

Abstract
Selective macroautophagy (hereafter autophagy) can eliminate large cytotoxic structures that are designated for degradation by autophagy receptors. In our recent paper, we showed that a key function of target-bound autophagy receptors is to activate the autophagy kinase, Atg1, via interactions with the scaffold protein Atg11. Our work thus reveals a mechanism by which target recognition coordinates the earliest steps in autophagosome biogenesis.
Keywords: Ape1, Atg1, Atg11, Atg19, Atg36, autophagosome formation, pexophagy, phagophore, selective autophagy
Selective autophagy is a cellular quality control process that can degrade large unwanted cytoplasmic structures such as protein aggregates and damaged organelles. Defects in the clearance of selective autophagy targets kill post-mitotic cells, such as neurons, due to the accumulation of cytotoxic targets in the absence of cell division.
Targets of selective autophagy are recognized by autophagy receptor proteins. Autophagy receptors link targets to a membrane structure called the phagophore by binding phagophore-associated Atg8-family proteins. Phagophores expand around targets, maturing into sealed vesicles called autophagosomes. Autophagosomes fuse with lysosomes, leading to target degradation and subsequent recycling of the resulting degradation products (e.g. amino acids).
Target degradation thus depends in part on the ready supply of phagophores. A fundamental unresolved question is whether the rate of phagophore synthesis is responsive to variations in target abundance. The Atg1 kinase controls early steps in phagophore formation and is thus well-positioned as a control node for phagophore production. Indeed, a related process called nonselective autophagy—in which starvation signals induce the proliferation of phagophores specialized to recycle bulk cytoplasm—relies on Atg1 stimulation to drive increased phagophore synthesis. However, it remains unclear whether Atg1 activity is regulated during selective autophagy, or if basal Atg1 activity drives a constant rate of phagophore synthesis sufficient for target elimination.
We hypothesized that autophagy receptors activate Atg1 to link target detection with phagophore initiation. To test this idea, we examined Atg1 activity in yeast, first confirming that fewer Atg1 molecules are active in fed cells than in cells treated with rapamycin (a starvation signal mimic). By further characterizing Atg1 activity in fed cells, we discovered that this “basal” activity of Atg1 is in fact dependent on the presence of a constitutive autophagy target, the aminopeptidase I precursor (prApe1) complex, and its autophagy receptor, Atg19.
To gain insight into the mechanism by which Atg19 and prApe1 control Atg1 activity, we affinity-purified Atg1 from fed yeast cells and identified copurified proteins by mass spectrometry. Strikingly, Atg1 was isolated as a complex that contains prApe1 and Atg19, as well as a third protein, Atg11. To delineate the function of Atg11, we isolated Atg1 from cells lacking Atg11, and found that in these cells Atg1 is both inactive and does not copurify with Atg19 or prApe1. These data suggest that Atg11 functions as a scaffold protein that allows the Atg19-prApe1 complex to control the activity of the Atg1 kinase.
To more rigorously test the idea that Atg11 enables receptor-bound targets to activate Atg1, we developed a cell-free assay for Atg1 activation that is amenable to reconstitution with purified components. Because recombinant Atg1 is refractory to purification, we constructed a chemical genetic reporter of endogenously expressed Atg1 in cell extracts. Briefly, we mutated the Atg1 ATP-binding pocket such that Atg1 alone among all kinases could use bulky ATP analogs to label its substrates. To validate this system, we showed that analog-sensitive Atg1 labels all known substrates and that its activity in extracts depends on both Atg11 and Atg19.
Using this system, we generated several lines of evidence showing that receptor-bound targets activate Atg1 using Atg11. First, purified Atg11 restores activity to extracts lacking Atg11, but not extracts that additionally lack Atg19. Second, affinity-purified Atg19 complexes activate Atg1 in extracts lacking both Atg11 and Atg19, but only when added with purified Atg11 and when prApe1 is present in the complex. Third, mutant versions of Atg11 and Atg19 unable to bind each other do not drive Atg1 activation. Finally, a second autophagy target, damaged peroxisomes, activates Atg1 both in vivo and when added to extracts in vitro, but only when the peroxisome autophagy receptor, Atg36, is present.
Overall, our work shows that receptors not only link targets to phagophores, but exert direct control over the formation of a new phagophore by using Atg11 to activate Atg1. Our data thus support a unifying model for autophagy regulation in which Atg1 drives phagophore formation in response to at least two types of autophagic cues (targets and nutrient depletion) via recruitment of two scaffold proteins (Atg11 and Atg17, respectively) (Fig. 1) .
Figure 1.
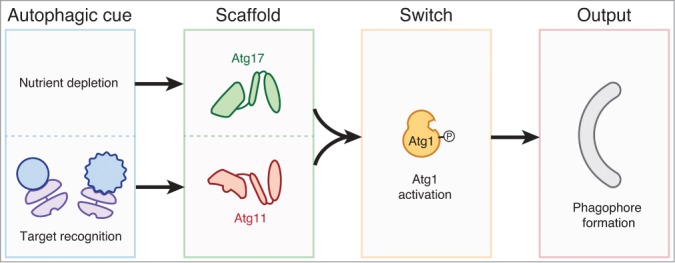
Two autophagic cues, nutrient depletion and the recognition of an autophagy target (blue) by an autophagy receptor (purple), are detected by two Atg1-binding scaffold proteins (Atg17 and Atg11, respectively). Both scaffolds activate the Atg1 kinase as a molecular switch to drive a single output: phagophore production. Not shown is the downstream maturation of a phagophore into either a selective or nonselective autophagosome; specialization into a selective autophagosome is dictated by interactions between receptors and Atg8-family proteins.
Previously, Atg1 kinase activation had been proposed to play a role in converting between selective and nonselective modes of autophagy. Our findings argue against this model, as we find that regulated activation of Atg1 is crucial for both types of autophagy. We also note that our in vitro assay for Atg1-mediated phosphorylation reveals no discernible differences in the pattern of Atg1 substrates whether Atg1 is activated by receptor-bound targets or by rapamycin. While it remains possible that the set of Atg1 phosphorylation sites differs subtly but importantly depending on the scaffold used to activate Atg1, the more parsimonious interpretation of our data is that Atg1 phosphorylates a common set of substrates to drive phagophore synthesis in response to multiple stimuli. Figuring out how these phosphorylation events drive phagophore formation remains a central goal in the field. Notably, dissection of these events, particularly phosphorylation of Atg9 vesicles, which are difficult to purify, should benefit greatly from the cell-free assay for Atg1-mediated phosphorylation we have developed.
How, at a molecular level, autophagy receptors use Atg11 to activate Atg1 is an exciting question raised by our work. One possibility, given that Atg1 must autophosphorylate to become fully active, is that receptors clustered on the surface of a target promote Atg1 dimerization and trans-autophosphorylation. Appealingly, an enforced dimerization model for Atg1 activation could also explain how binding of Atg17—which is structurally unrelated to Atg11 but exists as a pre-formed dimer—activates Atg1. An alternative, but not mutually exclusive, possibility is that both scaffolds could activate Atg1 using allosteric mechanisms. Testing these speculative models will be facilitated by structural characterization of Atg1 bound to its activators combined with detailed biochemical analysis.
Perhaps the most pressing next step is to test the generality of the mechanism we uncovered in yeast by determining whether receptor-bound targets activate the mammalian homolog of Atg1, ULK1. Intriguingly, ULK1 has recently been shown be recruited to mammalian targets in a receptor-dependent manner. In addition, several mammalian proteins with sequence homology to Atg11 bind both receptors and ULK1. Most notably, HTT (huntingtin; mutations in which cause Huntington disease) was recently suggested to be a human homolog of Atg11, heightening the urgency of studying these mechanisms in human cells.