloning bispecific H variants, a modular system was generated, where the linker connecting both DARPins is easily exchangeable via XmaI/BstBI restriction sites. On the basis of the plasmid pCR2.1-Ec4-G3 (ref. 36), the αEpCAM-DARPin coding sequence was elongated by polymerase chain reaction (PCR) (Expand High FidelityPLUS PCR System, Roche Diagnostics, Mannheim, Germany) to introduce a SfiI site to the 5’ end and adjacent XmaI/BstBI sites in 3’. PCR products were directly ligated into pCR2.1-TOPO (TOPO-TA cloning kit, Life Technologies, Darmstadt, Germany) and sequenced (Eurofins MWG Operon, Ebersberg, Germany). The resulting SfiI-Ec4-BstBI fragment was inserted into pCR2.1-Ec4-G3 via SfiI/BstBI (NEB, Frankfurt a.M., Germany) to generate pCR2.1-Ec4-X/B-G3 with a single Gly-Ser encoding linker sequence between the adjacent XmaI/BstBI restriction sites. The different linker peptide-encoding sequences were subsequently ligated into pCR2.1-Ec4-X/B-G3 via XmaI/BstBI (NEB) using preannealed (2 minutes 95°C, then slow cool-down to RT) synthetic oligonucleotides (Eurofins MWG Operon) with sticky ends. The different bispecific DARPin-linker-DARPin cassettes were then inserted into pCG-HmutXαCD20 (ref. 65) via SfiI/NotI (NEB) to yield expression plasmids for bispecifically retargeted H (pCG-Hmut-Ec4-X/B-G3, pCG-Hmut-Ec4-αHelix2-G3, pCG-Hmut-Ec4-αHelix2-hi-G3, pCG-Hmut-Ec4-αHelix4-G3, pCG-Hmut-Ec4-αHelix4-hi-G3, pCG-Hmut-Ec4-(G4S)1-G3, pCG-Hmut-Ec4-(G4S)2-G3, pCG-Hmut-Ec4-(G4S)3-G3, pCG-Hmut-Ec4-(G4S)4-G3, pCG-Hmut-Ec4-Pro9-G3, pCG-Hmut-Ec4-Pro9-hi-G3, pCG-Hmut-Ec4-Pro18-G3, and pCG-Hmut-Ec4-Pro18-hi-G3). The H-DARPin-linker-DARPin-encoding genes were transferred via PacI/SpeI into plasmid p(+)PolII-MVNSe-GFP(N)36 to yield p(+)PolII-MVNSe-GFP(N)-Ec4-(G4S)2-G3, p(+)PolII-MVNSe-GFP(N)-Ec4-αHelix2-G3, and p(+)PolII-MVNSe-GFP(N)-Ec4-Pro9-G3. As the cassettes in pCG-Hmut-Ec4-αHelix2-G3 and pCG-Hmut-Ec4-Pro9-G3 were not conforming to the ‘rule of six’, their terminal His6-tag was elongated before subcloning by an additional His via NotI/SpeI (NEB) using appropriate synthetic oligonucleotides (Eurofins MWG Operon). Detailed sequences of primers, PCR programs and oligonucleotides are available upon request. For the expression of DARPin-Fc proteins, different DARPin cassettes were excised by SfiI/NotI (NEB) and fused in-frame to a human IgG1-Fc-tag in the plasmid phuFc-(SfiI-NotI)66 to yield phuFc-G3, phuFc-Ec4, phuFc-Ec4-G3, phuFc-Ec4-αHelix2-G3, and phuFc-Ec4-Pro9-G3.
DARPin-Fc expression and purification
DARPin-Fc proteins were expressed and purified as described before.66 In short, phuFc-derived plasmids were transfected into 293T cells and subsequently medium was replaced with serum-free PANSERIN 293A (Pan-Biotech, Aidenbach, Germany). 48 and 72 hours post-transfection, medium was cleaned using a 0.22-µm filter and pooled. Fc-tagged proteins were purified by a Sartobind Protein A column (Sartorius, Göttingen, Germany) on a high performance liquid chromatography (HPLC) system (Bioline, Knauer, Berlin, Germany) according to the manufacturer’s instructions. Purity was analyzed by Coomassie blue staining (GelCode Blue Safe Protein Stain, Thermo Scientific, Dreieich, Germany) after separation using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Protein concentration was analyzed by the absorption at 280 nm (NanoDrop, Thermo Scientific).
Viruses
For rescue of recombinant MV, the previously described PolII rescue system67 was used with modifications. In short, the plasmids encoding recombinant MV genomes were cotransfected with expression plasmids pCA-MV-N, pCA-MV-P, and pCA-MV-L into 293T cells using Lipofectamine 2000 (Life Technologies). Two days after transfection, transfected 293T cells were overlaid onto 50% confluent Vero-αHis cells allowing rescue of retargeted viruses. The viruses were propagated in Vero-αHis cells and titers were determined by 50% tissue culture infective dose (TCID50) titration on Vero-αHis cells.68,69
Virus particle purification
Vero-αHis cells were seeded in 10 cm dishes and infected with respective MV (multiplicity of infection (MOI) = 0.03). The culture supernatants were collected 2 days after infection, clarified and pelleted in an SW28 rotor (25,000 rpm, 2.5 hours) through 20% sucrose onto a 60% sucrose cushion in STE buffer (10 mmol/l Tris (pH 8.0), 100 mmol/l NaCl, and 1 mmol/l ethylene diamine tetraacetic acid (EDTA) (pH 8.0)). Purified viral particles were pelleted in an SW41 rotor (35,000 rpm, 1.5 hours) through 20% sucrose, resuspended in RIPA lysis buffer, and subjected to immunoblot analysis.
Transfection of mammalian cells
For expression analysis, the different pCG-H expression plasmids were transfected into 293T cells using lipofectamine-2000 (Life Technologies). 8 × 105 293T cells were seeded in six-well plates (Nunc Delta Surface; Thermo Scientific) 1 day prior to transfection. For transfection, 5 μg DNA were mixed with 12.5 μl lipofectamine-2000 in OptiMEM and transfected according to the manufacturer’s protocol. The transfection mix was changed after 4 hours to culture medium. 48 to 72 hours post-transfection, transfected cells were used for further analysis. Transient transfection to assess fusion helper function was done by seeding 3.5 × 105 CHO-HER2, CHO-EpCAM, or CHO-K1 cells plated in 12-well plates (Nunc Delta Surface) 1 day prior to transfection. 1 μg of pCG-F70 and 1 μg of a plasmid encoding one H variant were mixed by vortexing with 3 μl FuGENE HD Transfection Reagent (Roche Diagnostics) in OptiMEM and incubated for 15 minutes at room temperature. Transfection mix was added drop-wise to cells without exchanging the medium afterwards.
Infection of mammalian cells
Cells were seeded in an appropriate plate (Nunc Delta Surface) and infected 1 to 4 hours after seeding when reaching 50–70% confluence by a defined MOI via simple addition of virus suspension. Infected cells were cultivated and analyzed 48–72 hours after infection at the peak of viral infection. For analysis of receptor specificity, 3 × 105 of respective receptor-transgenic CHO cells were seeded in six-well plates and infected at an MOI of 0.3. To assess tumor heterogeneity, 1 × 105 LN-308red and 2 × 105 MCF-7 were seeded in 12-well plates or 2 × 105 of each cell line separately, and infected at an MOI of 0.1.
Immunoblotting
Forty-eight hours post-transfection or infection with viruses at an MOI of 0.1, cells were lysed in radio immunoprecipitation assay (RIPA) lysis buffer (50 mmol/l Tris, 150 nmol/l NaCl, 1% NP40 (w/v), 0.5% sodiumdesoxycholate (w/v), 0.1% SDS (w/v), pH 8.0 + Protease Inhibitor Cocktail Complete (Roche Diagnostics)). Lysates were denatured in an equal volume of urea buffer (200 mmol/l Tris-HCl (pH 6.8), 8 mol/l urea, 5% SDS (w/v), 0.1 mmol/l EDTA, 0.03% bromophenol blue (w/v), 1.5% DTT (w/v)) for 10 minutes at 95 °C and separated by SDS-PAGE (Bio-Rad, München, Germany). Proteins were transferred to a polyvinylidene difluoride membrane (Hybond-P; GE Healthcare, München, Germany) by the semi-dry method (TransBlot SD; Bio-Rad) and blocked by 5% milk powder (Roth, Karlsruhe, Germany) in TBS-T (50 mmol/l Tris-HCl (pH 7.4), 150 mmol/l NaCl, H2O, 0.1% Tween 20 (v/v)) for a least 1 hour at room temperature. Proteins were detected using as primary antibodies αMV-H (1:20,000),71 αMV-N (clone ab23974; 1:25,000; Abcam, Cambridge, UK) or α-β-actin (clone AC-15; 1:5,000; Abcam) over-night at 4 °C, and donkey-α-rabbit-HRP sera (1:10,000; Rockland, Limerick, PA) or rabbit-α-mouse-HRP sera (1:10,000; Life Technologies) as secondary Abs for at least 45 minutes at room temperature. Finally, membranes were analyzed using ECL+ (GE Healthcare) detection reagent.
Flow cytometry analysis
Expression of H-DARPin-linker-DARPin proteins was analyzed using directly fluorescently-labeled αHis6-PE (clone GG11-8F3.5.1; 1:11; Miltenyi Biotec, Bergisch Gladbach, Germany). Surface expression of HER2/neu, EpCAM, or CD46 was detected using fluorescence-labeled mAbs αHER2-PE (clone Neu 24.7; 1:80; BD Bioscience, Heidelberg, Germany), αEpCAM-PE (clone HEA-125; 1:100; Miltenyi Biotec), or αCD46-AlexaFluor700 (clone MEM-258; 1:100; EXBIO, Vestec, Czech Republic), respectively. Viability of cells was analyzed using viability dye (Fixable Viability Dye eFluor 450; 1:1,000; eBioscience, Frankfurt a.M., Germany). Tumor cells were stained either directly after cell isolation or after isolated cells formed a confluent layer in cell culture. To analyze DARPin-Fc binding, 2 × 105 cells were incubated with the respective concentration of DARPin-Fc for 1 hour and stained with α-human-IgG1-Fcγ-AlexaFluor647 (Jackson ImmunoResearch, Suffolk, UK; 1:100) antibody for 45 minutes. For determining the ratio for an equal number of LN-308 and MCF-7 in coculture, LN-308 were labeled by CFSE, mixed with unstained MCF-7, and analyzed by flow cytometry. For CFSE labeling, cells were incubated with CFSE for 10 minutes at 37 °C in PBS containing 10% FCS. The reaction was then stopped with five volumes of cold PBS on ice for 5 minutes and cells were washed once with culture medium. Flow cytometry analyses were made using a LSRII flow cytometer (BD Bioscience).
Fusion assay
Four hours after transfection of respective cell lines with expression plasmids encoding the H variants to be analyzed together with F, 200 μmol/l fusion inhibitor peptide (FIP72; BACHEM, Bubendorf, Switzerland) was added and cells were incubated for 24 hours. Fusion was started by removal of FIP and cultures were fixed with 10% paraformaldehyde in PBS after formation of syncytia. Fixed cultures were subjected to microscopic analysis and the number of nuclei per syncytium was determined for 25 independent syncytia for each retargeted H variant.
Virus growth kinetics
2 × 105 SK-OV-3, Vero-αHis, or 3 × 105 Caco-2 cells were seeded in 12-well plates (Nunc Delta Surface). Cells were infected at an MOI of 0.03 in a total of 1 ml medium. At the indicated time points, supernatants were clarified by centrifugation and stored in aliquots at −80 °C. Infected cells were scraped into 1 ml OptiMEM and subjected to a freeze-thaw cycle. After thawing, supernatants containing released particles were also clarified by centrifugation and stored in aliquots at −80 °C. Cell-associated virus titers and titers of virus in supernatants were determined by TCID50 titration. To analyze the effect of DARPins on MV replication, 10 µmol/l of the respective DARPin-Fc was added to the SK-OV-3 cells twice: once during the infection and 24 hours after infection. Viral titers were analyzed 48 hours after infection.
Cytotoxicity assay
1 × 104 cells were seeded in in flat-bottom 96-well plates (Nunc Delta Surface) and infected with recombinant MV (MOI = 1), or left uninfected (mock), for each sample and time-point in quadruplicates. Viability of the cells after infection was determined using MTT assay (Cell Proliferation Kit I; Roche Diagnostics) according to manufacturer´s instructions. 24, 48, 72, 96, or 168 hours after infection, cells were incubated with MTT solution for 4 hours. Then, solubilization solution was added. Following overnight incubation, formation of formazan dye was quantified by 570 nm using an ELISA reader (Multiskan RC, Thermo Labsystems).
Animal experiments
Experimental mouse work was carried out in compliance with the regulations of the German animal protection law. To evaluate the oncolytic efficacy against orthotopic tumors, 5 × 106 SK-OV-3-luc cells were intraperitoneally (i.p.) injected into 6- to 12-week-old female Hsd:Athymic Nude-Foxn1nu mice (Harlan, Rossdorf, Germany). The course of tumor development was monitored by in vivo imaging (IVIS Spectrum; Perkin Elmer, Rodgau, Germany) at indicated time points by detecting bioluminescence 8 minutes after i.p. injection of 150 μg D-Luciferin (Perkin Elmer) per gm body weight. Two days after injection of tumor cells, mice were assigned into groups according to luciferase signal intensities ensuring an even tumor burden in each group. Animals were euthanized when the mice lost more than 20% of body weight or showed symptoms of tumor related illness like ascites or cachexia. Representative tumors were prepared from sacrificed mice and tumor cells were isolated using GentleMACS (Miltenyi Biotec). In short, the tumor mass was cut into small pieces, 120 U/ml Collagenase I (Sigma) were added, and the tumor suspension was transferred to a “C-tube” (Miltenyi Biotec). After processing by the GentleMACS automat employing the program “m_impTumor_02”, the suspensions were incubated for 1 hour at 37 °C and 180 rpm. Subsequently, a further GentleMACS processing step with “m_impTumor_03” followed. The cell suspension was afterwards filtered using a 70 μm cell strainer (ThermoFisher Scientific). Cells were collected by centrifugation (300 g, 5 minutes, 4 °C), resuspended in culture medium, and cultivated for 3–7 days to 90–100% confluence before analysis.
Acknowledgments
The authors thank U. Schneider for providing the PolII Rescue System for measles viruses, Y. Yanagi for providing CHO-hSLAM, R. Cattaneo for CHO-CD46, M. Lopez for CHO-nectin4, A. Muth for CHO-EpCAM, S. Russell for Vero-αHis, U. Naumann for LN-308, J. Hartmann for modification of phuFc-(SfiI-NotI) with the SfiI site, and J. H. Schnotz for technical assistance. This work has been supported by grants from the German Cancer Aid (109614) to M.D.M. and the Deutsche Forschungsgemeinschaft to C.J.B. (BU 1301/3-1).
The authors declare no conflict of interest.
This work has been done in Langen, Hesse, Germany.
Footnotes
This work has been done in Langen, Hesse, Germany.
References
- Ferlay, J, Soerjomataram, I, Ervik, M, Dikshit, R, Eser, S, Mathers, C et al. (2013). GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. GLOBOCAN. http://globocan.iarc.fr. Accessed 27 October 2014. [Google Scholar]
- Goldenberg, MM (1999). Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin Ther 21: 309–318. [DOI] [PubMed] [Google Scholar]
- Eroles, P, Bosch, A, Pérez-Fidalgo, JA and Lluch, A (2012). Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev 38: 698–707. [DOI] [PubMed] [Google Scholar]
- Slamon, DJ, Clark, GM, Wong, SG, Levin, WJ, Ullrich, A and McGuire, WL (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182. [DOI] [PubMed] [Google Scholar]
- Meden, H and Kuhn, W (1997). Overexpression of the oncogene c-erbB-2 (HER2/neu) in ovarian cancer: a new prognostic factor. Eur J Obstet Gynecol Reprod Biol 71: 173–179. [DOI] [PubMed] [Google Scholar]
- Vu, T, Sliwkowski, MX and Claret, FX (2014). Personalized drug combinations to overcome trastuzumab resistance in HER2-positive breast cancer. Biochim Biophys Acta 1846: 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, TA, Carden, CP and Kaye, SB (2009). Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 9: 167–181. [DOI] [PubMed] [Google Scholar]
- Nahta, R, Yu, D, Hung, MC, Hortobagyi, GN and Esteva, FJ (2006). Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 3: 269–280. [DOI] [PubMed] [Google Scholar]
- Jordan, CT, Guzman, ML and Noble, M (2006). Cancer stem cells. N Engl J Med 355: 1253–1261. [DOI] [PubMed] [Google Scholar]
- Meacham, CE and Morrison, SJ (2013). Tumour heterogeneity and cancer cell plasticity. Nature 501: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz, M, Baeuerle, PA and Gires, O (2009). The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 69: 5627–5629. [DOI] [PubMed] [Google Scholar]
- Visvader, JE and Lindeman, GJ (2008). Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8: 755–768. [DOI] [PubMed] [Google Scholar]
- Gires, O, Klein, CA and Baeuerle, PA (2009). On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer 9: 143; author reply 143. [DOI] [PubMed] [Google Scholar]
- Gastl, G, Spizzo, G, Obrist, P, Dünser, M and Mikuz, G (2000). Ep-CAM overexpression in breast cancer as a predictor of survival. Lancet 356: 1981–1982. [DOI] [PubMed] [Google Scholar]
- Spizzo, G, Obrist, P, Ensinger, C, Theurl, I, Dünser, M, Ramoni, A et al. (2002). Prognostic significance of Ep-CAM AND Her-2/neu overexpression in invasive breast cancer. Int J Cancer 98: 883–888. [DOI] [PubMed] [Google Scholar]
- Spizzo, G, Went, P, Dirnhofer, S, Obrist, P, Simon, R, Spichtin, H et al. (2004). High Ep-CAM expression is associated with poor prognosis in node-positive breast cancer. Breast Cancer Res Treat 86: 207–213. [DOI] [PubMed] [Google Scholar]
- Spizzo, G, Went, P, Dirnhofer, S, Obrist, P, Moch, H, Baeuerle, PA et al. (2006). Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol Oncol 103: 483–488. [DOI] [PubMed] [Google Scholar]
- Greig, SL (2016). Talimogene Laherparepvec: first global approval. Drugs 76: 147–154. [DOI] [PubMed] [Google Scholar]
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miest, TS and Cattaneo, R (2014). New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol 12: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US National Institutes of Health. Keywords: Measles Virus & Tumor. US NIH. http://www.clinicaltrials.gov. Accessed 10 February 2015. [Google Scholar]
- Russell, SJ, Federspiel, MJ, Peng, KW, Tong, C, Dingli, D, Morice, WG et al. (2014). Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc 89: 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, WJ and Griffin, DE (2006). Global measles elimination. Nat Rev Microbiol 4: 900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörig, RE, Marcil, A, Chopra, A and Richardson, CD (1993). The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75: 295–305. [DOI] [PubMed] [Google Scholar]
- Naniche, D, Varior-Krishnan, G, Cervoni, F, Wild, TF, Rossi, B, Rabourdin-Combe, C et al. (1993). Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol 67: 6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuo, H, Ono, N, Tanaka, K and Yanagi, Y (2000). SLAM (CDw150) is a cellular receptor for measles virus. Nature 406: 893–897. [DOI] [PubMed] [Google Scholar]
- Noyce, RS, Bondre, DG, Ha, MN, Lin, LT, Sisson, G, Tsao, MS et al. (2011). Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7: e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlebach, MD, Mateo, M, Sinn, PL, Prüfer, S, Uhlig, KM, Leonard, VH et al. (2011). Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480: 530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishelson, Z, Donin, N, Zell, S, Schultz, S and Kirschfink, M (2003). Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol 40: 109–123. [DOI] [PubMed] [Google Scholar]
- Anderson, BD, Nakamura, T, Russell, SJ and Peng, KW (2004). High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res 64: 4919–4926. [DOI] [PubMed] [Google Scholar]
- Navaratnarajah, CK, Oezguen, N, Rupp, L, Kay, L, Leonard, VH, Braun, W et al. (2011). The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat Struct Mol Biol 18: 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongpunsawad, S, Oezgun, N, Braun, W and Cattaneo, R (2004). Selectively receptor-blind measles viruses: Identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J Virol 78: 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T, Peng, KW, Harvey, M, Greiner, S, Lorimer, IA, James, CD et al. (2005). Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol 23: 209–214. [DOI] [PubMed] [Google Scholar]
- Schneider, U, Bullough, F, Vongpunsawad, S, Russell, SJ and Cattaneo, R (2000). Recombinant measles viruses efficiently entering cells through targeted receptors. J Virol 74: 9928–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallak, LK, Merchan, JR, Storgard, CM, Loftus, JC and Russell, SJ (2005). Targeted measles virus vector displaying echistatin infects endothelial cells via alpha(v)beta3 and leads to tumor regression. Cancer Res 65: 5292–5300. [DOI] [PubMed] [Google Scholar]
- Friedrich, K, Hanauer, JR, Prüfer, S, Münch, RC, Völker, I, Filippis, C et al. (2013). DARPin-targeting of measles virus: unique bispecificity, effective oncolysis, and enhanced safety. Mol Ther 21: 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plückthun, A (2015). Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol 55: 489–511. [DOI] [PubMed] [Google Scholar]
- Zahnd, C, Wyler, E, Schwenk, JM, Steiner, D, Lawrence, MC, McKern, NM et al. (2007). A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J Mol Biol 369: 1015–1028. [DOI] [PubMed] [Google Scholar]
- Stefan, N, Martin-Killias, P, Wyss-Stoeckle, S, Honegger, A, Zangemeister-Wittke, U and Plückthun, A (2011). DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J Mol Biol 413: 826–843. [DOI] [PubMed] [Google Scholar]
- Steiner, D, Forrer, P and Plückthun, A (2008). Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J Mol Biol 382: 1211–1227. [DOI] [PubMed] [Google Scholar]
- Calain, P and Roux, L (1993). The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol 67: 4822–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, KW, TenEyck, CJ, Galanis, E, Kalli, KR, Hartmann, LC and Russell, SJ (2002). Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res 62: 4656–4662. [PubMed] [Google Scholar]
- Heidmeier, S, Hanauer, JR, Friedrich, K, Prüfer, S, Schneider, IC, Buchholz, CJ et al. (2014). A single amino acid substitution in the measles virus F₂ protein reciprocally modulates membrane fusion activity in pathogenic and oncolytic strains. Virus Res 180: 43–48. [DOI] [PubMed] [Google Scholar]
- Winkler, J, Martin-Killias, P, Plückthun, A and Zangemeister-Wittke, U (2009). EpCAM-targeted delivery of nanocomplexed siRNA to tumor cells with designed ankyrin repeat proteins. Mol Cancer Ther 8: 2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz, HK, Stumpp, MT, Forrer, P, Amstutz, P and Plückthun, A (2003). Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol 332: 489–503. [DOI] [PubMed] [Google Scholar]
- Buchholz, CJ, Schneider, U, Devaux, P, Gerlier, D and Cattaneo, R (1996). Cell entry by measles virus: long hybrid receptors uncouple binding from membrane fusion. J Virol 70: 3716–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley, MA, Chaudhury, S and Plemper, RK (2015). Measles virus glycoprotein complexes preassemble intracellularly and relax during transport to the cell surface in preparation for fusion. J Virol 89: 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, B, Dey, A, Ghorani, E, Kumar, S, Malam, Y, Rai, L et al. (2011). Differential cytopathology and kinetics of measles oncolysis in two primary B-cell malignancies provides mechanistic insights. Mol Ther 19: 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Breton, C, Russell, LO, Russell, SJ and Peng, KW (2014). Faster replication and higher expression levels of viral glycoproteins give the vesicular stomatitis virus/measles virus hybrid VSV-FH a growth advantage over measles virus. J Virol 88: 8332–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, K, Hu, C, Nakamura, T, Marks, JD, Russell, SJ and Peng, KW (2007). Affinity thresholds for membrane fusion triggering by viral glycoproteins. J Virol 81: 13149–13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost, C, Schilling, J, Tamaskovic, R, Schwill, M, Honegger, A and Plückthun, A (2013). Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 21: 1979–1991. [DOI] [PubMed] [Google Scholar]
- Galanis, E, Atherton, PJ, Maurer, MJ, Knutson, KL, Dowdy, SC, Cliby, WA et al. (2015). Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res 75: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, K, Nakamura, T, Harvey, M, Ikeda, Y, Oberg, A, Figini, M et al. (2006). The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res 12(20 Pt 1): 6170–6178. [DOI] [PubMed] [Google Scholar]
- Zielinski, R, Lyakhov, I, Hassan, M, Kuban, M, Shafer-Weaver, K, Gandjbakhche, A et al. (2011). HER2-affitoxin: a potent therapeutic agent for the treatment of HER2-overexpressing tumors. Clin Cancer Res 17: 5071–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubalek, M, Brunner, C, Matthä, K and Marth, C (2010). Resistance to HER2-targeted therapy: mechanisms of trastuzumab resistance and possible strategies to overcome unresponsiveness to treatment. Wien Med Wochenschr 160: 506–512. [DOI] [PubMed] [Google Scholar]
- Valabrega, G, Montemurro, F and Aglietta, M (2007). Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol 18: 977–984. [DOI] [PubMed] [Google Scholar]
- Lesniak, D, Sabri, S, Xu, Y, Graham, K, Bhatnagar, P, Suresh, M et al. (2013). Spontaneous epithelial-mesenchymal transition and resistance to HER-2-targeted therapies in HER-2-positive luminal breast cancer. PLoS One 8: e71987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchester, M, Liszewski, MK, Atkinson, JP and Oldstone, MB (1994). Multiple isoforms of CD46 (membrane cofactor protein) serve as receptors for measles virus. Proc Natl Acad Sci USA 91: 2161–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre-Lafay, S, Garrido-Urbani, S, Reymond, N, Gonçalves, A, Dubreuil, P and Lopez, M (2005). Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J Biol Chem 280: 19543–19550. [DOI] [PubMed] [Google Scholar]
- Münch, RC, Mühlebach, MD, Schaser, T, Kneissl, S, Jost, C, Plückthun, A et al. (2011). DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther 19: 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch, RC, Muth, A, Muik, A, Friedel, T, Schmatz, J, Dreier, B et al. (2015). Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat Commun 6: 6246. [DOI] [PubMed] [Google Scholar]
- Studer, A, de Tribolet, N, Diserens, AC, Gaide, AC, Matthieu, JM, Carrel, S et al. (1985). Characterization of four human malignant glioma cell lines. Acta Neuropathol 66: 208–217. [DOI] [PubMed] [Google Scholar]
- Funke, S, Maisner, A, Mühlebach, MD, Koehl, U, Grez, M, Cattaneo, R et al. (2008). Targeted cell entry of lentiviral vectors. Mol Ther 16: 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach, P, Abel, T, Hoffmann, C, Gal, Z, Braun, G, Voelker, I et al. (2013). Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer Res 73: 865–874. [DOI] [PubMed] [Google Scholar]
- Ungerechts, G, Springfeld, C, Frenzke, ME, Lampe, J, Johnston, PB, Parker, WB et al. (2007). Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res 67: 10939–10947. [DOI] [PubMed] [Google Scholar]
- Friedel, T, Hanisch, LJ, Muth, A, Honegger, A, Abken, H, Plückthun, A et al. (2015). Receptor-targeted lentiviral vectors are exceptionally sensitive toward the biophysical properties of the displayed single-chain Fv. Protein Eng Des Sel 28: 93–106. [DOI] [PubMed] [Google Scholar]
- Martin, A, Staeheli, P and Schneider, U (2006). RNA polymerase II-controlled expression of antigenomic RNA enhances the rescue efficacies of two different members of the Mononegavirales independently of the site of viral genome replication. J Virol 80: 5708–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert, J. (1984). Spearman-Karber Method. Bioassays. 2nd edn. Hunt Publishing, Dubuque, IA: 65–66. [Google Scholar]
- Kärber, G. (1931). Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch Exp Pathol Pharmakol: 480–483.
- Cathomen, T, Buchholz, CJ, Spielhofer, P and Cattaneo, R (1995). Preferential initiation at the second AUG of the measles virus F mRNA: a role for the long untranslated region. Virology 214: 628–632. [DOI] [PubMed] [Google Scholar]
- Cathomen, T, Naim, HY and Cattaneo, R (1998). Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol 72: 1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, CD, Scheid, A and Choppin, PW (1980). Specific inhibition of paramyxovirus and myxovirus replication by oligopeptides with amino acid sequences similar to those at the N-termini of the F1 or HA2 viral polypeptides. Virology 105: 205–222. [DOI] [PubMed] [Google Scholar]
- Gerlier, D, Varior-Krishnan, G and Devaux, P (1995). CD46-mediated measles virus entry: a first key to host-range specificity. Trends Microbiol 3: 338–345. [DOI] [PubMed] [Google Scholar]
- Cattaneo, R (2010). Paramyxovirus entry and targeted vectors for cancer therapy. PLoS Pathog 6: e1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz, T, Wetzel, SK, Firbank, S, Plückthun, A, Grütter, MG and Mittl, PR (2008). Stabilizing ionic interactions in a full-consensus ankyrin repeat protein. J Mol Biol 376: 232–240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.