

Abstract
Novel therapeutic strategies are needed for the treatment of hematologic malignancies; and bispecific antibody-derived molecules, such as dual-affinity re-targeting (DART) proteins, are being developed to redirect T cells to kill target cells expressing tumor or viral antigens. Here we present our findings of specific and systemic human B-cell depletion by a CD19xCD3 DART protein in humanized BLT mice. Administration of the CD19xCD3 DART protein resulted in a dramatic sustained depletion of human CD19+ B cells from the peripheral blood, as well as a dramatic systemic reduction of human CD19+ B-cell levels in all tissues (bone marrow, spleen, liver, lung) analyzed. When human CD8+ T cells were depleted from the mice, no significant B-cell depletion was observed in response to CD19xCD3 DART protein treatment, confirming that human CD8+ T cells are the primary effector cells in this in vivo model. These studies validate the use of BLT humanized mice for the in vivo evaluation and preclinical development of bispecific molecules that redirect human T cells to selectively deplete target cells.
Introduction
Therapies using targeted monoclonal antibodies have proven safe and effective against hematologic malignancies.1 In particular, rituximab, which targets the B-cell marker CD20, has greatly enhanced outcomes in patients with non-Hodgkin’s lymphoma or chronic lymphocytic leukemia. However, not all patients respond to rituximab, and many of those who do eventually experience disease relapse.2–5 Monoclonal antibody therapies directed against other B-cell antigens, such as CD19, CD22, CD30, CD37, CD40, or CD52, are in development at different stages of preclinical/clinical testing.5–11 B-cell–targeted therapies with novel mechanisms of action are still necessary in order to improve cure rates, and innovative therapies could prove cost-effective in the treatment of hematologic malignancies.12
Existing monoclonal antibody therapies rely on the action of complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity,13,14 or utilize a conjugated toxin or radiolabeled isotope.13 Other strategies harness the ability of cytotoxic T lymphocytes (CTLs) to kill target cells, relying on ex vivo manipulation to expand tumor-specific CTLs15 or to express chimeric antigen receptors16; but these approaches are limited by major histocompatibility (MHC) restriction in tumor-specific CTLs, as well as scalability and risks involved.17
Recently, the development of bispecific T-cell-redirecting antibody-derived molecules has made possible treatment strategies that bypass the requirement for MHC matching or ex vivo manipulation and expansion of CTLs. These bispecific molecules bind simultaneously to a receptor on T cells and to a specific antigen on a target cell, thus redirecting T cells to kill the target cells. One example is blinatumomab, a bispecific T cell engager (BiTE) molecule targeting CD19, which demonstrated complete responses in 72% of patients with persistent or relapsed minimal residual disease and a median overall survival of 9 months.18
To build upon this success, newer generations of bispecific molecules have been developed, like the dual-affinity re-targeting (DART) molecules. DART molecules differ from BiTE molecules in two ways: there is no intervening linker sequence between the V regions of DART molecules, and there are two cysteine residues at the C-terminus of each chain which form a disulfide bridge.19,20 In a previous report comparing DART molecules with BiTE molecules, DART molecules seemed to perform better than BiTE molecules with respect to antigen binding, ability to crosslink target/effector cells, induction of T-cell activation markers, EC50 for target cell lysis, and maximal target cell lysis.20,21 A CD123xCD3 DART protein (directed against human CD3 and human CD123) was active against human AML cell line engraftments in NSG/β2m-/- mice reconstituted with human peripheral blood mononuclear cells (PBMCs), and, due to its crossreactivity to both antigens from cynomolgus monkeys, depleted CD123+ cells when administered to the monkeys.22 A CD19xTCR DART protein (directed against human CD19 and human T-cell receptor αβ subunit) was active against human B-cell lymphoma xenografts in NOD/SCID mice reconstituted with human PBMCs.20 A CD19xCD3 DART protein in an extended half-life format was active against B-cell lymphoma xenografts in mice reconstituted with human PBMCs, and, due to its crossreactivity to both antigens from cynomolgus monkeys, depleted CD19+ B cells in peripheral blood and lymph nodes when administered to the monkeys.23 However, there has yet to be a systemic in vivo evaluation of the effect of CD19xCD3 DART molecules on human immune cells generated de novo from hematopoietic stem cells.
Bone marrow–liver–thymus (BLT) humanized mice could serve as an excellent preclinical model for the in vivo evaluation of CD19xCD3 DART molecules. BLT mice are generated by implanting human thymus and liver tissue into sublethally irradiated NOD/SCID-γ chain null mice, followed by transplanting autologous human CD34+ hematopoietic stem cells.24,25 BLT mice develop robust levels of human hematopoietic cells throughout the body, including T cells, B cells, monocytes/macrophages, and dendritic cells26; and this model has been utilized in the study of B cells, immune reconstitution, and HIV infection.27–37
In this manuscript, we evaluated the efficacy of human B-cell depletion by a CD19xCD3 DART protein, in which the binding arms are equivalent to those utilized in blinatumomab,21,38 in BLT humanized mice. Our results demonstrate that this DART molecule is effective at depleting human B cells in peripheral blood and tissues. We also show that this effect occurred on mature B cells and that progenitor cells remain functionally capable of generating new human B cells in the bone marrow. Finally, we demonstrate that B-cell depletion by this CD19xCD3 DART molecule is dependent on human CD8+ T cells. Together, our results provide in vivo evidence of the functional capacity of T-cell–redirecting DART proteins and validate the utilization of the BLT humanized mouse model for the preclinical evaluation of these molecules.
Results
Depletion of human CD19+ B cells in peripheral blood after administration of CD19xCD3 DART protein
In order to evaluate the efficacy of CD19xCD3 DART proteins to deplete human B cells in vivo, we utilized BLT humanized mice. BLT mice were constructed as previously described.32 The mice used for all experiments were 16–20 weeks post humanization procedure, and they were well reconstituted with human CD45+ cells (63.5% ± 13.6 SD). Of the human CD45+ cells present in peripheral blood at the beginning of the experiments, 39% ± 9.3 SD expressed human CD19, and 53.3% ± 10.7 SD expressed CD3 on their cell surface. 78.3% ± 6.5 SD of human CD3+ cells expressed human CD4 (data not shown).
For our experiments, we used a CD19xCD3 DART molecule in which the CD19 arm was derived from anti-human CD19 antibody HD37, and the CD3 arm was derived from anti-human CD3 antibody TR66, as described by Moore et al.20; these binding arms are equivalent to those of blinatumomab.38 This bispecific molecule binds to human CD3 on T cells and to human CD19 on B cells, and it recruits the cytotoxic activity of the T cells to kill the target B cells. The CD19xCD3 DART molecule was administered to BLT mice (n = 3) i.v. (1 mg/kg) in two doses, 7 days apart. A similar group of BLT mice was administered vehicle (normal saline, n = 3). Peripheral blood was collected for flow cytometric analysis on days 0, 1, 3, 7, and 11 after the first injection (Figure 1a, gating scheme in Supplementary Figure S1a).
Figure 1.
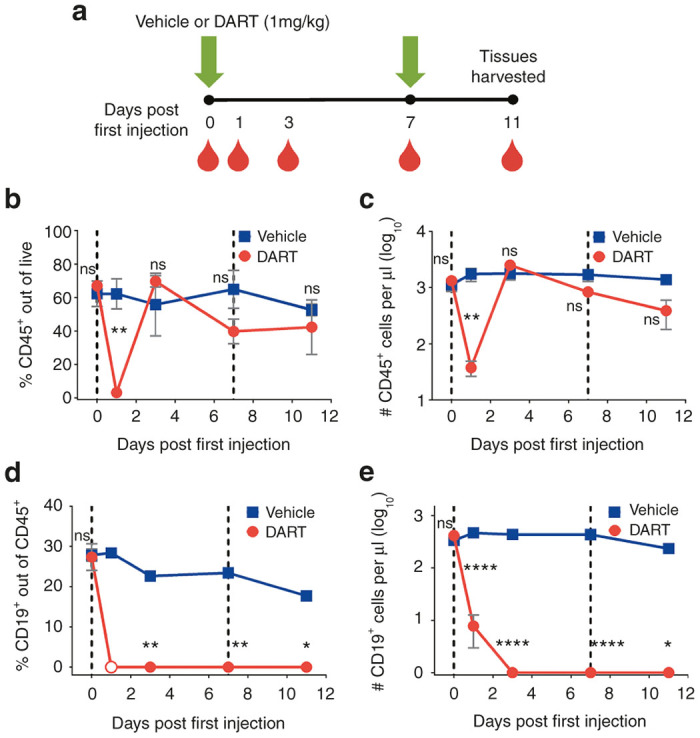
CD19xCD3 DART protein administration depletes human CD19+ B cells from the peripheral blood. (a) Experimental outline. NSG/BLT mice were administered CD19xCD3 (n = 3, 1 mg/kg i.v.) or vehicle (n = 3) at day 0 and day 7. Peripheral blood (PB) was collected and analyzed at days 0, 1, 3, 7, 11. (b) Percent CD45+ cells out of live cells and (c) number CD45+ cells per microliter detected in PB by flow cytometry (vehicle group in blue, CD19xCD3 group in red). (d) Percent CD19+ cells out of CD45+ cells and (e) number CD19+ cells per microliter detected in PB by flow cytometry (open circle = below limit of quantitation). (Mean ± SEM plotted. P values were calculated by repeated-measures two-way analysis of variance with Sidak’s multiple comparisons test, comparing vehicle versus CD19xCD3. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.)
Initially after CD19xCD3 DART molecule administration (day 1), the levels of total human CD45+ cells detected from the peripheral blood dropped to 3.2% ± 1.3 SEM (or 38 ± 11 CD45+ cells per microliter of blood); but this decrease was transient, as the levels of CD45+ cells returned to approximately normal levels by day 3 (Figure 1b,c). With regard to the target human CD19+ B cells, the pretreatment (day 0) levels of CD19+ cells in peripheral blood were 27.8% ± 1.5 CD19+ out of CD45+. By day 1 after CD19xCD3 administration, the percentage of CD19+ cells out of CD45+ cells fell to below limit of quantitation by flow cytometry. The levels of CD19+ cells in the peripheral blood remained lower than 0.1% on days 3, 7, and 11 after CD19xCD3 administration (Figure 1d). This decrease in human CD19+ B cells was also reflected in the absolute numbers of cells. Specifically, the number of CD19+ cells per microliter in the peripheral blood of the CD19xCD3-treated animals was reduced from 413 ± 67 CD19+ cells per microliter to <1 CD19+ cell per microliter of blood (Figure 1e).
In contrast to the extensive decreases in the levels of human CD19+ B cells in the CD19xCD3-treated mice, the animals treated with vehicle maintained relatively high levels of CD19+ cells. It should be noted that the decrease observed in percentage of human CD19+ cells out of CD45+ cells is consistent with the increases in human T-cell reconstitution that occur over time in this animal model (Supplementary Figure S2). However, the number of human CD19+ cells per microliter did not change significantly between day 0 and day 11 in the animals receiving vehicle (Figure 1e). All differences in the levels of human B cells between the CD19xCD3 group and the vehicle group were statistically significant at days 3, 7, and 11.
To evaluate the specificity of the effect of the CD19xCD3 DART molecule, we administered a control DART protein (4420xCD3), with one arm directed against an irrelevant target fluorescein, to BLT mice (n = 4). We did not observe a decrease in the levels of human CD19+ B cells. An increase in the percentage of CD19+ levels was noted 1 day after DART protein administration, but levels returned to normal by day 3 (Supplementary Figure S3).
We also measured the levels of human inflammatory cytokines (IL-1β, IL-6, IL-8, IL-10, IL-12p70, and TNF) in plasma samples from mice treated with vehicle, with control DART molecule, or with CD19xCD3 DART molecule (n = 4 each group). Before treatment, the levels of all cytokines tested were below the limit of quantitation (20 pg/ml) in all mice. One day after treatment, the levels remained below limit of quantitation in the mice treated with vehicle or control DART molecule. In contrast, 1 day after CD19xCD3 treatment, we noted a significant increase in the plasma levels of IL-6, IL-8, and IL-10: 337 ± 122 pg/ml IL-6, 528 ± 110 pg/ml IL-8, 1,800 ± 622 pg/ml IL-10 (mean ± SEM, P < 0.0001 with two-way repeated-measures analysis of variance and Tukey’s multiple comparisons test). By three days after treatment, the cytokine levels in the CD19xCD3-treated mice decreased back to basal levels. Together, these results show (i) a transient increase in cytokine levels in response to CD19xCD3 DART molecule treatment that resolves by 72 hours and (ii) the efficient and specific depletion of human CD19+ B cells from the peripheral blood of BLT humanized mice.
Depletion of human CD19+ B cells in tissues after administration of CD19xCD3 DART protein
In order to determine the systemic effects of the CD19xCD3 DART molecule, we harvested and isolated cells from the bone marrow, spleen, liver, and lung of the animals in the experiment described in Figure 1 at day 11, and measured levels of human CD19+ B cells in each tissue. We observed a marked depletion of CD19+ cells in all the tissues analyzed of the CD19xCD3-treated animals, down to <0.1% (Figure 2a). In contrast, the percentages of CD19+ cells in the tissues from animals that received vehicle remained normal (61.6% ± 0.9 in the bone marrow, 48.3%± 1.0 in the spleen, 14.2% ± 1.6 in the liver, and 12.1% ± 0.6 in the lung). The absolute numbers of CD19+ cells were reduced over 2,100-fold in the bone marrow of the CD19xCD3-treated animals and over 7,600-fold in the spleen, as compared to vehicle-treated animals. We could not detect CD19+ cells by flow cytometry in the livers and lungs of the mice that received the CD19xCD3 DART molecule (Figure 2b). These differences in CD19+ cell levels were statistically significant between the vehicle group and the CD19xCD3-treated group across all tissues analyzed. Together, these results demonstrate that the CD19xCD3 DART molecule can effectively deplete human CD19+ B cells from tissues.
Figure 2.
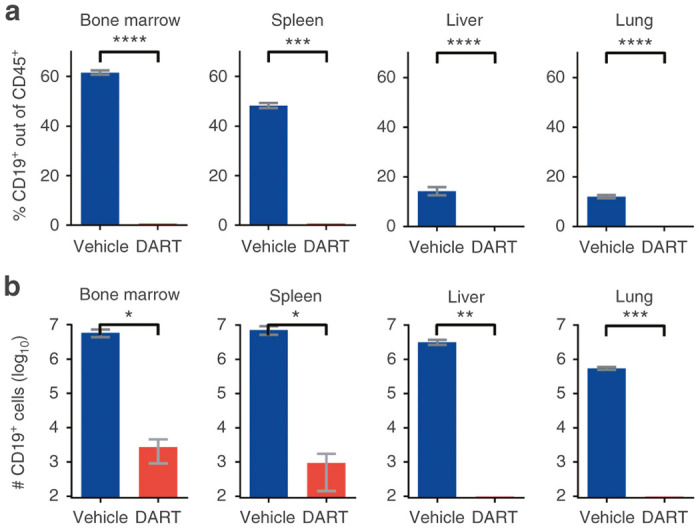
CD19xCD3 DART protein administration depletes human CD19+ B cells from the tissues. NSG/BLT mice were administered CD19xCD3 (n = 3, 1 mg/kg intravenously) or vehicle (n = 3) at day 0 and day 7. Tissues were harvested and analyzed at day 11. (a) Percent CD19+ cells out of CD45+ cells and (b) number CD19+ cells detected in tissues by flow cytometry (vehicle group in blue; CD19xCD3 group in red). (Mean ± SEM plotted. P values were calculated by unpaired t-test, comparing vehicle vs. CD19xCD3. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.)
Regeneration of human CD19+ B cells after administration of CD19xCD3 DART protein
Next, we sought to investigate the durability of depletion as well as potential for regeneration of human CD19+ B cells after administration of the CD19xCD3 DART molecule. We treated BLT mice with CD19xCD3 or vehicle (n = 3 for each group). We monitored the levels of human CD19+ B cells in peripheral blood at days 1, 3, 7, 14, 21, and 28 after the first injection (Figure 3a).
Figure 3.
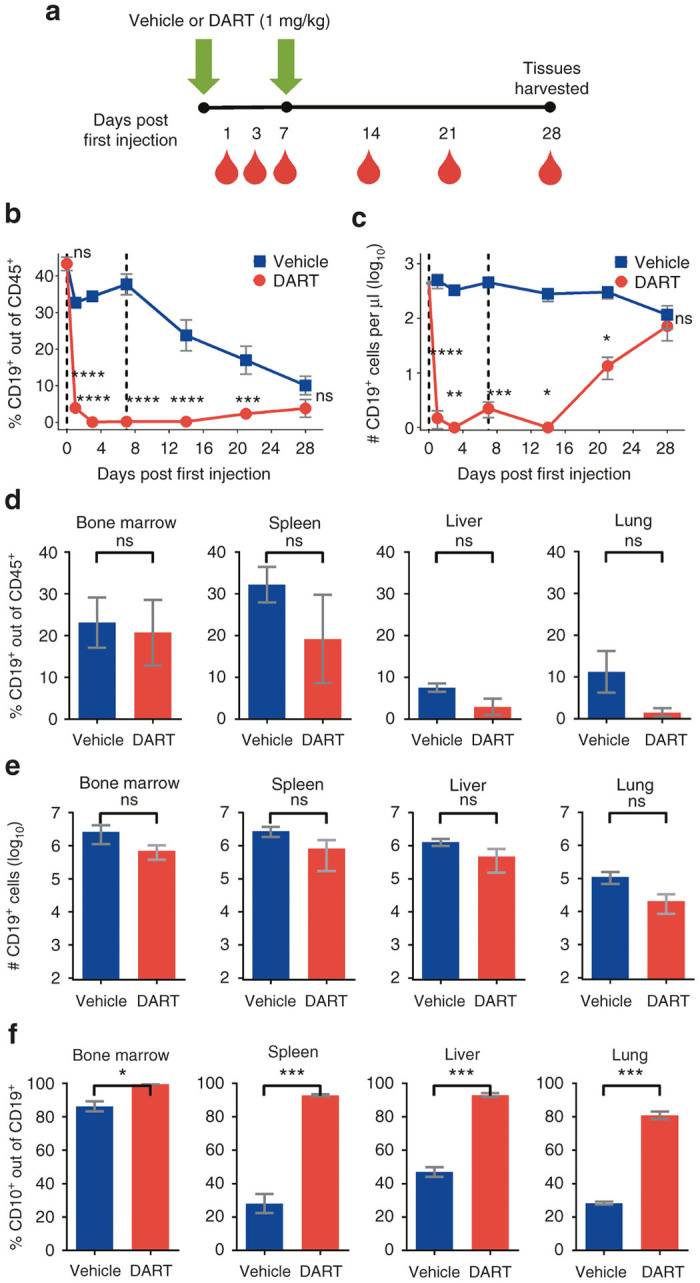
Immature human CD19+ B cells regenerate in NSG/BLT mice after CD19xCD3 DART protein administration. (a) Experimental outline. NSG/BLT mice were administered CD19xCD3 (n = 3, 1 mg/kg i.v.) or vehicle (n = 3) at day 0 and day 7. Peripheral blood (PB) was collected and analyzed at days 0, 1, 3, 7, 14, 21, and 28; tissues were harvested and analyzed at day 28. (b) Percent CD19+ cells out of CD45+ cells and (c) number CD19+ cells per microliter detected in PB by flow cytometry (vehicle group in blue; CD19xCD3 group in red). (d) Percent CD19+ cells out of CD45+ cells and (e) number CD19+ cells detected in tissues by flow cytometry. (f) Percent immature CD10+ cells out of CD19+ cells detected in tissues by flow cytometry. (Mean ± SEM plotted. P values were calculated in b and c by repeated-measures two-way analysis of variance with Sidak’s multiple comparisons test, comparing vehicle versus CD19xCD3. P values were calculated in d–f by unpaired t-test, comparing vehicle versus CD19xCD3. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Gray diamond indicates data from age-matched NSG/BLT mice, n = 4.)
As in the previous experiment in Figure 1b,c, we observed a depletion of CD19+ cells in the peripheral blood of the CD19xCD3-treated animals over the first 2 weeks. At day 21 after the first injection, the levels of CD19+ cells in the peripheral blood began to increase in the CD19xCD3-treated group. By day 28, the differences in the levels of CD19+ cells were not statistically significant between the vehicle-treated mice and the CD19xCD3-treated mice (Figure 3b,c). Analysis of the levels of CD19+ cells in the tissues at this time point were not significantly different between the animals that received vehicle versus CD19xCD3, indicating that regeneration of the B-cell population had occurred in the DART protein-treated animals (Figure 3d,e).
To confirm that the human CD19+ B cells that were present at day 28 in the CD19xCD3-treated animals were not residual from the pretreatment period, we assessed the developmental stage of the CD19+ cells appearing after CD19xCD3 administration. Specifically, we measured the levels of immature B cells as determined by human CD10 cell surface expression (gating scheme in Supplementary Figure S1b). The percentage of CD10+ cells out of the CD19+ population was significantly higher in the CD19xCD3-treated mice across all tissues analyzed, reaching >90% in the bone marrow, spleen, and liver, and 80% in the lung (Figure 3f). These results demonstrate the production of new B cells in mice previously treated with the CD19xCD3 DART protein.
Effect of CD19xCD3 DART protein administration on the levels of human T cells in vivo
To examine the effect of the CD19xCD3 DART molecule on human T cells in vivo, in particular CD8+ cytotoxic T cells, we measured the levels of human CD45+, CD3+, and CD8+ cells in peripheral blood after two administrations of vehicle or CD19xCD3 (1 mg/kg) 7 days apart, over a period of 4 weeks after the first administration.
Similar to the results presented above (Figure 1), we observed a sharp decline at day 1 of human CD45+ lymphocytes (both percentage and absolute number) in the peripheral blood of CD19xCD3-treated animals (Figure 4a,b). However, this decline was only transient: by day 3, there was no longer a significant difference in the levels of human hematopoietic cells between the vehicle- and the CD19xCD3-treated mice. The levels were again significantly lower in the CD19xCD3-treated group at day 14 (7 days after the second administration), but there was not a significant difference at day 28.
Figure 4.
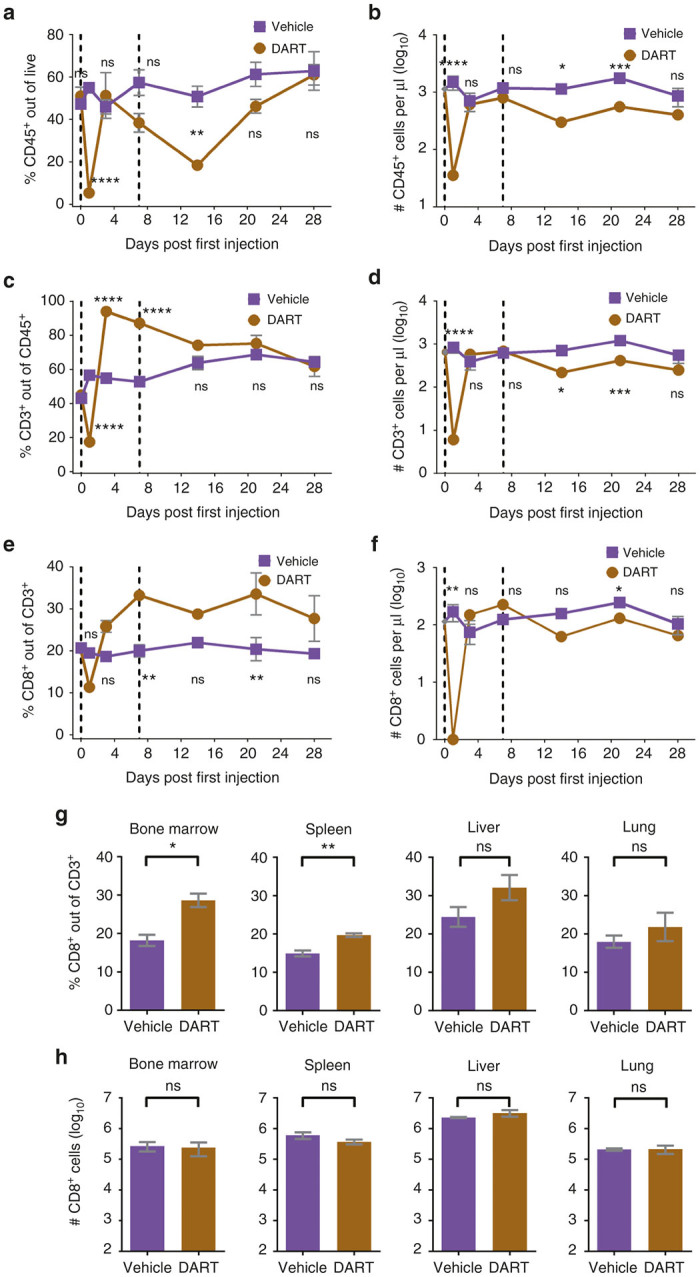
CD19xCD3 DART protein administration results in transient differences in the levels of human T cells in the peripheral blood of NSG/BLT mice, and over time there are no significant differences in absolute numbers of human CD8+ T cells in the peripheral blood or tissues as compared to vehicle-treated mice. NSG/BLT mice were administered CD19xCD3 (n = 3, 1 mg/kg i.v.) or vehicle (n = 3) at day 0 and day 7. Peripheral blood (PB) was collected and analyzed at days 0, 1, 3, 7, 14, 21, and 28; tissues were harvested and analyzed at day 28. (a) Percent CD45+ cells out of live cells and (b) number CD45+ cells per microliter detected in peripheral blood by flow cytometry (vehicle group in purple, CD19xCD3 group in brown). (c) Percent CD3+ cells out of CD45+ cells and (d) number CD3+ cells per microliter. (e) Percent CD8+ cells out of CD3+ cells and (f) number CD8+ cells per microliter. (g) Percent CD8+ cells out of CD3+ cells and (h) number CD8+ T cells detected in tissues by flow cytometry. (Mean ± SEM plotted. P values were calculated in a–f by repeated-measures two-way analysis of variance with Sidak’s multiple comparisons test, comparing vehicle versus CD19xCD3. P values were calculated in g and h by unpaired t-test, comparing vehicle versus CD19xCD3. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Gray diamond indicates data from age-matched NSG/BLT mice, n = 4.)
With regard to T cells, we observed the same sharp decline at day 1 in the percentage and absolute number of human CD3+ and CD8+ T cells in the peripheral blood. There followed an initial increase in the percent of CD3+ cells out of CD45+ cells on day 3 and day 7 in the CD19xCD3-treated group, although this increase was not reflected in the absolute numbers of CD3+ cells per microliter of blood (Figure 4c,d). The percentages of CD8+ cells out of CD3+ cells were also higher in the CD19xCD3-treated animals on days 3, 7, and 14. However, there was not a significant difference in the absolute numbers of CD8+ cells at day 3 or day 7, and the CD19xCD3-treated animals had lower numbers of CD8+ cells on day 14 (Figure 4e,f). By day 28, there were no significant differences between the groups in the percentages or absolute numbers of peripheral blood human CD45+, CD3+, or CD8+ cells.
At harvest on day 28, there was a small but statistically significant increase in the percentage of CD8+ cells out of CD3+ cells in the bone marrow and spleen of the CD19xCD3-treated animals: 18.2% ± 1.5 (vehicle) versus 28.6% ± 1.8 (CD19xCD3) (P < 0.05) and 14.9% ± 1.3 (vehicle) versus 19.7% ± 0.5 (CD19xCD3) (P < 0.01), respectively (Figure 4g). The percentages of CD8+ cells were not significantly different in the livers or the lungs of these mice. We observed no difference in the total number of CD8+ cells in any of the tissues analyzed (Figure 4h). These results demonstrate that exposure to the CD19xCD3 DART molecule does not have a significant effect on the levels of human CD8+ T cells in peripheral blood or tissues.
Dependence of CD19xCD3 DART protein-mediated depletion on the presence of human CD8+ T cells
To investigate the mechanism of action in vivo of the CD19xCD3 DART molecule, we evaluated the necessity of human CD8+ T cells for target B-cell depletion. One group of BLT mice was administered a CD8-depleting antibody (anti-CD8α antibody MT807R1, kindly provided by Dr Guido Silvestri)39,40 i.v. at 3 mg/kg (CD8-depleted group, n = 8), and one group was administered vehicle (normal saline, CD8-intact group, n = 7). Four days later, both groups were administered CD19xCD3 one time i.v. (1 mg/kg). Peripheral blood was collected and analyzed at 4 days and 1 day prior to and at days 3, 7, and 14 after administration of CD19xCD3 DART protein; tissues were harvested and analyzed at day 15 (Figure 5a).
Figure 5.

Human CD8+ T cells are depleted after administration of CD8-depleting antibody. (a) Experimental outline. NSG/BLT mice were administered CD8-depleting antibody (CD8-depleted, n = 8, 3 mg/kg i.v.) or vehicle (CD8-intact, n = 7) 4 days prior to administration of CD19xCD3 DART protein to all mice (1 mg/kg i.v.). Peripheral blood (PB) was collected and analyzed at 4 days and 1 day prior to and at days 3, 7, and 14 after administration of CD19xCD3 DART protein; tissues were harvested and analyzed at day 15. (b) Percent CD8+ cells out of CD3+ cells and (c) number CD8+ cells per microliter detected in PB by flow cytometry (CD8-depleted in orange, CD8-intact in green). (d) Percent CD8+ cells out of CD3+ cells and (e) number CD8+ cells detected in tissues by flow cytometry. (Mean ± SEM plotted. P values were calculated in b and c by repeated-measures two-way analysis of variance with Sidak’s multiple comparisons test, comparing CD8-depleted versus CD8-intact. P values were calculated in d and e by unpaired t-test, comparing CD8-depleted versus CD8-intact. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.)
To confirm depletion of human CD8+ T cells, we measured the levels of CD8+ cells in the peripheral blood and tissues. By 3 days after administration of the CD8-depleting antibody (or 1 day prior to CD19xCD3 administration), the percentage of CD8+ cells out of CD3+ cells decreased to <0.3% ± 0.1, and the number of CD8+ cells decreased to 2 ± 0.7 CD8+ cells per microliter peripheral blood in the CD8-depleted group. The levels of CD8+ cells in the CD8-intact group were 19% CD8+ out of CD3+ and 528 CD8+ cells per microliter at this time point (Figure 5b,c). In the tissues (harvested at day 15 after CD19xCD3 administration), the percentages of CD8+ out of CD3+ were <1% in the bone marrow and <0.5% in the spleen, liver, and lung of the CD8-depleted mice (Figure 5d); the number of CD8+ cells were reduced 28-fold in the bone marrow, 89-fold in the spleen, 186-fold in the liver, and 110-fold in the lung in the CD8-depleted animals as compared to the CD8-intact group (Figure 5e).
Similar to previous experiments, the human CD19+ B cells were virtually completely depleted in the peripheral blood of the CD8-intact mice (Figure 6a,b). In contrast, in the CD8-depleted mice, we observed only a transient decline in the levels of CD19+ cells at day 3 followed by a recovery to near normal CD19+ levels by day 7. The differences in the levels of CD19+ cells between the CD8-intact animals and the CD8-depleted animals were statistically significant at days 7 and 14.
Figure 6.

Human CD19+ B-cell depletion by CD19xCD3 DART protein is dependent on the presence of human CD8+ T cells. NSG/BLT mice were administered CD8-depleting antibody (CD8-depleted, n = 8, 3 mg/kg i.v.) or vehicle (CD8-intact, n = 7) 4 days prior to administration of CD19xCD3 DART protein to all mice (1 mg/kg i.v.). Peripheral blood (PB) was collected and analyzed at 4 days and 1 day prior to and at days 3, 7, and 14 after administration of CD19xCD3 DART protein; tissues were harvested and analyzed at day 15. (a) Percent CD19+ cells out of CD45+ cells and (b) number CD19+ cells per microliter detected in PB by flow cytometry (CD8-depleted in orange, CD8-intact in green). (c) Percent CD19+ cells out of CD45+ cells and (d) number CD19+ cells detected in tissues by flow cytometry. (Mean ± SEM plotted. P values were calculated in a and b by repeated-measures two-way analysis of variance with Sidak’s multiple comparisons test, comparing CD8-depleted vs. CD8-intact. P values were calculated in c and d by unpaired t-test, comparing CD8-depleted versus CD8-intact. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.)
Consistent with the depletion of the human CD19+ B cells from the peripheral blood, the levels of CD19+ cells in the tissues were also significantly reduced in the CD8-intact group as compared to the CD8-depleted group (Figure 6c). Even though there was no difference between the groups in number of total bone marrow CD19+ cells, there was at least a 10-fold statistically significant reduction in the total number of CD19+ cells in the spleen, liver, and lung of the CD8-intact versus CD8-depleted animals (Figure 6d). Together, these results demonstrate that the depletion of human CD19+ B cells by CD19xCD3 DART molecule is dependent on the presence of human CD8+ T cells.
Discussion
Building upon the success of monoclonal antibody therapies in the treatment of hematologic malignancies, novel strategies can still be useful toward improving cure rates. Bispecific molecules like DART molecules are promising in their ability to recruit T cells to kill target cells without the need for ex vivo expansion or MHC matching. The in vivo effects of T-cell–redirecting DART molecules have been evaluated in xenograft-bearing mice reconstituted with human PBMCs and in nonhuman primates, and several DART candidates are currently being evaluated in human clinical trials for oncology indications (NCT02152956, NCT02248805, NCT02454270).
Humanized BLT mice present an excellent preclinical model for the in vivo evaluation of CD19xCD3 DART molecules, as they harbor HLA-matched target and effector cells (human CD19+ B cells and human CD3+ T cells, respectively). BLT mice also allow us to assess (i) the extent of target cell depletion systemically, (ii) the effect on other relevant human hematopoietic cells, and (iii) the role of cytotoxic T cells after DART protein administration in vivo.
In our study, we observed a profound depletion of target human CD19+ B cells in both the peripheral blood and the tissues (bone marrow, spleen, liver, and lung) after two i.v. administrations of 1 mg/kg CD19xCD3 (Figures 1d,e and 2a,b). This effect was specific to the CD19-targeting arm, as a fluorescein-directed 4420xCD3 DART molecule did not result in similar CD19+ cell depletion (Supplementary Figure S3).
CD19+ cells began to return in the peripheral blood of CD19xCD3-treated BLT mice by day 21 after the first injection (or by day 14 after the second injection) (Figure 3b,c); and by day 28, the levels of B cells were not significantly lower in the tissues of CD19xCD3-treated mice as compared to vehicle-treated mice (Figure 3d,e). A vast majority of the human CD19+ cells appearing in the tissues had an immature phenotype (Figure 3f,g), as indicated by CD10 staining, suggesting that the B cells present are the result of de novo generation and development rather than of homeostatic proliferation.
A recent report evaluating an anti-CD20/CD3 T cell-dependent bispecific antibody reported initial increases in human CD8+ T cell counts followed by decreases to baseline or lower in the peripheral blood and spleen of CD3ϵ/CD20-transgenic mice or NSG mice humanized by CD34+ cell transplant.41 We investigated the effect of the CD19xCD3 DART molecules on human lymphocytes and T cells in humanized BLT mice. Interestingly, we observed a transient decrease in virtually all the human lymphocytes in the peripheral blood on day 1 after the first injection (Figures 1b,c and 4a–f). This initial decline is followed by a recovery to near normal levels of the peripheral blood human lymphocytes by day 3, with the remarkable absence of virtually all the B cells that had been depleted (Figure 1d,e). A similar transient lymphopenia has been observed in chimpanzees following administration of a bispecific anti-CD19/anti-CD3 single-chain BiTE construct at 10 hours followed by recovery at 24–72 hours.42 This transient lymphopenia could be explained by redistribution of lymphocytes that are adhering to blood vessel walls or migrating into tissues. Despite temporary differences, peripheral blood human CD45+, CD3+, and CD8+ cell levels in the CD19xCD3 DART protein-treated animals returned to similar levels as the vehicle group by day 28 after the first injection (Figure 4a,f). Notably, the number of human CD8+ T cells were not significantly different between the vehicle- and CD19xCD3 DART protein-treated animals in the bone marrow, spleen, liver, and lung (Figure 4h).
Finally, we evaluated the dependence of target human CD19+ B-cell depletion by CD19xCD3 DART molecules on the presence of human CD8+ T cells. BLT mice that were CD8-depleted prior to CD19xCD3 administration did not exhibit the reduction of CD19+ cells in the peripheral blood that was observed in CD8-intact animals (Figure 6b,c), and the CD8-depleted group had significantly higher systemic levels of CD19+ cells, except in the bone marrow (Figure 6d,e). These data suggest that human CD8+ T cells are required for depletion of human CD19+ B cells by CD19xCD3 DART molecules.
This study validates the humanized BLT mouse as a preclinical model for the use and evaluation of bispecific reagents, in particular DART molecules. The BLT mouse model provides opportunities to study the in vivo effects of bispecific molecules on human target cells both in the peripheral blood and the tissues. Future directions could include the evaluation of DART molecules directed against other target immune cells or against viral antigens expressed on infected cells. A recent paper from Sung et al.43 showed data on the efficacy ex vivo of a DART molecule directed against HIV envelope protein, thus providing a rationale for further in vivo studies in BLT humanized mice of DART molecules as a targeted cytotoxic therapy against HIV-infected cells, as we have previously demonstrated using an immunotoxin strategy.44
Materials and Methods
Ethics statement
All animal experiments were conducted following NIH guidelines for housing and care of laboratory animals and in accordance with University of North Carolina at Chapel Hill (UNC Chapel Hill) regulations after review and approval by the UNC Chapel Hill Institutional Animal Care and Use Committee (permit number 12–171).
Generation of BLT humanized mice
BLT mice were generated as described previously.32 NOD/SCID-γ chain null mice (NSG, Stock #5557, The Jackson Laboratory, Bar Harbor, ME) were sublethally irradiated, implanted with human thymus and liver tissue, and transplanted with autologous human liver CD34+ cells (Advanced Bioscience Resources, Rockville, MD); then they were monitored for human reconstitution in peripheral blood by flow cytometry.25,28,32 All BLT mice (n = 31) used for these experiments contained an average of 63.5% ± 13.6 SD human CD45+ cells in the peripheral blood, of which 39% ± 9.3 SD expressed human CD19, and 53.3% ± 10.7 SD expressed CD3 on their cell surface. 78.3% ± 6.5 SD of human CD3+ cells expressed human CD4.
DART molecules
As described by Moore et al.,20 the CD19xCD3 DART molecule, in basic format, was constructed using anti-human CD19 Fv sequences from HD37 (ref. 45) and anti-human CD3 Fv sequences from TR66 (refs. 38,46); and the irrelevant arm of the control DART molecule (4420xCD3) was constructed from anti-fluorescein Fv sequences from 4-4-20 (ref. 47). The binding arms of this CD19xCD3 DART protein do not crossreact with murine antigens. The DART proteins were produced in CHO-S cells and purified as described.20
Treatment of BLT mice
BLT mice were injected intravenously with CD19xCD3 DART molecule (MacroGenics, Rockville, MD) at a dose of 1 mg/kg either one time or two times 7 days apart, with 4420xCD3 DART molecule at a dose of 1 mg/kg two times 7 days apart, or with vehicle (normal saline, 0.9% sodium chloride, #0409-4888-10, Hospira, Lake Forest, IL). For CD8+ T-cell depletion, BLT mice were injected i.v. with CD8-depleting antibody (anti-CD8α antibody MT807R1, gift from Dr Guido Silvestri, Atlanta, GA) at a dose of 3 mg/kg or with vehicle (normal saline).
Analysis of BLT mice
Peripheral blood and tissues were collected and cells isolated as previously described for flow cytometric analysis.25,28 Antibodies used in these experiments included anti-human CD45 APC (clone HI30, catalog #555485), CD45 FITC (2D1, 347463), CD3 FITC (HIT3a, 555339), CD8 APC-Cy7 (SK1, 557834), CD8 PerCP (SK1, 347314), CD19 PE-Cy7 (SJ25C1, 557835), CD10 APC (HI10a, 340923) (BD Biosciences, San Jose, CA). Flow cytometry data were acquired using a BD FACSCanto Cytometer and analyzed using BD FACSDiva software (version 6.1.3) with the following gating strategy: live cells → human CD45+ → human CD19+ (→ human CD10+) or live → human CD45+ → human CD3+ → human CD8+. The numbers of cells per microliter peripheral blood were calculated by dividing the number of positive events acquired by the number of microliters of peripheral blood stained. The numbers of cells per tissue were calculated by multiplying the total number of live cells isolated from the tissue, by the fraction of positive events over live events. Cytokine analysis was performed on plasma samples using a Human Inflammatory Cytokine Kit (551811, BD Biosciences, San Jose, CA); these data were acquired using a BD LSR II Cytometer and analyzed using FCAP Array Infinite (Soft Flow, St Louis Park, MN).
Statistical analysis
All statistical tests were performed with an alpha level of 0.05. Repeated-measures two-way analysis of variance with Sidak’s multiple comparisons test was used to generate the P values in Figures 1b–e, 3b,c, 4a–f, 5b,c, and 6a,b. Unpaired t-test was utilized to generate the P values in Figures 2, 3d,f, 4g,h, 5d,e, and 6c,d. Graphs were generated in Graphpad Prism (version 6).
Acknowledgments
This work was supported in part by grants AI112487-02, AI096113, AI111899, and by the UNC Center for AIDS Research P30 AI50410. We thank Tia Morgan and Christopher Nixon (UNC) for reading the manuscript. We thank former and current lab members as well as the UNC Division of Laboratory Animal Medicine for their assistance with aspects of this work. We also thank Kay Shah, Yinhua Yang, and Vatana Long (MacroGenics) for their assistance in DART molecule construction and analysis.
Footnotes
P.T., W.O.T., G.S., and J.V.G. have no conflicts of interest to declare. L.L. and J.L.N. are employees of Macrogenics, Inc.
References
- Vasekar, M, Rizvi, S, Liu, X, Vrana, KE and Zheng, H. (2015). Novel immunotherapies for hematological malignancies. Curr Mol Pharmacol (epub ahead of print). [DOI] [PubMed]
- Griffin, MM and Morley, N (2013). Rituximab in the treatment of non-Hodgkin’s lymphoma–a critical evaluation of randomized controlled trials. Expert Opin Biol Ther 13: 803–811. [DOI] [PubMed] [Google Scholar]
- Plosker, GL and Figgitt, DP (2003). Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs 63: 803–843. [DOI] [PubMed] [Google Scholar]
- Lim, SH and Levy, R (2014). Translational medicine in action: anti-CD20 therapy in lymphoma. J Immunol 193: 1519–1524. [DOI] [PubMed] [Google Scholar]
- Bauer, K, Rancea, M, Roloff, V, Elter, T, Hallek, M, Engert, A et al. (2012). Rituximab, ofatumumab and other monoclonal anti-CD20 antibodies for chronic lymphocytic leukaemia. Cochrane Database Syst Rev 11: CD008079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan, SB, Sørensen, JF, Olsen, BN and Pedersen, AE (2014). Anti-CD40-mediated cancer immunotherapy: an update of recent and ongoing clinical trials. Immunopharmacol Immunotoxicol 36: 96–104. [DOI] [PubMed] [Google Scholar]
- Siddiqi, T, Thomas, SH and Chen, R (2014). Role of brentuximab vedotin in the treatment of relapsed or refractory Hodgkin lymphoma. Pharmgenomics Pers Med 7: 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, X, LaVallee, T and Lechleider, R (2011). CD22 as a target for cancer therapy. J Exp Ther Oncol 9: 241–248. [PubMed] [Google Scholar]
- Skoetz, N, Bauer, K, Elter, T, Monsef, I, Roloff, V, Hallek, M et al. (2012). Alemtuzumab for patients with chronic lymphocytic leukaemia. Cochrane Database Syst Rev 2: CD008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak, T and Robak, P (2014). Anti-CD37 antibodies for chronic lymphocytic leukemia. Expert Opin Biol Ther 14: 651–661. [DOI] [PubMed] [Google Scholar]
- Castillo, J, Winer, E and Quesenberry, P (2008). Newer monoclonal antibodies for hematological malignancies. Exp Hematol 36: 755–768. [DOI] [PubMed] [Google Scholar]
- Saret, CJ, Winn, AN, Shah, G, Parsons, SK, Lin, PJ, Cohen, JT et al. (2015). Value of innovation in hematologic malignancies: a systematic review of published cost-effectiveness analyses. Blood 125: 1866–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheson, BD and Leonard, JP (2008). Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma. N Engl J Med 359: 613–626. [DOI] [PubMed] [Google Scholar]
- Cheson, BD (2010). Ofatumumab, a novel anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. J Clin Oncol 28: 3525–3530. [DOI] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Robbins, PF, Yang, JC, Hwu, P, Schwartzentruber, DJ et al. (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Yu, Z, Frasheri, D, Restifo, NP and Rosenberg, SA (2010). Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116: 3875–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus, MV, Grupp, SA, Porter, DL and June, CH (2014). Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123: 2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp, MS, Gökbuget, N, Zugmaier, G, Degenhard, E, Goebeler, ME, Klinger, M et al. (2012). Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood 120: 5185–5187. [DOI] [PubMed] [Google Scholar]
- Johnson, S, Burke, S, Huang, L, Gorlatov, S, Li, H, Wang, W et al. (2010). Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol 399: 436–449. [DOI] [PubMed] [Google Scholar]
- Moore, PA, Zhang, W, Rainey, GJ, Burke, S, Li, H, Huang, L et al. (2011). Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 117: 4542–4551. [DOI] [PubMed] [Google Scholar]
- Rader, C (2011). DARTs take aim at BiTEs. Blood 117: 4403–4404. [DOI] [PubMed] [Google Scholar]
- Chichili, GR, Huang, L, Li, H, Burke, S, He, L, Tang, Q et al. (2015). A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med 7: 289ra82. [DOI] [PubMed] [Google Scholar]
- Liu L, Alderson R, Yang Y, Li H, Long V, et al. (2014). MGD011, humanized CD19 x CD3 DART(R) protein with enhanced pharmacokinetic properties, demonstrates potent T-cell mediated anti-tumor activity in preclinical models and durable B-cell depletion in cynomolgus monkeys following once-a-week dosing. Blood 124: 1775. [Google Scholar]
- Lan, P, Tonomura, N, Shimizu, A, Wang, S and Yang, YG (2006). Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 108: 487–492. [DOI] [PubMed] [Google Scholar]
- Melkus, MW, Estes, JD, Padgett-Thomas, A, Gatlin, J, Denton, PW, Othieno, FA et al. (2006). Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med 12: 1316–1322. [DOI] [PubMed] [Google Scholar]
- Wege, AK, Melkus, MW, Denton, PW, Estes, JD and Garcia, JV (2008). Functional and phenotypic characterization of the humanized BLT mouse model. Curr Top Microbiol Immunol 324: 149–165. [DOI] [PubMed] [Google Scholar]
- Sun, Z, Denton, PW, Estes, JD, Othieno, FA, Wei, BL, Wege, AK et al. (2007). Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med 204: 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, PW, Estes, JD, Sun, Z, Othieno, FA, Wei, BL, Wege, AK et al. (2008). Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med 5: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, PW, Krisko, JF, Powell, DA, Mathias, M, Kwak, YT, Martinez-Torres, F et al. (2010). Systemic administration of antiretrovirals prior to exposure prevents rectal and intravenous HIV-1 transmission in humanized BLT mice. PLoS One 5: e8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, PW, Othieno, F, Martinez-Torres, F, Zou, W, Krisko, JF, Fleming, E et al. (2011). One percent tenofovir applied topically to humanized BLT mice and used according to the CAPRISA 004 experimental design demonstrates partial protection from vaginal HIV infection, validating the BLT model for evaluation of new microbicide candidates. J Virol 85: 7582–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen, R, Wahl, A, Denton, PW and Garcia, JV (2011). Immune reconstitution of the female reproductive tract of humanized BLT mice and their susceptibility to human immunodeficiency virus infection. J Reprod Immunol 88: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, PW, Nochi, T, Lim, A, Krisko, JF, Martinez-Torres, F, Choudhary, SK et al. (2012). IL-2 receptor γ-chain molecule is critical for intestinal T-cell reconstitution in humanized mice. Mucosal Immunol 5: 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, PW, Olesen, R, Choudhary, SK, Archin, NM, Wahl, A, Swanson, MD et al. (2012). Generation of HIV latency in humanized BLT mice. J Virol 86: 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, A, Swanson, MD, Nochi, T, Olesen, R, Denton, PW, Chateau, M et al. (2012). Human breast milk and antiretrovirals dramatically reduce oral HIV-1 transmission in BLT humanized mice. PLoS Pathog 8: e1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateau, ML, Denton, PW, Swanson, MD, McGowan, I and Garcia, JV (2013). Rectal transmission of transmitted/founder HIV-1 is efficiently prevented by topical 1% tenofovir in BLT humanized mice. PLoS One 8: e60024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nochi, T, Denton, PW, Wahl, A and Garcia, JV (2013). Cryptopatches are essential for the development of human GALT. Cell Rep 3: 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Torres, F, Nochi, T, Wahl, A, Garcia, JV and Denton, PW (2014). Hypogammaglobulinemia in BLT humanized mice–an animal model of primary antibody deficiency. PLoS One 9: e108663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle, EJ (2006). MT-103 micromet/medImmune. Curr Opin Mol Ther 8: 62–68. [PubMed] [Google Scholar]
- Schmitz, JE, Simon, MA, Kuroda, MJ, Lifton, MA, Ollert, MW, Vogel, CW et al. (1999). A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. Am J Pathol 154: 1923–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury, A, Hayes, TL, Bosinger, SE, Lawson, BO, Vanderford, T, Schmitz, JE et al. (2015). Differential impact of in vivo CD8+ T lymphocyte depletion in controller versus progressor simian immunodeficiency virus-infected macaques. J Virol 89: 8677–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, LL, Ellerman, D, Mathieu, M, Hristopoulos, M, Chen, X, Li, Y et al. (2015). Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med 7: 287ra70. [DOI] [PubMed] [Google Scholar]
- Schlereth, B, Quadt, C, Dreier, T, Kufer, P, Lorenczewski, G, Prang, N et al. (2006). T-cell activation and B-cell depletion in chimpanzees treated with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Cancer Immunol Immunother 55: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, JA, Pickeral, J, Liu, L, Stanfield-Oakley, SA, Lam, CY, Garrido, C et al. (2015). Dual-Affinity Re-Targeting proteins direct T cell-mediated cytolysis of latently HIV-infected cells. J Clin Invest 125: 4077–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, PW, Long, JM, Wietgrefe, SW, Sykes, C, Spagnuolo, RA, Snyder, OD et al. (2014). Targeted cytotoxic therapy kills persisting HIV infected cells during ART. PLoS Pathog 10: e1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipriyanov, SM, Kupriyanova, OA, Little, M and Moldenhauer, G (1996). Rapid detection of recombinant antibody fragments directed against cell-surface antigens by flow cytometry. J Immunol Methods 196: 51–62. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia, A and Scheidegger, D (1987). The use of hybrid hybridomas to target human cytotoxic T lymphocytes. Eur J Immunol 17: 105–111. [DOI] [PubMed] [Google Scholar]
- Kranz, DM and Voss, EW Jr (1981). Partial elucidation of an anti-hapten repertoire in BALB/c mice: comparative characterization of several monoclonal anti-fluorescyl antibodies. Mol Immunol 18: 889–898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.