

Abstract
A reciprocal translocation of the ABL1 gene to the BCR gene results in the expression of the oncogenic BCR-ABL1 fusion protein, which characterizes human chronic myeloid leukemia (CML), a myeloproliferative disorder considered invariably fatal until the introduction of the imatinib family of tyrosine kinase inhibitors (TKI). Nonetheless, insensitivity of CML stem cells to TKI treatment and intrinsic or acquired resistance are still frequent causes for disease persistence and blastic phase progression experienced in patients after initial successful therapies. Here, we investigated a possible role for the MAPK15/ERK8 kinase in BCR-ABL1-dependent autophagy, a key process for oncogene-induced leukemogenesis. In this context, we showed the ability of MAPK15 to physically recruit the oncogene to autophagic vesicles, confirming our hypothesis of a biologically relevant role for this MAP kinase in signal transduction by this oncogene. Indeed, by modeling BCR-ABL1 signaling in HeLa cells and taking advantage of a physiologically relevant model for human CML, i.e. K562 cells, we demonstrated that BCR-ABL1-induced autophagy is mediated by MAPK15 through its ability to interact with LC3-family proteins, in a LIR-dependent manner. Interestingly, we were also able to interfere with BCR-ABL1-induced autophagy by a pharmacological approach aimed at inhibiting MAPK15, opening the possibility of acting on this kinase to affect autophagy and diseases depending on this cellular function. Indeed, to support the feasibility of this approach, we demonstrated that depletion of endogenous MAPK15 expression inhibited BCR-ABL1-dependent cell proliferation, in vitro, and tumor formation, in vivo, therefore providing a novel “druggable” link between BCR-ABL1 and human CML.
Keywords: autophagy, BCR-ABL1, LC3B, MAP kinases, signal transduction
Abbreviations
- ATG
autophagy related
- Baf
bafilomycin A1
- BCR-ABL1
breakpoint cluster region–ABL proto-oncogene 1, nonreceptor tyrosine kinase
- CML
chronic myeloid leukemia
- CQ
chloroquine
- Doxy
doxycycline
- EGFP
enhanced green fluorescent protein
- ERK
extracellular signal-regulated kinase
- FM
full medium
- GST
glutathione S-transferase
- HA
hemagglutinin tag
- LIR
LC3-interacting region
- MAP
mitogen-activated protein
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- PE
phosphatidylethanolamine
- aa
amino acid
- Rap
rapamycin
- ROS
reactive oxygen species
- SCR
scrambled
- SH3
SRC homology 3
- SQSTM1
sequestrosome 1
- ST
starvation
- TKI
tyrosine kinase inhibitor
- WB
western blot
- WM
wortmannin.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder characterized by excessive accumulation of apparently normal myeloid cells, molecularly characterized by the presence of the Philadelphia (Ph) chromosome, resulting from a (9;22)(q34;q11) reciprocal translocation.1-4 Expression of the resulting BCR-ABL1 oncogene is usually considered the initiating event in the genesis of this disease and is sufficient to induce leukemia.5 Thanks to its constitutively active tyrosine kinase activity, BCR-ABL1 is, indeed, able to mimic growth factors stimulation by activating many signaling pathways, leading to increased proliferation, decreased apoptosis, reduced growth factor-dependence, and abnormal interaction with extracellular matrix and stroma.6,7
Most CML patients are usually diagnosed in the initial, chronic phase of the disease and treated with first and/or second generation drugs designed to block the enzymatic activity of the BCR-ABL1 tyrosine kinase, namely imatinib, dasatinib, and nilotinib.8 Still, approximately 20% of patients in chronic phase fail to respond to both imatinib and to subsequent second generation tyrosine kinase inhibitors (TKIs), with very poor prognosis once progressed to the advanced blastic phase.8 Therefore, while these TKIs have clearly revolutionized therapy for the disease, there is still need for supplementary or alternative options to “integrate” current pharmacological approaches. In this context, autophagy has been demonstrated as necessary for BCR-ABL1-induced leukemogenesis,7,9,10 as well as to protect cancer cells from apoptosis induced by antineoplastic drugs such as imatimib.11-16 Based on these evidences, an inhibitor of autophagy, hydroxychloroquine, has been already successfully used to potentiate TKI-induced cell death in Ph chromosome-positive cells, including primary CML stem cells.7,14 Importantly, new clinical trials are also investigating the effect of adding hydroxychloroquine to Imatinib treatment for CML (CHOICES trial, http://www.cancerresearchuk.org/about-cancer/find-a-clinical-trial/a-trial-hydroxychloroquine-with-imatinib-for-choices).
MAPK15 is currently the last identified member of the MAP kinase family of proteins.17 Its activity can be modulated by nutrient deprivation,18,19 and by important human oncogenes, such as RET-PTC3, RET-MEN2B, and BCR-ABL1.20 Still, very limited information is available about the role of this MAP kinase in cell proliferation and transformation, sometimes with opposite results depending on the experimental system used. Indeed, while MAPK15 activity is important for transformation of human colon cancer cells,21 its mouse orthologous gene negatively regulates cell growth of Cos7 cells.22 Importantly, we have recently described a role for MAPK15 in the regulation of autophagy, and have demonstrated the feasibility of pharmacologically interfering with this process by modulating the activity of this MAP kinase.19
Here, we show that BCR-ABL1 was able to modulate autophagy and that MAPK15 mediated this effect in an LIR-dependent manner. Moreover, not only artificial depletion of the endogenous MAP kinase inhibited BCR-ABL1-dependent autophagy but, also, we demonstrate that it was possible to pharmacologically interfere with this process by using a MAPK15 inhibitor. Importantly, based on the role of autophagy in BCR-ABL1-dependent transformation, we show that MAPK15 and its ability to control the autophagic process was required for cell proliferation and in vivo tumor development induced by this oncogene, therefore establishing MAPK15 as a novel potential and feasible therapeutic target for human CML.
Results
BCR-ABL1 interacts with MAPK15 and colocalizes with it at phagophores
We have previously shown that the BCR-ABL1 oncogene stimulates MAPK15 activity and that the ABL1 proto-oncogene interacts with this MAP kinase and mediates its activation by RET-PTC320 (Fig. S1). Expanding these results, we therefore tested the interaction between MAPK15 and BCR-ABL1 and demonstrated that they readily coimmunoprecipitated (Fig. 1A). In this context, sequence analysis of MAPK15 has already revealed the presence of 2 potential SH3-domain binding motifs, PXXP, within its C-terminal region and Abe and collaborators have demonstrated that such motifs associate with the SRC SH3 domain.17 We therefore tested whether MAPK15 could associate also with the ABL1 SH3 domain. To this aim, we cotransfected MAPK15 and an activated form of ABL1 (Act. ABL1) in which the SH3 domain has been deleted.23 In these settings, MAPK15 was still able to coimmunoprecipitate with the mutated protein, even in the absence of ABL1 SH3 (Fig. 1A), suggesting the existence of additional domains in ABL1 able to mediate interaction with the MAP kinase.
Figure 1.

BCR-ABL1 interacts with MAPK15. (A) 293T cells were transfected with HA-MAPK15 and with plasmids encoding the empty vector or the indicated ABL1 and BCR-ABL1-expressing vectors. Total lysates were collected 24 h later and, after immunoprecipitation, western blot analysis was performed. Similar results were obtained in 3 independent experiments. (B) K562 cells were harvested and total lysates were collected. Lysates were subjected to immunoprecipitation with preimmune IgG, as control, or with anti-MAPK15 IgG. Then, immunoprecipitated protein complexes were analyzed by SDS-PAGE and western blot. Similar results were obtained in 3 independent experiments. (C) HeLa cells were transfected with HA-MAPK15 and BCR-ABL1 plasmids and then subjected to immunofluorescence analysis. MAPK15 is visualized in green, BCR-ABL1 in red, and DAPI-stained nuclei in blue. White arrows indicate colocalization spots. The Pearson correlation between MAPK15 and BCR-ABL1 was calculated (P = 0.658 ± 0 .071). The colocalization rate of MAPK15 and BCR-ABL1 was obtained by analyzing at least 400 cells from 3 different experiments (n = 3). Scale bars, 10 μm. (D) GFP-LC3 HeLa cells were transfected with HA-MAPK15 and BCR-ABL1 plasmids and then subjected to immunofluorescence analysis. GFP-LC3 is visualized in green, BCR-ABL1 in red, MAPK15 in cyan and DAPI-stained nuclei in blue. White arrows indicate colocalization spots. a, anti. Scale bars, 10 μm.
In order to define a physiological context for MAPK15 in BCR-ABL1 signaling, we analyzed a panel of myeloid and lymphoid cell lines for the expression of the MAP kinase (Fig. S2). Interestingly, all BCR-ABL1-encoding CML cell lines (KCL-22, LAMA84 and K562) expressed MAPK15 at high levels, whereas immortalized T lymphocyte cells (Jurkat) and other BCR-ABL1-negative leukemic cell lines (U937, REH and HL60) showed much lower MAPK15 expression. Among BCR-ABL1 positive CML cell lines, exhibiting high levels of MAPK15, we then choose K562 cells to investigate the interaction between this kinase and BCR-ABL1, in their physiological context and at endogenous levels. In these conditions, BCR-ABL1 readily coimmunoprecipitated with endogenous MAPK15 (Fig. 1B), which appeared as 2 bands possibly representing alternatively phosphorylated or cleaved forms of the protein.
The BCR-ABL1 oncoprotein has been extensively localized to the cytoplasm and to the nucleus.24,25 In particular, BCR-ABL1 is restrained to the cytosol through its F-actin binding domain,26 but can translocate to the nucleus upon imatinib treatment, inducing cellular apoptosis.27,28 Therefore, we examined BCR-ABL1 and MAPK15 localization in our cellular model and demonstrated, by an immunofluorescence approach, that the 2 proteins colocalized with a very high Pearson correlation (P = 0.658 ± 0 .071, Fig. 1C), further supporting, in vivo, the evidence of their interaction. Interestingly, available data from cell fractionation and immunofluorescence experiments have shown that MAPK15, besides being localized to the cytoplasm and the nucleus, is also on vesicles identified as autophagic in nature.19,29 Remarkably, MAPK15 and BCR-ABL1 colocalization was also evident on intracellular vesicles (Fig. 1C), confirming a punctate pattern already described for BCR-ABL1.26 We, therefore, took advantage of HeLa cells stably expressing GFP-LC3B to investigate the localization of both these molecules on autophagic vesicles and confirmed their colocalization with LC3B, a marker of phagophores and autophagosomes (Fig. 1D). Altogether, our data demonstrate physical interaction, at the endogenous levels, of the BCR-ABL1 oncoprotein with MAPK15, on vesicles of autophagic origin, suggesting their interplay as functionally important to control oncogene-dependent functions.
BCR-ABL1 induces autophagy
Some data indicate that BCR-ABL1 is involved in the regulation of autophagy in leukemic cells.9,11-16 However, its precise role in this process is still the object of controversy. Indeed, on one hand imatinib, the most used BCR-ABL1 inhibitor, has been described to increase autophagy in leukemic cells,14,30 suggesting this oncogene as a negative regulator of autophagy whereas, on the other hand, it has been demonstrated that BCR-ABL1 cellular transformation is dependent on autophagy for cancer cell survival and proliferation.9,31,32 In this context, we decided to examine BCR-ABL1 effect in a well-established model for the study of autophagy, i.e. the HeLa cancer cell line.19,33 To this aim, we created an inducible cell line for BCR-ABL1, HeLa-T-Rex-BCR-ABL1, in order to circumvent potential biases arising from stress induced by transient transfection.19,33 In this cellular system, BCR-ABL1 was expressed only upon doxycycline treatment (Fig. 2A). Next, we analyzed the effect of BCR-ABL1 expression on autophagy, by monitoring the average number of autophagosomes per cell. In HeLa-T-Rex-BCR-ABL1 cells, the expression of the oncogenic fusion protein led to a significant increase in the amount of LC3B dots per cell, compared to control sample (Fig. 2B). In order to confirm these data, we also examined the autophagic flux in HeLa cell by analyzing the amount of the lipidated, autophagosome-associated form of LC3B (LC3B-II) in cells treated with an inhibitor of lysosomal proteases, bafilomycin A1 (Baf). Indeed, induction of autophagy is associated to an increase of LC3B-II33 that can be monitored by using anti-LC3B antibodies specifically recognizing this modified protein.19,33 As shown in Figure. 2C, BCR-ABL1 increased the amount of LC3B-II in HeLa cells when compared to control samples. Moreover, to specifically determine the autophagic flux,33 BCR-ABL1-expressing cells were treated with Baf, revealing a further increase in LC3B-II amount when compared to untreated controls. Thus, the BCR-ABL1 oncoprotein led to an increase of autophagic flux in HeLa cells, indicating that BCR-ABL1 is a positive regulator of autophagy. As a further control, we also overexpressed a dominant negative construct encoding for a kinase dead ABL1 isoform. Unlike BCR-ABL1, this construct caused a reduction of the autophagic flux compared to control samples (Fig. 2C), indicating that tyrosine kinase activity is implicated in BCR-ABL1-dependent regulation of autophagy. Taken together, these data suggested that BCR-ABL1 is a bona fide inducer of the autophagic process.
Figure 2.

BCR-ABL1 induces autophagy in HeLa cells. (A) HeLa T-Rex BCR-ABL1 cells were treated with doxycycline for the indicated periods. Then, total lysates were collected and subjected to western blot analysis. Similar results were obtained in 3 independent experiments. (B) HeLa T-Rex BCR-ABL1 cells were treated with doxycycline for 24 h. Cells were stained for LC3B with a specific antibody, in green, and with DAPI for nuclei, in blue. LC3B-positive dots were counted using a specific protocol by Volocity software (see graph on the right). Similar results were obtained in 3 independent experiments (n = 3). Scale bars, 10 μm. (C) HeLa cells were transfected with HA-MAPK15, BCR-ABL, GFP-ABLKD or control plasmids. After 24 h, cells were treated with 100 nM Baf for 1 h and total lysates were collected for western blotting analysis. Densitometric analysis of bands was also performed on this experiment, representative of 3 independent experiments (n = 3).
MAPK15 mediates BCR-ABL1-induced autophagy
We have recently described MAPK15 as a regulator of the autophagic process.19 As BCR-ABL1 was able to induce autophagy, to stimulate MAPK15 activity,20 and to interact with this MAP kinase (see above), we decided to examine whether MAPK15 was able to modulate BCR-ABL1-induced autophagy. We therefore exploited a RNA interference approach, by using already validated commercial MAPK15-specific siRNAs,18,19,29 custom-made siRNAs (Fig. S3), and commercial shRNAs (Fig. S4), in HeLa cells expressing BCR-ABL1. In these settings, using MAPK15 siRNAs, we observed a significant decrease in the number of autophagosomes in BCR-ABL1-expressing cells depleted for MAPK15, comparing with those treated with control siRNA (Fig. 3A). Furthermore, we also assessed the autophagic activity of cells stably expressing BCR-ABL1 and MAPK15-specific shRNAs, by monitoring LC3B and SQSTM1 protein levels under full medium, starvation, and Baf treatment conditions. As expected,19 interfering with MAPK15 expression strongly reduced autophagic flux and starvation-induced autophagy (Fig. 3B). BCR-ABL1, when expressed, stimulated the autophagic flux, whereas depletion of MAPK15 in BCR-ABL1-expressing cells caused a reduction in this flux and a decrease in the starvation-induced autophagic response (Fig. 3B). Once established that MAPK15 controlled BCR-ABL1-induced autophagy in HeLa model cell lines, we next sought to confirm the existence of this specific pathway in K562 CML cells endogenously expressing both these kinases (see above). Autophagic flux was, therefore, evaluated by western blot in K562 cells, comparing shSCR with shMAPK15. MAPK15-depleted K562 cells had, indeed, a decreased autophagic flux with a higher basal level for SQSTM1 and a reduced increase for LC3B-II upon Baf treatment (Fig. 4A). To corroborate this data, we also evaluated autophagy by immunofluorescence, using specific shRNA for MAPK15 in K562 cells, demonstrating a strong reduction in the amount of autophagic vesicles per cell, in both basal conditions and upon Baf treatment (Fig.4B). Altogether these data further confirm that BCR-ABL1 was able to induce autophagy and that MAPK15 was important for proper basal autophagic response, while ultimately demonstrating a role for this endogenous MAP kinase in mediating BCR-ABL1-induced autophagy.
Figure 3.

MAPK15 mediates BCR-ABL1-induced autophagy. (A) HeLa T-Rex BCR-ABL1 cells were transfected with scrambled or MAPK15 siRNA. After 48 h cells were treated with doxycycline for 24 h and subjected to immunofluorescence analysis. In these representative images, LC3B is visualized in green and DAPI-stained nuclei in blue. LC3B-positive dots were counted using a specific protocol by Volocity software (see graph on the right). Scale bars, 25 μm. (B) HeLa cells, stable for the indicated plasmids (empty vector, BCR-ABL1, shSCR or shMAPK15), were treated as indicated: starvation was performed in Hank's medium for 20 min, 100 nM Baf was added for 1 h. Then, total lysates were harvested and subjected to western blot analysis. One experiment, representative of 3 independent experiments is shown (n = 3). Densitometric analysis of bands was also performed.
Figure 4.

MAPK15 mediates BCR-ABL1-induced autophagy in CML cells. (A) K562 cells, stably expressing the indicated plasmids (shSCR or shMAPK15), were treated as indicated: 100 nM WM or 100 nM Baf was added for 1 h. Then, total lysates were harvested and subjected to western blot analysis. One experiment, representative of 3 independent experiments is shown (n = 3). Densitometric analysis of bands was also performed. (B) K562 cells, stably expressing the indicated plasmids (shSCR or shMAPK15), were treated with 100 nM Baf (1 h), and then cells were subjected to immunofluorescence analysis. In these representative images, LC3B is visualized in green and DAPI-stained nuclei in blue. LC3B positive dots were counted using a specific protocol by Volocity software (see graph on the right). Scale bars, 7.5 μm.
Localization of BCR-ABL1 to autophagic vesicles is mediated by the ability of MAPK15 to interact with LC3-family proteins, in an LIR-dependent manner
Since MAPK15 is localized to autophagic vesicles thanks to LC3-interacting region (LIR)-dependent interactions,19 we investigated whether this domain was also necessary for BCR-ABL1 localization to autophagic vesicles. Indeed, BCR-ABL1 colocalized with LC3B-positive dots in cells expressing wild-type (WT) MAPK15 (Fig. 5A), whereas no localization of BCR-ABL1 was observed in cells expressing a LIR-defective MAPK15 mutant (MAPK15AXXA; Fig. 5B). To confirm the formation of a complex between BCR-ABL1 and LC3B, mediated by MAPK15 and its LIR motif, we next precipitated a GST-fused BCR-ABL1 isoform by affinity purification together with WT and LIR-defective MAPK15 and evaluated endogenous LC3B coprecipitation. Interestingly, BCR-ABL1 was able to precipitate LC3B only in presence of WT MAPK15 whereas the expression of the MAPK15AXXA mutant completely prevented LC3 precipitation by BCR-ABL1 (Fig. 5C and Fig. S5). Based on these results, we also investigated whether correct localization of BCR-ABL1 to autophagic vesicles was required for oncoprotein-induced autophagy. Hence, we evaluated autophagic flux and autophagosome amount in WT MAPK15- and MAPK15AXXA-expressing cells. These experiments highlighted the ability of the MAPK15 LIR mutant, which prevented BCR-ABL1 relocalization on autophagic vesicles, to strongly inhibit BCR-ABL1-induced autophagy and autophagic flux, as scored by accumulation of LC3B-II protein (Fig. 5D) and by the decreased number of autophagosomes (Fig. 5E). Altogether, these data demonstrated that BCR-ABL1 is localized on autophagic vesicles by MAPK15-dependent interaction with LC3B and that such localization is required for BCR-ABL1 induction of autophagy.
Figure 5.

Localization of BCR-ABL1 to autophagic vesicles is mediated by the ability of MAPK15 to interact with LC3-family proteins, in an LIR-dependent manner. (A-B) GFP-LC3B HeLa cells were transfected with BCR-ABL1 and WT HA-MAPK15 or HA-MAPK15AXXA plasmids and then subjected to immunofluorescence analysis. GFP-LC3 is visualized in green, BCR-ABL1 in red, MAPK15 in cyan, and DAPI-stained nuclei in blue. In these representative images, white arrows indicate colocalization spots. Scale bars, 10 μm. (C) 293T cells were transfected with the indicated plasmids (empty vector, GST, GST-BCR-ABL, BCR-ABL, WT HA-MAPK15 or HA-MAPK15AXXA) and harvested after 24 h. Total lysates were then subjected to GST affinity purification and analyzed by western blot. Similar results were obtained in 3 independent experiments (n = 3). (D) HeLa cells were transfected with BCR-ABL1, WT HA-MAPK15, HA-MAPK15AXXA or control plasmids. After 24 h, cells were treated with 100 nM Baf for 1 h and total lysates were harvested for western blotting analysis. One experiment, representative of 3 independent experiments is shown. Densitometric analysis of bands was also performed. (E) GFP-LC3B Hela cells were transfected with BCR-ABL, HA-MAPK15AXXA or control plasmids. After 48 h, cells were treated with 100 nM Baf for 1 h and fixed. Then cells were immunolabeled for BCR-ABL1 and MAPK15. Positive cells for BCR-ABL1, MAPK15 or both were then analyzed for the number of LC3B dots using a specific protocol by Volocity software. The accompanying histogram was obtained by analyzing at least 400 cells/sample from 3 different experiments (n = 3).
MAPK15 catalytic activity is required for BCR-ABL1-dependent induction of autophagy
Over the past decade, protein kinases have become the pharmaceutical industry’s most popular drug targets, especially in the field of cancer. Consequently, based on the potential role of MAPK15 in autophagy and cancer,19,21,34-36 we decided to investigate whether its enzymatic activity was required for BCR-ABL1-induced autophagy. Thus, we overexpressed the kinase inactive mutant of MAPK15 (MAPK15KD, Asp154Ala (D154A) mutation) in BCR-ABL1-positive cells and monitored the expression levels of autophagic markers such as LC3-II and SQSTM1. In these settings, the MAPK15KD mutant reduced both basal and BCR-ABL1-induced autophagy (Fig. 6A). A further control of autophagy constraint by MAPK15KD came from analysis of the autophagic flux. Indeed, this analysis evidenced that this mutant reduced flux of both basal and BCR-ABL1-induced autophagy (Fig. 6B). Moreover, we monitored autophagy in BCR-ABL1-expressing cells treated with the MAPK15 kinase inhibitor, Ro318220.18,19 Also in this case, inhibition of MAPK15 catalytic activity led to a reduction of BCR-ABL1-induced autophagy (Fig. 6C) and autophagic flux of both basal and BCR-ABL1-induced autophagy (Fig. 6D). Altogether, these data indicate that MAPK15 kinase activity is necessary to mediate BCR-ABL1-dependent autophagy.
Figure 6.
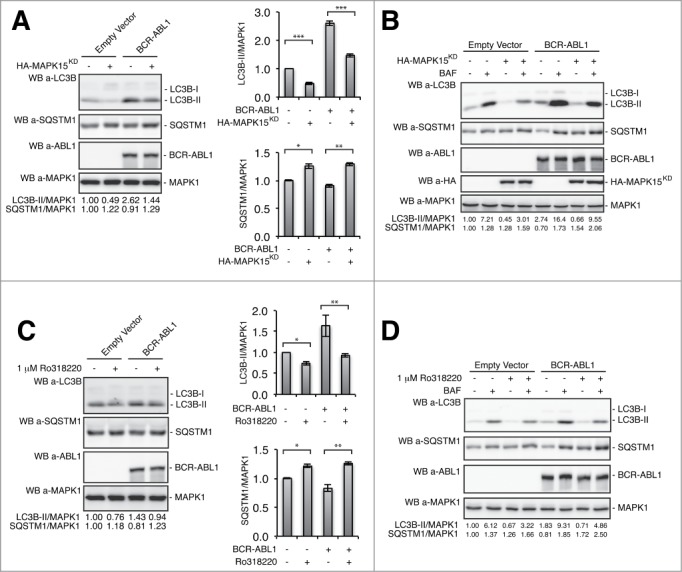
MAPK15 catalytic activity is required for BCR-ABL1-dependent induction of autophagy. (A) HeLa cells were transfected with BCR-ABL1 and HA-MAPK15KD plasmids. After 24 h, total lysates were collected for western blotting analysis. Representative western blot shows levels of the LC3B and SQSTM1 autophagic markers in cells expressing BCR-ABL1 with or without MAPK15KD. Densitometric analysis of bands was also performed. Histograms show means ± SD of densitometric analysis for LC3B-II, SQSTM1 and MAPK1 from 3 independent experiments (n = 3). (B) HeLa cells were transfected with BCR-ABL1, HA-MAPK15KD or control plasmids. After 24 h, cells were treated with 100 nM Baf for 1 h and total lysates were harvested for western blotting analysis. One experiment, representative of 3 independent experiments is shown (n = 3). Densitometric analysis of bands was also performed. (C) HeLa cells were transfected with BCR-ABL1 or control plasmids. After 24 h, cells were treated with 1uM Ro318220 for 1 h and total lysates were collected for western blotting analysis. Similar results were obtained in 3 independent experiments. Densitometric analysis of bands was also performed. Charts show means ± SD of densitometric analysis for LC3B-II, SQSTM1, and MAPK1 from 3 independent experiments (n = 3). (D) HeLa cells were transfected with BCR-ABL1 or control plasmids. After 24 h, cells were treated with 1uM Ro318220 and 100 nM Baf for 1 h, as indicated, and total lysates were harvested for western blotting analysis. Similar results were obtained in 3 independent experiments (n = 3). Densitometric analysis of bands was also performed.
MAPK15 depletion inhibits BCR-ABL1-dependent cell proliferation and in vivo tumor formation
BCR-ABL1 expression confers cells the ability to proliferate in the absence of growth signals and to escape apoptosis.6,37 Indeed, we confirmed the ability of this oncogene to increase proliferation also in our model system, HeLa cells (Fig. S6). Based on the role of MAPK15 in autophagy induced by BCR-ABL1 and on the already demonstrated role of this cellular process in cell survival and leukemogenesis controlled by this oncogene,9,15 we next asked whether MAPK15 may have a role in BCR-ABL1-dependent cell proliferation. Using a MAPK15 depletion approach (see above), we indeed demonstrated that interfering for the expression of the endogenous MAP kinase caused a significant reduction in HeLa cell proliferation induced by BCR-ABL1 overexpression (Fig. 7A), without affecting apoptosis in these cells (Fig. S7).
Figure 7.

MAPK15 depletion inhibits BCR-ABL1-dependent cell proliferation and transformation. (A) HeLa cells were transfected with SCR siRNA or MAPK15 specific siRNA. After 8 h cells were transfected with BCR-ABL1 or control plasmids. After further 64 h, cells were harvested and counted. (B) HeLa cells were transfected with SCR siRNA or MAPK15 specific siRNA. After 24 h cells were transfected with BCR-ABL1 or control plasmids. Cells were then assessed for anchorage-independent growth by seeding them into agar medium. After 14 d, colonies were counted. All quantitative data shown represent the means ± SD of 3 independent experiments (n = 3). (C) K562 cells, stable for the indicated shRNA-encoding plasmids, were seeded in 6-well plates at 2 × 105 cells per well in triplicate. After 72 h, cells were harvested and counted. One-way ANOVA test comparing each shSCR cell line with each shMAPK15 cell line resulted in a P value significance lower than 0.001 (***). (D) Growth curves of tumors generated by K562 cells stably expressing 2 independent shRNA SCR (#509 and #543) and 2 independent shRNA for MAPK15 (#547 and #549), after injection on the flank of athymic nude-FOXN1nu mice. Each point represents the mean volume ± SEM of 10 tumors. One-way ANOVA test for tumor volume at time points of 13, 16, 19 d comparing each shSCR xenograft group with each shMAPK15 xenograft group resulted in a P value significance lower than 0.001 (***).
Neoplastic transformation occurs via a series of genetic and epigenetic alterations that give rise to a cell population capable of proliferating independently of both external and internal signals. Anchorage-independent growth is one of the hallmarks of cell transformation and in vitro assays monitoring this phenomenon are considered among the most accurate and stringent for detecting malignant transformation of cells.38 Indeed, we performed soft-agar assays with HeLa cells overexpressing BCR-ABL1 and, demonstrated that the oncogene increased the number of transformed colonies (Fig. S8). To establish a role for MAPK15 in BCR-ABL1-dependent cellular transformation, we therefore evaluated colony formation ability demonstrating that MAPK15 depletion caused a reduction in the number of anchorage-independent colonies from BCR-ABL1-expressing cells (Fig. 7B).
To confirm that MAPK15 is necessary for BCR-ABL1 biological effects in a system expressing this oncogene at physiologically relevant levels, we next evaluated cell proliferation of K562 CML cells depleted for MAPK15 expression. Indeed, K562 cells expressing 2 different MAPK15 specific shRNA (#547 and #549) showed significantly reduced proliferation rates as compared to K562 cells expressing 2 independent control shRNAs (scrambled, SCR, #509 and #543) (Fig. 7C).
Ultimately, K562-dependent tumor formation was assessed, in vivo, by a xenograft approach in nude mice. Two lines for K562 expressing control shRNAs (SCR, #509 and #543) and 2 for K562 cells expressing MAPK15 specific shRNAs (#547 and #549) were inoculated in athymic nude-FOXN1nu/nu mice and tumor growth was monitored over a time span of approximately 3 wk. Tumor growth rates in the 4 experimental groups significantly diverged early on during the period of observation, with most of the inoculi generated with MAPK15-depleted K562 cells being completely unable to form appreciable tumors at the endpoint of the experiment (Fig. 7D). Overall, our data therefore demonstrated a key role of the endogenous MAPK15 protein in mediating BCR-ABL1-dependent transformation signals, both in a model system (HeLa cells) exogenously expressing BCR-ABL1 and in a physiologically relevant model for CML, i.e. K562 cells endogenously expressing the BCR-ABL1 human oncogene.
MAPK15-dependent regulation of autophagy is necessary for BCR-ABL1-dependent cell proliferation and tumor formation
Based on previously described results, we finally aimed at demonstrating that MAPK15-dependent autophagy was indeed crucial for BCR-ABL1-dependent cell proliferation and tumor formation in K562 cells, in which a pharmacological inhibitor of autophagy, chloroquine, has already been demonstrated to inhibit cell proliferation even when associated to low doses of imatinib.12 For this, we took advantage of the previously described LIR-defective and autophagy-incompetent MAPK15AXXA mutant, that nonetheless maintains its kinase activity.19 We, therefore, generated K562 cells stably expressing this mutant and, as additional controls, an empty vector, WT MAPK15 and a MAPK15 kinase dead mutant (MAPK15KD). Their proliferation rate was evaluated by Trypan blue exclusion assay, demonstrating that both the MAPK15AXXA and the MAPK15KD mutants caused, respectively, a 43% and 58% decrease in cell amount compared to WT MAPK15 (Fig. 8A).
Figure 8.
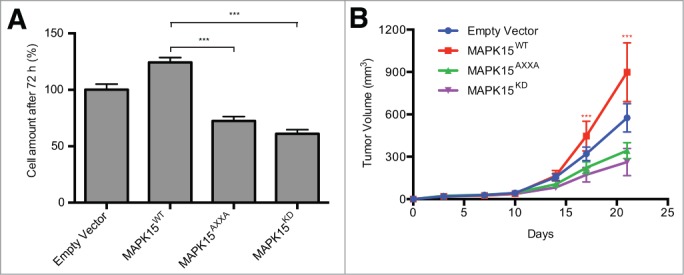
MAPK15-dependent autophagy controls BCR-ABL1-dependent cell proliferation and tumor formation. (A) K562 cells stably expressing empty vector, WT MAPK15, MAPK15AXXA and MAPK15KD were seeded in 6-well plates at 2 × 105 cells per well in triplicate. After 72 h, cells were harvested and counted. (B) Growth curves of tumors generated by K562 cells stably expressing empty vector, WT MAPK15, MAPK15AXXA and MAPK15KD, after injection on the flank of athymic nude-FOXN1nu mice. Each point represents the mean volume ± SEM of 10 tumors. One-way ANOVA test for tumor volume at time points of 17 and 21 d comparing each MAPK15 mutant xenograft group (MAPK15AXXA and MAPK15KD) with WT MAPK15 xenograft group resulted in a P value significance lower than 0.001 (***).
Ultimately, to evaluate the role of autophagy controlled by MAPK15 in endogenous BCR-ABL1-dependent in vivo tumor formation, we injected K562 cells stably expressing empty vector and WT, AXXA, and KD MAPK15 mutants, in athymic nude-FOXN1nu/nu mice and monitored their tumor growth over a time span of 3 wk. At the endpoint of observations, the MAPK15AXXA mutant (with impaired autophagic activity) and the MAPK15KD (with impaired kinase and autophagic activity) mutant significantly affected K562-dependent tumor growth, as compared to WT MAPK15 (Fig. 8B). Altogether these data, therefore, demonstrated that the ability of MAPK15 to control the autophagic process is necessary for mediating BCR-ABL1-dependent oncogenic effects. Importantly, they also confirm that targeting MAPK15 kinase activity impairs the oncogenic potential of BCR-ABL1 in CML cells suggesting this MAP kinase as a novel pharmacological target for the therapy of human CML.
Discussion
Autophagy has been demonstrated as necessary for BCR-ABL1-induced leukemogenesis.7,9,10 In this study, we show that MAPK15 mediated BCR-ABL1-induced autophagy and that this signaling pathway is necessary for proliferation and transformation sustained by this human oncogene (Fig. 9). At the molecular level, BCR-ABL1 interacted with and activated MAPK15 and their binding promoted BCR-ABL1 relocalization to autophagic vesicles. Furthermore, MAPK15 depletion or pharmacological inhibition limited BCR-ABL1-induced autophagy, supporting the possibility that specific MAPK15 inhibitors may be beneficial for the therapy of human CML. Although, at the moment, we have no experimental evidence sustaining a similar role for this MAP kinase in controlling autophagy by other human oncogenes, available data from our laboratory suggest that MAPK15 mediates activation of autophagy also by starvation, while rapamycin, another typical stimulus, seems not to affect its activity.19 Our hypothesis is, therefore, that MAPK15 will likely transduce autophagy signals by other stimuli, possibly oncogenes, and we are currently investigating this issue for its potentially general importance on cellular transformation and human cancer.
Figure 9.

MAPK15 mediates BCR-ABL1-induced autophagy and cellular transformation. (A) BCR-ABL1 interacts with MAPK15 inducing autophagy, resulting in cell proliferation and transformation. (B) In cells depleted for MAPK15 expression or kinase activity (or even expressing a LIR-deficient mutant, MAPK15AXXA), BCR-ABL1 is unable to trigger autophagy, cell proliferation, and transformation.
It is interesting to notice that all BCR-ABL1-expressing CML cell lines analyzed expressed MAPK15 at very high levels, whereas immortalized T lymphocyte cells and other BCR-ABL1 negative leukemic cells showed much lower MAPK15 expression (Fig. S2). A possible explanation for this result is that the BCR-ABL1 oncogene itself is able to control MAPK15 expression, which, in turn, actively participates in the leukemogenic process, thanks to its ability to control autophagy, as demonstrated in this report. Moreover, besides BCR-ABL1, other human oncogenes with constitutive tyrosine kinase activity have been reported to control MAPK15 activity,17,20 while no data is currently available regarding the ability of these oncogenes to control its expression. Still, as this MAP kinase is devoid of upstream MEKs (ref. 22 and our observations), and it can be controlled at the level of expression and/or stability by different stimuli,18,19,39-41 we are now actively pursuing the idea that expression of the kinase is under the control of specific oncogenic signaling pathways that are, in turn, able to take advantage of MAPK15 ability to stimulate autophagy and, consequently, cell proliferation, and tumor formation.
Interestingly, the BCR-ABL1 oncoprotein has been extensively localized to the cytoplasm and to the nucleus,24,25 although only recently an F-actin binding domain in BCR-ABL1 has been identified and its localization on the cytoskeleton has been intensely investigated.26 Simple disruption of the BCR-ABL1 F-actin binding domain causes loss of cytoskeletal distribution and relocalization to cytoplasmatic puncta of BCR-ABL1.26 A similar distribution of BCR-ABL1 has been also shown in leukemic cells upon chemotherapeutic treatment, and such BCR-ABL1 cytoplasmic dots have been recognized as autophagosomes.42,43 Here, we confirm that BCR-ABL1 is localized on autophagic vesicles by demonstrating its colocalization with 2 previously described markers, MAPK15 and LC3B.19 In addition, we also established that BCR-ABL1 localization to autophagic vesicles is dependent on its interaction with MAPK15 and on the presence, in MAPK15, of an intact LIR motif. Indeed, overexpression of a LIR-defective mutant of MAPK15 prevented BCR-ABL1 localization to LC3B-positive structures. It is therefore tempting to speculate that MAPK15, while transducing signals upon BCR-ABL1 activation, may also be important for specific localization of this oncoprotein and, possibly, of the wild-type ABL1 protein, on autophagic vesicles. This, in turn, might allow BCR-ABL1 to access specific sets of autophagic vesicle-localized proteins, strongly sustaining the recent interest in autophagy for the treatment of CML.
While the role of autophagy in BCR-ABL1-dependent leukemogenesis is now established, the molecular mechanisms by which the leukemic driver controls this process are still mostly unclear. In particular, few papers adopt genetic approaches to investigate the direct effect of BCR-ABL1 on autophagy and try to dissect downstream signals to the autophagic process. Conversely, here we show that ectopic addition of the BCR-ABL1 oncogene could affect cell autophagic rates (Fig. 2 and Fig. 3) and that depletion of endogenous MAPK15 inhibits this effect, even in a physiologically relevant system in which the oncogene is endogenously expressed. Interestingly, the use of a MAPK15 mutant specifically devoid of a LIR domain necessary for autophagy regulation but still proficient for kinase activity,19 strongly interfered with the ability of BCR-ABL1 i) to stimulate autophagy, ii) to localize to autophagic vesicles, iii) to stimulate cell proliferation and, iv) to sustain tumor formation in vivo, supporting a key role of MAPK15-dependent autophagy in CML. Importantly, our data confirm previous observations describing leukemogenesis as a process highly dependent on autophagy.9,15 Altogether, our hypothesis is that BCR-ABL1 encodes for a constitutive active tyrosine kinase that controls the autophagic process in a MAPK15-dependent fashion, lowering cell stress and allowing leukemogenesis. Based on this, we predict that inhibition of autophagy by pharmacologically acting on MAPK15 might have therapeutic effects on CML, in association with specific BCR-ABL1 kinase inhibitors. In this context, the identification of MAPK15 as a new target for the development of small drug inhibitors,44 able to affect CML through regulation of the autophagic process elicited by BCR-ABL1, surely deserves attention and further investigation.
Materials and Methods
Reagents and antibodies
Bafilomycin A1 (Baf) (Santa Cruz Biotechnology, sc-201550) and Ro318220 (VWR International, 557521–500) were dissolved in DMSO. Hank's medium (H15–010), used as starvation medium, was obtained by PAA. The following primary antibody was used for immunoprecipitations: anti-HA (Covance, MMS-101R). For western blots, the following primary antibodies were used: anti-MAPK15 (custom preparation),19,29,44 anti-HA (Covance, MMS-101R), anti-LC3B (Nanotools, 0231–1000), anti-phospho-MAPK1/3 (Thr202/Tyr204) (Cell Signaling Technology, 9101), anti-SQSTM1/p62 (BD Biosciences, 610833), anti-ABL1 (BD Pharmingen, 554148) anti-MAPK1 (Santa Cruz Biotechnology, sc-154). For confocal microscopy experiments, the following primary antibodies were used: anti-MAPK15 (custom preparation),19,29,44 anti-HA (Santa Cruz Biotechnology, sc-7392), anti-LC3B (MBL, M152–3), anti-ABL1 (BD PharMingen, 554148), anti-ABL1 (Santa Cruz Biotechnology, sc-131). The following secondary antibodies were used for western blot experiments: anti-mouse (Santa Cruz Biotechnology, sc-2004) and anti-rabbit (Santa Cruz Biotechnology, sc-2005) HRP-conjugated IgGs. The following secondary antibodies were used for confocal microscopy experiments: anti-mouse Alexa Fluor 488-conjugated (Life Technologies, A21202), anti-rabbit Alexa Fluor 488-conjugated (Life Technologies, A21206), anti-mouse Alexa Fluor 555-conjugated (Life Technologies, A31570), anti-rabbit Alexa Fluor 555-conjugated (Life Technologies, A31572), anti-mouse Alexa Fluor 647-conjugated (Life Technologies, A31571), anti-rabbit Alexa Fluor 647-conjugated (Life Technologies, A31573).
Expression vectors
pCEFL-HA-MAPK15 and all its mutants have already been described.19,20 ABL1 encoding plasmids: pCEFL-c-ABL1, pCEFL-EGFP-ABL1, pCEFL-ABL1KD (kinase dead), pCEFL-BCR-ABL1 are already described.20 pcDNA4-Tet-On-BCR-ABL1 was generated by subcloning with EcoRI restriction sites the BCR-ABL1 gene from the MIG-210 BCR-ABL1 plasmid, kindly provided from Ricardo Sanchez Prieto (Universidad de Castilla-La Mancha),45 into the pcDNA4 Tet-inducible vector, kindly provided from Francesca Carlomagno (Università degli Studi di Napoli). The plasmid encoding for GST-tagged isoform of BCR-ABL1, pLEF GST-BCR-ABL1(p210) was obtained from Addgene (plasmid 38158, deposited by Nora Heisterkamp).46 pCEFL-EGFP-LC3B is already described.19 pGIPZ plasmids encoding for shRNA scrambled and shRNA MAPK15 were purchased from Open Biosystem (RHS4531-EG225689). pCEFL-MAPK15-IRES-GFP and all its mutants (AXXA; KD) were generated by subcloning the MAPK15 gene, excised from pCEFL-HA-MAPK15, upstream the IRES sequence, with BamHI and XbaI restriction sites.
Cell culture and transfections
293T and HeLa cells were maintained in DMEM (PAA, E15–009) supplemented with 10% fetal bovine serum (PAA, A15–151), 2 mM L-glutamine and 100 units/ml penicillin-streptomycin at 37°C in an atmosphere of 5% CO2/air. HeLa cells stably expressing GFP-LC3B were created transfecting HeLa cells with the previously described pCEFL GFP-LC3B plasmid,19 and selecting cells with 1 mg/ml G418 (Genespin, STS-G418). HeLa T-Rex BCR-ABL1 cells were created by transfecting HeLa T-Rex cells with the pcDNA4-Tet-On-BCR-ABL plasmid and selecting them with 200 ug/ml zeocin until cells were stabilized. HeLa T-Rex BCR-ABL1 cells were maintained in DMEM supplemented with 10% fetal bovine serum (Clonetech, 631106), 2 mM L-glutamine, 5 ug/ml blasticidin and 100 units/ml penicillin-streptomycin at 37°C in an atmosphere of 5% CO2/air. In HeLa T-Rex BCR-ABL1, the expression of the BCR-ABL1 protein is inducible by addition of doxycycline. HeLa cells stably expressing pCEFL and scrambled (SCR) shRNA, pCEFL and MAPK15 shRNA, BCR-ABL1 and SCR shRNA, or BCR-ABL1 and MAPK15 shRNA respectively were transfected with pCEFL empty vector, pCEFL-BCR-ABL, pGIPZ shSCR or pGIPZ shMAPK15 and selected for respective antibiotic resistances for G418 (1 mg/ml) and puromycin (2 ug/ml). The stably expressing cells were then plated at a limiting dilution in 96-well plates to obtain single cell clones.
293T and HeLa cells were maintained in RPMI 1640 (PAA, E15–039) supplemented with 10% fetal bovine serum (PAA, A15–151), 2 mM L-glutamine and 100 units/ml penicillin-streptomycin at 37°C in an atmosphere of 5% CO2/air. K562 cells stably expressing shSCR and shMAPK15 were generated by electroporating cells with respective shRNAs and by selecting with Puromycin (2 ug/ml) whereas K562 expressing WT as well as the AXXA and KD MAPK15 mutants were generated by electroporating cells with the above-described corresponding plasmids and selected with G418 (1 mg/ml).
For immunofluorescence experiments and western blot analysis, 5 × 104 cells were seeded in 12-well plates (2 × 105 cells in 6-well plates) and transfected with 200 ng (500 ng in 6-well plates) of each expression vector, using Lipofectamine LTX (Life Technologies, 15338500). All experiments were performed, unless specified, 24 h after transfection. For confocal microscopy experiments, 2.5 × 104 cells were seeded on coverslips placed in 12-well plates. Each sample was transfected with 200 ng of each plasmid using Lipofectamine LTX.
RNA interference
MAPK15-specific siRNA (target sequence for MAPK15 #01 siRNA 5′-GCTTGGAGGCTACTCCC-3′, for MAPK15 #02 siRNA 5′-GACAGATGCCCAGAGAACA-3′, for MAPK15 #03 siRNA 5′-CCTGGTGTTTGAGTTTATG-3′) and nonsilencing siRNA (scrambled, SCR; target sequence 5′-AATTCTCCGAACGTGTCACGT-3′) were obtained from Qiagen. HeLa cells were transfected with siRNA at a final concentration of 5 nM using HiPerFect (Qiagen, 301707), according to the manufacturer’s instructions. Samples were analyzed, unless specified, 72 h after transfection.
Western blots
Total lysates were obtained by resuspending washed cellular pellet fractions in MAPK lysis buffer (20 mM HEPES, pH 7.5, 10 mM EGTA, 40 mM β-glycerophosphate, 1% NP-40 (Sigma Aldrich, I3021), 2.5 mM MgCl2, 2 mM orthovanadate, 2 mM NaF, 1 mM DTT, Roche protease inhibitors cocktail (Roche Diagnostics, 05056489001). Proteins were quantified by the Bradford assay and, before loading, Laemmli 5X was added to the proteins which were incubated for 5 min at 95°C. Then, proteins were loaded on SDS-PAGE poly-acrylamide gel, transferred to Immobilon-P PVDF membrane (Millipore, IPVH00010), probed with the appropriate antibodies, and detected by enhanced chemoluminescence (ECL Prime; GE Healthcare, RPN2232). Images were then acquired with a LAS 4000 imager (GE Healthcare, Milan, Italy). Densitometric analysis of western blots was performed with NIH ImageJ 1.43u (National Institutes of Health).
Coimmunoprecipitations
Whole cell lysates were obtained by resuspending washed pellet fractions in the above-mentioned MAPK lysis buffer. Lysates were incubated with appropriate antibodies for 2 h at 4°C. Then, immunocomplexes were purified by incubating the lysates for 45 min with protein G Mag Sepharose Xtra (GE Healthcare, 28–9670–70). After 5 washes, the immunocomplexes were resuspended in 2X Laemmli buffer and subjected to western blot analysis. For endogenous coimmunoprecipitation experiments, 5 mg of K562 cell lysates were used.
Affinity purifications
Whole cell lysates were obtained by resuspending washed pellet fractions in the above-mentioned MAPK lysis buffer. Lysates were incubated with Glutathione Magnetic Beads (Pierce, 88822) for 2 h at 4°C. After 5 washes, protein-beads complexes were resuspended in 2X Laemmli buffer and subjected to western blot analysis.
Immunofluorescence (IF)
Cells were washed with phosphate-buffered saline (PBS; Oxoid, BR0014G), then fixed with 4% paraformaldehyde in PBS for 20 min and permeabilized with 0.2% Triton X-100 (Sigma Aldrich, T8787) solution or 100 μg/ml digitonin solution (Life Technologies, BN2006) for 20 min, as indicated. Cells were incubated with the appropriate primary antibodies for 1 h, washed 3 times with PBS, and then incubated for 30 min with appropriate Alexa Fluor 488-conjugated (Life Technologies, A21202), Alexa Fluor 555-conjugated (Life Technologies, A31272) or Alexa Fluor 647-conjugated (Life Technologies, A21245) secondary antibodies and then washed again 3 times in PBS. Nuclei were stained with a solution of 1.5 μM of 4′,6-diamidino-2-phenylindole (DAPI; Sigma Aldrich, D9542) in PBS for 5 min. Coverslips were mounted in Fluorescence Mounting Medium (Dako, S3023). Samples were visualized on a TSC SP5 confocal microscope (Leica Microsystems, Germany, Mannheim) installed on an inverted LEICA DMI 6000CS microscope (Leica Microsystems, Germany, Mannheim) and equipped with an oil immersion PlanApo 63X 1.4 NA objective. Images were acquired using the LAS AF acquisition software (Leica Microsystems).
Anchorage-independent growth
HeLa cells were transient transfected with the indicated DNA plasmid or siRNA. After recovery, cells were seeded at 300 cells per 9-cm dish in DMEM, supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 units/ml penicillin-streptomycin and 0.35% agar, on a more dense layer of the same medium (0.5% agar). The cultures were maintained in a 37°C, 5% CO2 incubator for 14 d. Colonies were stained with 4 mg/ml iodonitrotetrazolium chloride (Sigma Aldrich, I8377) and scored using a microscope.
Cell count
Briefly, cells were seeded in 6-well plates at 2 × 105 cells per well in triplicate. Then cells were transfected and 72 h post transfection cell number was determined by counting the viable cells in a hemocytometer by trypan blue dye exclusion assay.
Cell cycle analysis
Cells were harvested and counted. Then, 1 × 106 cells were pelleted and resuspended in 1 ml Nicoletti solution (propidium iodide 50 mg/liter in 0.1% sodium citrate plus 0.1% Triton X-100). Cells were incubated in the dark at 4°C for 60 min and analyzed in a flow cytometer (FACSCanto II, BD Biosciences, Italy, Milan).
Xenografts
All protocols involving animals have been approved by the internal Animal Welfare Body and by Italian Ministero della Salute. For each injection, 5 × 106 K562 cells were resuspended in 100 μl PBS. Six-week-old athymic nude female mice (Harlan Laboratories) were injected in both flanks using a 21-gauge needle. The mice were anesthetized with isoflurane throughout the procedure. Five mice were injected on both flanks, for a total of 10 xenograft tumors per group. The animals were then monitored for tumor growth and tumor size was measured twice a week with a caliper (2Biological Instruments). Tumor volumes were calculated using the formula V = W2 × L × 0.5, where W and L are tumor width and length, respectively.
Dot count, Pearson correlation, and statistical analysis
For the LC3B-positive dot count, we performed intensitometric analysis of fluorescence using the Quantitation Module of Volocity software (PerkinElmer Life Science). Pearson correlation was also measured by the Quantitation Module of Volocity software. Dot count and colocalization rate were subjected to statistical analysis. Measures were obtained by analyzing at least 400 cells/sample from 3 different experiments. Significance (P value) was assessed by one-way ANOVA test. Asterisks were attributed for the following significance values: P < 0 .05 (*), P < 0 .01 (**), P < 0 .001 (***).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Ricardo Sánchez-Prieto (Universidad de Castilla-La Mancha, CRIB/UCLM, Albacete, Spain) and Francesca Carlomagno (Università degli Studi di Napoli Federico II, Italy) for plasmids and Toscana Life Sciences Foundation for providing highly performing technological platforms.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by a start-up grant from Regione Toscana, by the Italian Association for Cancer Research (AIRC) and by a grant to Regione Toscana from Ministero della Salute, Italy, in the context of the “Programma per la Ricerca Sanitaria Finalizzata 2008.”
References
- 1.Rudkin CT, Hungerford DA, Nowell PC. DNA contents of chromosome Ph1 and chromosome 21 in human chronic granulocytic leukemia. Science 1964; 144:1229-31; PMID:14150328; http://dx.doi.org/ 10.1126/science.144.3623.1229 [DOI] [PubMed] [Google Scholar]
- 2.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243:290-3; PMID:4126434; http://dx.doi.org/ 10.1038/243290a0 [DOI] [PubMed] [Google Scholar]
- 3.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985; 315:550-4; PMID:2989692; http://dx.doi.org/ 10.1038/315550a0 [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nature Med 1996; 2:561-6; PMID:8616716; http://dx.doi.org/ 10.1038/nm0596-561 [DOI] [PubMed] [Google Scholar]
- 5.Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci U S A 1988; 85:9312-6; PMID:3143116; http://dx.doi.org/ 10.1073/pnas.85.23.9312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood 2000; 96:3343-56; PMID:11071626 [PubMed] [Google Scholar]
- 7.Helgason GV, Karvela M, Holyoake TL. Kill one bird with two stones: potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood 2011; 118:2035-43; PMID:21693757; http://dx.doi.org/ 10.1182/blood-2011-01-330621 [DOI] [PubMed] [Google Scholar]
- 8.Savona MR, Saglio G. Identifying the time to change BCR-ABL inhibitor therapy in patients with chronic myeloid leukemia. Acta Haematol 2013; 130:268-78; PMID:23949495; http://dx.doi.org/ 10.1159/000353163 [DOI] [PubMed] [Google Scholar]
- 9.Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, Ilkayeva O, Jia W, He YW, Rathmell JC. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30:1855-67; PMID:21151168; http://dx.doi.org/ 10.1038/onc.2010.561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekiz HA, Can G, Baran Y. Role of autophagy in the progression and suppression of leukemias. Crit Rev Oncol Hematol 2012; 81:275-85; PMID:21612942; http://dx.doi.org/ 10.1016/j.critrevonc.2011.03.009 [DOI] [PubMed] [Google Scholar]
- 11.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007; 110:313-22; PMID:17363733; http://dx.doi.org/ 10.1182/blood-2006-10-050260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mishima Y, Terui Y, Mishima Y, Taniyama A, Kuniyoshi R, Takizawa T, Kimura S, Ozawa K, Hatake K. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci 2008; 99:2200-8; PMID:18823378; http://dx.doi.org/ 10.1111/j.1349-7006.2008.00932.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamitsuji Y, Kuroda J, Kimura S, Toyokuni S, Watanabe K, Ashihara E, Tanaka H, Yui Y, Watanabe M, Matsubara H, et al.. The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and different modes of cell death execution in Bcr-Abl-positive leukemias. Cell Death Differ 2008; 15:1712-22; PMID:18617896; http://dx.doi.org/ 10.1038/cdd.2008.107 [DOI] [PubMed] [Google Scholar]
- 14.Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, et al.. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009; 119:1109-23; PMID:19363292; http://dx.doi.org/ 10.1172/JCI35660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crowley LC, Elzinga BM, O'Sullivan GC, McKenna SL. Autophagy induction by Bcr-Abl-expressing cells facilitates their recovery from a targeted or nontargeted treatment. Am J Hematol 2011; 86:38-47; PMID:21132731; http://dx.doi.org/ 10.1002/ajh.21914 [DOI] [PubMed] [Google Scholar]
- 16.Zhu S, Cao L, Yu Y, Yang L, Yang M, Liu K, Huang J, Kang R, Livesey KM, Tang D. Inhibiting autophagy potentiates the anticancer activity of IFN1@/IFNα in chronic myeloid leukemia cells. Autophagy 2013; 9:317-27; PMID:23242206; http://dx.doi.org/ 10.4161/auto.22923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe MK, Saelzler MP, Espinosa R, Kahle KT, Hershenson MB, Le Beau MM, Rosner MR. ERK8, a new member of the mitogen-activated protein kinase family. J Biol Chem 2002; 277:16733-43; PMID:11875070; http://dx.doi.org/ 10.1074/jbc.M112483200 [DOI] [PubMed] [Google Scholar]
- 18.Klevernic IV, Martin NM, Cohen P. Regulation of the activity and expression of ERK8 by DNA damage. FEBS Lett 2009; 583:680-4; PMID:19166846; http://dx.doi.org/ 10.1016/j.febslet.2009.01.011 [DOI] [PubMed] [Google Scholar]
- 19.Colecchia D, Strambi A, Sanzone S, Iavarone C, Rossi M, Dall'Armi C, Piccioni F, Verrotti di Pianella A, Chiariello M. MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy 2012; 8:1724-40; PMID:22948227; http://dx.doi.org/ 10.4161/auto.21857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iavarone C, Acunzo M, Carlomagno F, Catania A, Melillo RM, Carlomagno SM, Santoro M, Chiariello M. Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the RET proto-oncogene. J Biol Chem 2006; 281:10567-76; PMID:16484222; http://dx.doi.org/ 10.1074/jbc.M513397200 [DOI] [PubMed] [Google Scholar]
- 21.Xu YM, Zhu F, Cho YY, Carper A, Peng C, Zheng D, Yao K, Lau AT, Zykova TA, Kim HG, et al.. Extracellular signal-regulated kinase 8-mediated c-Jun phosphorylation increases tumorigenesis of human colon cancer. Cancer Res 2010; 70:3218-27; PMID:20395206; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4306 [DOI] [PubMed] [Google Scholar]
- 22.Abe MK, Kuo WL, Hershenson MB, Rosner MR. Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol Cell Biol 1999; 19:1301-12; PMID:9891064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayer BJ, Baltimore D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol Cell Biol 1994; 14:2883-94; PMID:8164650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wetzler M, Talpaz M, Van Etten RA, Hirsh-Ginsberg C, Beran M, Kurzrock R. Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation. J Clin Invest 1993; 92:1925-39; PMID:8408645; http://dx.doi.org/; http://dx.doi.org/ 10.1172/JCI116786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon AS, Constance JE, Tanaka T, Rabbitts TH, Lim CS. Changing the subcellular location of the oncoprotein Bcr-Abl using rationally designed capture motifs. Pharm Res 2012; 29:1098-109; PMID:22183511; http://dx.doi.org/ 10.1007/s11095-011-0654-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hantschel O, Wiesner S, Güttler T, Mackereth CD, Rix LL, Mikes Z, Dehne J, Görlich D, Sattler M, Superti-Furga G. Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell 2005; 19:461-73; PMID:16109371; http://dx.doi.org/ 10.1016/j.molcel.2005.06.030 [DOI] [PubMed] [Google Scholar]
- 27.Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med 2001; 7:228-34; PMID:11175855; http://dx.doi.org/ 10.1038/84683 [DOI] [PubMed] [Google Scholar]
- 28.Preyer M, Vigneri P, Wang JY. Interplay between kinase domain autophosphorylation and F-actin binding domain in regulating imatinib sensitivity and nuclear import of BCR-ABL. PLoS One 2011; 6:e17020; PMID:21347248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossi M, Colecchia D, Iavarone C, Strambi A, Piccioni F, Verrotti di Pianella A, Chiariello M. Extracellular signal-regulated kinase 8 (ERK8) controls estrogen-related receptor α (ERRα) cellular localization and inhibits its transcriptional activity. J Biol Chem 2011; 286:8507-22; PMID:21190936; http://dx.doi.org/ 10.1074/jbc.M110.179523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drullion C, Trégoat C, Lagarde V, Tan S, Gioia R, Priault M, Djavaheri-Mergny M, Brisson A, Auberger P, Mahon FX, et al.. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis 2012; 3:e373; PMID:22898871; http://dx.doi.org/ 10.1038/cddis.2012.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, Guan JL. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010; 116:4806-14; PMID:20716775; http://dx.doi.org/ 10.1182/blood-2010-06-288589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortensen M, Watson AS, Simon AK. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011; 7:1069-70; PMID:21552009; http://dx.doi.org/ 10.4161/auto.7.9.15886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henrich M, Smith A, Kitt D, Errington M, Nguyen B, Traish M, Lannigan A. Extracellular Signal-Regulated Kinase 7, a Regulator of Hormone-Dependent Estrogen Receptor Destruction. Mol Cell Biol 2003; 23:5979-88; PMID:12917323; http://dx.doi.org/ 10.1128/MCB.23.17.5979-5988.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groehler AL, Lannigan DA. A chromatin-bound kinase, ERK8, protects genomic integrity by inhibiting HDM2-mediated degradation of the DNA clamp PCNA. J Cell Biol 2010; 190:575-86; PMID:20733054; http://dx.doi.org/ 10.1083/jcb.201002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerone MA, Burgess DJ, Naceur-Lombardelli C, Lord CJ, Ashworth A. High-throughput RNAi screening reveals novel regulators of telomerase. Cancer Res 2011; 71:3328-40; PMID:21531765; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2734 [DOI] [PubMed] [Google Scholar]
- 37.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990; 247:1079-82; PMID:2408149; http://dx.doi.org/ 10.1126/science.2408149 [DOI] [PubMed] [Google Scholar]
- 38.Wang LH. Molecular signaling regulating anchorage-independent growth of cancer cells. Mt Sinai J Med 2004; 71:361-7; PMID:15592654 [PubMed] [Google Scholar]
- 39.Hasygar K, Hietakangas V. p53- and ERK7-Dependent Ribosome Surveillance Response Regulates Drosophila Insulin-Like Peptide Secretion. PLoS Genet 2014; 10:e1004764; PMID:25393288; http://dx.doi.org/ 10.1371/journal.pgen.1004764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyatake K, Kusakabe M, Takahashi C, Nishida E. ERK7 regulates ciliogenesis by phosphorylating the actin regulator CapZIP in cooperation with Dishevelled. Nat Commun 2015; 6:6666; PMID:25823377; http://dx.doi.org/ 10.1038/ncomms7666 [DOI] [PubMed] [Google Scholar]
- 41.Zacharogianni M, Kondylis V, Tang Y, Farhan H, Xanthakis D, Fuchs F, Boutros M, Rabouille C. ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. EMBO J 2011; 30:3684-700; PMID:21847093; http://dx.doi.org/ 10.1038/emboj.2011.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, Altman JK, Platanias LC. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012; 120:3555-62; PMID:22898604; http://dx.doi.org/ 10.1182/blood-2012-01-402578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elzinga BM, Nyhan MJ, Crowley LC, O'Donovan TR, Cahill MR, McKenna SL. Induction of autophagy by Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am J Hematol 2013; 88:455-62; PMID:23440701; http://dx.doi.org/ 10.1002/ajh.23428 [DOI] [PubMed] [Google Scholar]
- 44.Strambi A, Mori M, Rossi M, Colecchia D, Manetti F, Carlomagno F, Botta M, Chiariello M. Structure prediction and validation of the ERK8 kinase domain. PLoS One 2013; 8:e52011; PMID:23326322; http://dx.doi.org/ 10.1371/journal.pone.0052011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aceves-Luquero CI, Agarwal A, Callejas-Valera JL, Arias-González L, Esparís-Ogando A, del Peso Ovalle L, Bellón-Echeverria I, de la Cruz-Morcillo MA, Galán Moya EM, Moreno Gimeno I, et al.. ERK2, but not ERK1, mediates acquired and "de novo" resistance to imatinib mesylate: implication for CML therapy. PLoS One 2009; 4:e6124; PMID:19568437; http://dx.doi.org/ 10.1371/journal.pone.0006124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kweon SM, Cho YJ, Minoo P, Groffen J, Heisterkamp N. Activity of the Bcr GTPase-activating domain is regulated through direct protein/protein interaction with the Rho guanine nucleotide dissociation inhibitor. J Biol Chem 2008; 283:3023-30; PMID:18070886; http://dx.doi.org/ 10.1074/jbc.M705513200 [DOI] [PubMed] [Google Scholar]
- 47.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991; 139:271-9; PMID:1710634; http://dx.doi.org/ 10.1016/0022-1759(91)90198-O [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.