

Abstract
Macroautophagy is a cellular process that mediates degradation in the lysosome of cytoplasmic components including proteins and organelles. Previous studies have shown that macroautophagy is induced in activated T cells to regulate organelle homeostasis and the cell's energy metabolism. However, the signaling pathways that initiate and regulate activation-induced macroautophagy in T cells have not been identified. Here, we show that activation-induced macroautophagy in T cells depends on signaling from common γ-chain cytokines. Consequently, inhibition of signaling through JAK3, induced downstream of cytokine receptors containing the common γ-chain, prevents full induction of macroautophagy in activated T cells. Moreover, we found that common γ-chain cytokines are not only required for macroautophagy upregulation during T cell activation but can themselves induce macroautophagy. Our data also show that macroautophagy induction in T cells is associated with an increase of LC3 expression that is mediated by a post-transcriptional mechanism. Overall, our findings unveiled a new role for common γ-chain cytokines as a molecular link between autophagy induction and T-cell activation.
Keywords: common γ-chain, interleukin 2, JAK, macroautophagy, T cell
Abbreviations
- ATG
autophagy-related
- IFN
interferon
- IL
interleukin
- JAK
Janus kinase
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NL
ammonium chloride and leupeptin
- PtdIns3K
class-III phosphatidylinositol 3-kinase
- RPS6KB/p70S6K
ribosomal protein S6 kinase
- 70kDa STAT
signal transducer and activator of transcription
- TCR
T cell receptor
- TH
T helper cell
- Vb
vinblastine
Introduction
Macroautophagy (hereafter referred to as autophagy) is a ubiquitous process of degradation and recycling of cytosolic proteins and organelles in eukaryotic cells.1,2 Autophagy is initiated by the formation of a phagophore that will elongate and mature, engulfing cargo from the cytoplasm, to form a double-membrane vesicle called an autophagosome. Autophagosomes will then fuse with the lysosome to degrade their content. The initiation and maturation of autophagosomes require a well-conserved set of proteins encoded by the autophagy-related (ATG) genes, first discovered and extensively characterized in the Saccharomyces cerevisiae yeast model.3,4 Among them, Map1lc3, a mammalian ortholog family of yeast Atg8, that codes for the MAP1LC3/LC3 proteins is currently one of the most reliable markers of the autophagosome.5,6 Soon after its translation, pro-LC3 is cleaved to produce its cytosolic form (LC3-I), which can then be conjugated to phosphatidylethanolamine (LC3-II),7 to allow association with the double membrane of the autophagosomes. Regulation of canonical autophagy involves 2 major protein complexes that control the initiation of autophagosome formation.8 The mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) is a negative regulator of autophagy that is sensitive to various signals including growth factors, nutrient availability or oxidative stress. TORC1/MTORC1 regulates autophagy through its activity on the Atg1/ULK protein kinase complex, one of the core autophagy protein complexes that is found associated with the phagophore following autophagy activation.9,10 The second regulator, the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, consisting of PIK3C3/Vps34, PIK3R4/Vps15 and ATG14 associated with BECN1 (mammalian ortholog of yeast Vps30/Atg6), is required at an early stage of autophagosome formation for the recruitment of other ATG proteins to the phagophore.11-14
Autophagy is an essential process not only to maintain cell homeostasis but also to respond to different types of stressors and stimuli.15,16 Evidence has emerged supporting the idea that autophagy plays fundamental roles in the immune system.17 Autophagy is involved in the initial steps of the immune response by acting on the innate immune cells. For instance, endogenous and exogenous antigens have been found in autophagosomes of antigen-presenting cells, revealing a role for autophagy in antigen processing and crosspresentation.18 Moreover, induction of autophagy promotes pathogen elimination in macrophages.19 In this context, as well as in other cell types, cytokines have been shown to modulate the induction of autophagy. For example, IFNG (interferon, gamma), a classical T helper (TH) cell 1 cytokine that notably acts on macrophages, activates autophagy in these cells, which contributes to the elimination of Mycobacteria tuberculosis by the phagocytic infected cells.19 In contrast, IL4 (interleukin 4) and IL13 inhibit autophagy in macrophages.20 Previous work has shown that autophagy is also induced in CD4+ T cells following activation.21,22 Conversely, T cells lacking essential autophagy genes fail to properly activate and proliferate following activation.22-24 Autophagy genes have also been proposed to be essential for T lymphocyte development and survival in the periphery.24-26 Although the use of models with Atg gene deletion in CD4+ T cells has revealed the requirement of autophagy in both steady- and activated- states, little is still known about the signaling pathways that induce and regulate autophagy in activated T cells.
To study the underlying mechanisms of activation-induced autophagy in CD4+ T cells, we have established a robust experimental procedure to monitor the T cell receptor (TCR) engagement-induced autophagy flux in several CD4+ T cell subsets. Using this approach, here we report that common γ-chain cytokines play a major role on activation-induced autophagy in T helper cells, notably through the activation of the JAK1/3 (Janus kinase 1/3) pathway. Moreover, we found that activation-induced autophagy in T cells is associated with upregulation of LC3 expression that is mediated at a post-transcriptional level. Our data reveal a new mechanism of activation and maintenance of autophagy by specific cytokines that controls the regulation of autophagy induction following T cell activation.
Results
Autophagy is induced in activated naïve CD4+ and effector T helper cells
Activation of autophagy occurs in response to activation in CD4+ T cells. Several reports have shown that, when activated, T cells increased autophagosome formation and turnover.21,22 To establish that we could reliably assess this previously characterized process and generate a tool that would allow us to identify the signaling pathways that might regulate autophagy in T cells, we defined the conditions to measure activation-induced turnover of endogenous LC3-II, which has been established as an accurate method to quantify autophagy flux.27 Naïve CD4+ T cells were left resting or activated with anti-CD3 and anti-CD28 antibodies and treated with ammonium chloride and leupeptin (NL) to inhibit lysosomal proteases for the last 3 h of the experiment, which led to a clear accumulation of LC3-II, supporting increased activation-induced autophagosome turnover (Fig. 1A). To further characterize the underlying molecular mechanisms involved in activation-induced autophagy, we extended our study to determine whether a similar induction of autophagy would also occur in activated effector TH1 cells and TH2 cells. In line with our previous results, we found that TCR+CD28 stimulation induced autophagy flux not only in naïve but also in effector CD4+ T cell populations including TH1 and TH2 cells (Fig. 1B and C). To confirm that the induction of autophagy reflected increased autophagosome formation, we also analyzed LC3-II turnover using vinblastine, a microtubule depolymerizing agent that leads to the accumulation of newly formed autophagosomes by preventing fusion with lysosomes. Data from those experiments confirmed that autophagosome formation was induced in activated CD4+ T cells (Fig. 1A to C).
Figure 1.
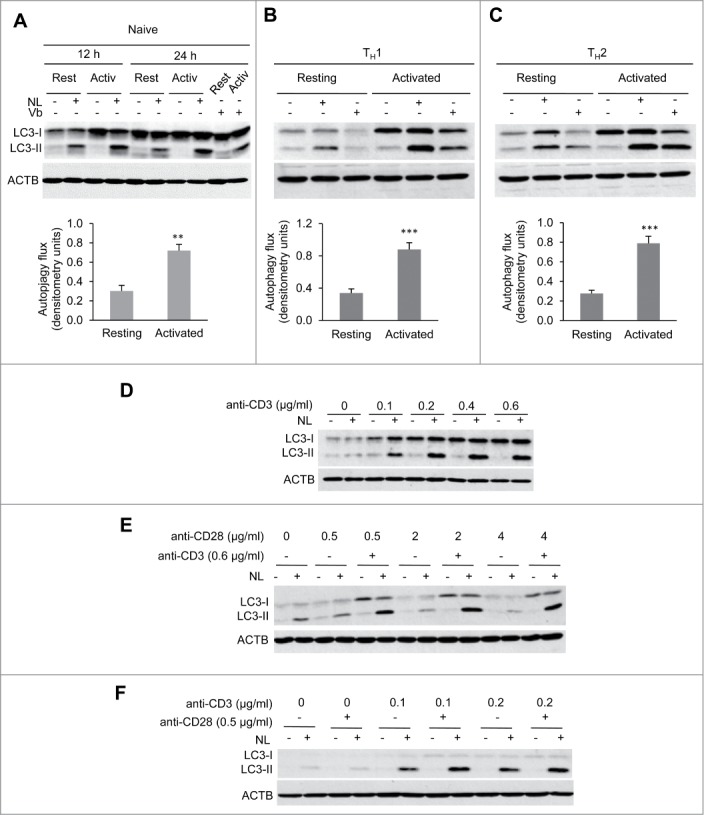
Autophagy is induced by TCR activation. Freshly isolated naïve mouse CD4+ T cells (A) or in vitro differentiated effector Th1 (B) and Th2 (C) cells were incubated in either media alone or stimulated with plate bound anti-CD3 and soluble anti-CD28 antibodies for 12 to 24 h. To measure autophagy flux, ammonium chloride and leupeptin (NL) or vinblastine 100 µM (Vb) were added for the last 3 h of culture. Accumulation of LC3-II in the presence of inhibitors was measured by immunoblot on whole cell lysates. Bar graph represent mean+SEM of autophagy flux, measured as the difference between the intensity of the LC3-II band in cells cultured in the presence or absence of NL, from 6 (naïve) 12 (TH1) or 10 (TH2) independent experiments after 20 to 24 h of activation (paired 2-tailed Student t test. **, P<0.01; ***, P<0.001). (D) Th1 cells were stimulated with increasing concentrations of plate-bound anti-CD3 antibodies. Autophagy flux was assessed by immunoblot on whole protein lysates after 20 h of stimulation and the addition of NL for the last 3 h. (E) TH1 cells were incubated with different doses of soluble anti-CD28 in the absence or presence of plate-bound anti-CD3. Autophagy flux was assessed as in (A). (F) Th1 cells were left unstimulated or stimulated with 2 different low doses of plate-bound anti-CD3 in the presence or absence of anti-CD28 and autophagy flux was assessed as in (A).
Overall, our data showed that autophagy was robustly induced following TCR + CD28 stimulation in all CD4+ T cell populations studied: naive, TH1 and TH2 cells, suggesting that autophagy activation in CD4+ T helper cells might respond to a common pathway. In our subsequent studies, TH1 cells were used as a preferred model, though in several cases naive and/or TH2 cells were also analyzed to determine whether mechanisms were shared among different T helper cell populations.
TCR stimulation triggers autophagy induction in T-helper cells
TCR as well as costimulatory signals are required for full T-cell activation. Accordingly, we sought to determine the individual role that each of those signals might play in the induction of autophagy during T-cell activation. To that aim, we first activated TH1 cells with different doses of plate-bound anti-CD3 without any costimulation. Increased autophagy flux was detected at all anti-CD3 concentrations tested, and a clear dose-dependence was observed (Fig. 1D). Similar experiments performed using different doses of anti-CD28 in the absence TCR stimulation failed, however, to modulate autophagy (Fig. 1E). Since costimulatory molecules are known to act in synergy with TCR signaling, we tested whether the presence of costimulation could enhance TCR-induced autophagy. At our working concentration of anti-CD3 dose (0.6 μg/ml), we could not detect any synergistic effect of CD28 engagement (Fig. 1E). However, as we had observed that autophagy was already highly induced with this dose of anti-CD3 alone, we thought that the role of CD28 could be masked in those conditions. Therefore, we determined whether using a suboptimal dose of anti-CD3 would permit us to examine the effects of the CD28-mediated costimulation. Interestingly, at low doses of anti-CD3, engagement of CD28 was able to clearly enhance autophagy flux induction in TH1 cells (Fig. 1F). Altogether, our results support that TCR stimulation is required and sufficient to induce autophagy in effector T-helper cells and that CD28 costimulatory signal acts in synergy with the TCR pathway to potentiate TCR-induced autophagy.
Interestingly, the increase in LC3-II turnover detected in response to TCR engagement, while dependent on BECN1/Beclin 1, did not appear to require MTOR inhibition (Fig. S1). TCR engagement induced, as expected, MTOR activation which occurred concomitant with increased LC3 flux in Th1 and Th2 cells (Fig. S1A). Moreover, even though rapamycin clearly inhibited MTOR-induced RPS6KB/p70S6K phosphorylation, it had no effect on the levels of activation-induced macroautophagy flux observed on T cells (Fig. S1A and S1B). These results indicated that macroautophagy induction occurring during T-cell activation was likely not dependent on MTOR inactivation. However, we wondered whether phospho-RPS6KB measured at the end of the experiment could reflect early MTOR activity, which would be later downregulated to allow induction of macroautophagy. To ascertain whether MTOR activity was maintained throughout the whole experiment, we added rapamycin at different times following activation. If MTOR was inactivated early after T-cell activation to induce macroautophagy activity, the late addition of rapamycin should not modify the phosphorylation status of RPS6KB. We found, however, that RPS6KB phosphorylation was inhibited when rapamycin was added even 17 h after activation, indicating a continuous turnover of RPS6KB phosphorylation and supporting that MTOR activation was maintained during the whole time course of the experiment (Fig. S1C). Altogether, these data indicate that MTOR and macroautophagy are both activated during T-cell activation and support that activation-induced macroautophagy in T-helper cells does not require MTOR inactivation.
LC3 expression is post-transcriptionally upregulated in activated T cells
Our data showed that activation of T cells not only increased LC3-II turnover but it also induced an upregulation of the levels of LC3-I (Fig. 2A) even when the turnover of this protein was accelerated due to the activation of autophagy (Fig. 1). It has been suggested that the magnitude of autophagy may depend, at least in part, on the amount of LC3 available in the cell. Under conditions that induce high autophagosome turnover, increased LC3 expression could help support rapidly augmented autophagosome formation and degradation. To gain insight into the regulation of LC3 expression in activated CD4+ T cells, we first investigated whether the expression of mRNA transcripts encoding for LC3 were affected by TCR and CD28 stimulation. Surprisingly, we found that Map1lc3b (coding for LC3B) mRNA levels not only failed to increase in activated cells, but even showed some decrease early after stimulation (Fig. 2B). Even though mammalian autophagy is generally associated with LC3B, we also quantified mRNA levels for the Map1lc3a gene (encoding LC3A) that has an 83% identity with LC3B, and found a very similar expression profile to the one detected for Map1lc3b (data not shown). Thus, changes in mRNA levels did not appear to be responsible for the increase in LC3 protein expression that happened in activated T cells. mRNA abundance does not always reflect the transcriptional activity of a gene or the levels of the protein encoded by the mRNA. We first determined if the lack of an increase in Map1lc3b mRNA in the presence of higher cellular levels of LC3 protein could respond to changes in mRNA processing or stability. We assessed the impact of T-cell activation on the levels of Map1lc3b pre-mRNA and found that, as we had seen with the total mRNA levels, Map1lc3b pre-mRNA did not increase after TCR and CD28 stimulation (Fig. 2B). Moreover, mRNA to pre-mRNA ratio did not significantly change over the activation time course, supporting that Map1lc3b mRNA stability was not affected by the stimulation. These data indicated, thus, that the increase in LC3 protein level was neither due to upregulation of transcriptional activity of the Map1lc3b gene nor to increased-mRNA processing or stabilization.
Figure 2 (See previous page).
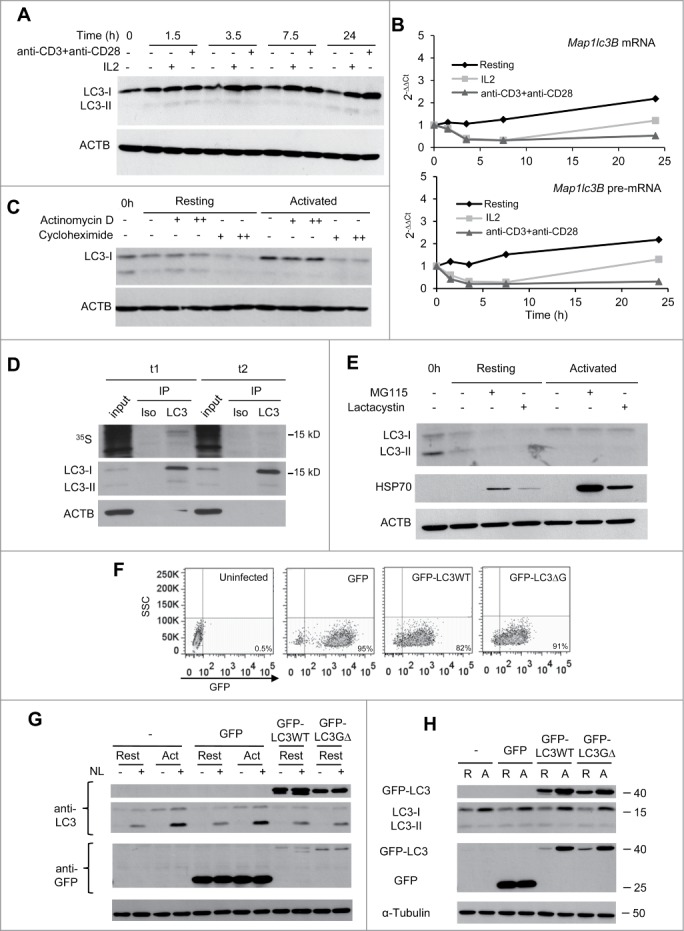
LC3 is post-transcriptionally upregulated following TCR and CD28 engagement in Th1 cells. TH1 cells were incubated with media alone or stimulated with either IL2 or plate-bound anti-CD3 and soluble anti-CD28 antibodies. Cells were harvested at indicated times after stimulation and samples were processed for protein and RNA analysis. (A) LC3 expression was analyzed by immunoblot on whole cell protein extracts and (B) Lc3b mRNA and pre-mRNA were quantified by real-time PCR. Graphs show one representative kinetic of RNA quantification out of 4 independent experiments with similar results analyzing RNA levels at different time points from 30 min to 24 h poststimulation. (C) TH1 cells were left resting or activated with plate-bound anti-CD3 and soluble anti-CD28 antibodies for 8 h. 0.1 µg/mL (+) or 1 µg/mL (++) of actinomycin D; or 10 µg/mL (+) or 100 µg/mL (++) of cycloheximide was added for the last 3 h. Whole protein extracts were analyzed by immunoblot to quantify LC3 expression. ACTB was used as loading control. (D) TH1 cells were activated with plate-bound anti-CD3 and soluble anti-CD28 antibodies in media without Met or Cys supplemented with 35S-Met+Cys. After 3.5 h of labeling, protein extracts were obtained from half of the cell in the culture (t1). Excess of cold Met+Cys were added to the remaining cells for 3.5 additional h (t2). Both protein extracts were immunoprecipitated (IP) with anti-LC3 antibody (LC3) or isotype control (Iso). Ten µg of input and the IP products were separated by SDS-PAGE and transferred to nitrocellulose membrane. 35S incorporation into proteins was measure by autoradiography (upper panel) and the same membrane was then immunoblotted for LC3 and ACTB (lower panels). (E) TH1 cells were incubated in media alone or activated with plate-bound anti-CD3 and soluble anti-CD28 antibodies for 7 h. Proteasome inhibitors (MG-115 10 µM or lactacystin 5 µM) were added for the last 4 h. Whole cell lysates were analyzed by immunoblot for the indicated proteins. HSP70 accumulation in the presence of proteasome inhibitors was used as a positive control. (F and G) TH1 cells were left uninfected (-) or transduced using lentiviruses containing either GFP alone or GFP-LC3 (wt or G∆) and sorted for GFP-positive cells. TH1 were then left resting or activated for 16 to 18 h, NL was added for the last 3 h. Transduction efficiency (percentage of GFP+ cells) was evaluated by FACS and expression of GFP-LC3 and autophagy flux were analyzed by immunoblot. (H) TH1 cells were obtained as described in (F), and left resting (R) or activated (A) for 16 to 18 h. Expression of endogenous and exogenous LC3 was followed using anti-LC3 and anti-GFP antibodies. TUBA/αTubulin was used as loading control in F and G because ACTB band overlapped with the size of GFP-LC3. Each blot from this figure is representative of at least 3 independent experiments.
We next sought to determine whether the higher level of LC3 protein in stimulated cells could respond to either an increase in translation or to a reduction of the rate of LC3 degradation. To that goal, we first compared the effect of transcription or translation inhibition, using actinomycin D or cycloheximide, respectively, on LC3 protein levels in resting and activated TH1 cells. In order to minimize any artifact due to the global effect of these inhibitors, cells were stimulated for 5 h to ensure adequate activation of early signaling pathways and then treated with each inhibitor for 3 additional h. Interestingly, while blockade of transcription did not affect LC3 protein levels, translation inhibition with cycloheximide severely reduced LC3 levels in activated cells (Fig. 2C). This showed that, in line with our analysis of mRNA levels, upregulation of LC3 expression in activated T cells was not dependent on transcriptional activity, but instead translation of LC3 was the one induced by TCR and CD28 engagement. In order to confirm this result, we performed a pulse-and-chase assay to determine the rate of LC3 turnover. Even after just 3.5 h of activation in the presence of 35S-labeled amino acids, we observed a significant accumulation of 35S incorporated into LC3, confirming that LC3 was newly translated in activated cells. Moreover, following a 3.5 h chase with media containing excess cold amino acid, most of the radioactive LC3 disappeared while total LC3 protein levels detected by immunoblot remained high (Fig. 2D), indicating that LC3 was rapidly synthesized in response to activation but also rapidly degraded and replaced by newly translated LC3. Although at least a part of the LC3 cellular pool should be degraded in the autolysosome, we sought to determine whether the proteasome, normally associated with the degradation of short-lived proteins, could also be involved in the regulation of LC3 turnover. Using 2 different proteasome inhibitors, we found that inhibition of proteasome activity did not affect the levels of LC3 in activated cells, while clearly resulting in the accumulation of HSP70, a known proteasome substrate (Fig. 2E). We next asked if the increase of LC3 protein could not only supply the required LC3 to maintain a high activity of autophagy but could play a role in the regulation of autophagy flux induction. In order to answer this question we used lentiviral vectors to overexpress in TH1 cells exogenous GFP-tagged wild-type LC3 or a mutant LC3 protein that cannot be conjugated to phosphatidylethanolamine and therefore fails to incorporate to the autophagosome membrane. Infected GFP+ cells were sorted and analyzed for their response to activation (Fig. 2F). When compared to the autophagy flux detected in activated uninfected T cells or cells expressing GFP alone, we observed that expression of GFP-LC3 alone, at least at the levels used in our study, was not sufficient to upregulate endogenous autophagy flux in resting cells (Fig. 2G). Interestingly, when transduced TH1 cells were activated, we observed not only an increase in endogenous LC3 but also in the levels of the exogenously expressed GFP-LC3 protein but not of GFP alone, both under the control of the PGK promoter, confirming that LC3 expression was upregulated at a post-transcriptional level (Fig. 2H).
Altogether, these results support the idea that following T-cell activation not only the rate of LC3 degradation is accelerated but LC3 synthesis is rapidly upregulated by a mechanism that reflects post-transcriptional modulation of LC3 expression.
Common γ-chain cytokines induce autophagy in T cells
One of the main consequences of CD4+ T cell activation is the production of cytokines that can in turn modulate these cells' own responses. We decided, thus, to investigate if cytokine-modulated signaling pathways could interfere or participate in the induction of autophagy in activated T cells. Several pieces of evidence suggested that IL2 might play a role in regulation of autophagy in TH1 cells: first we have observed that after being cultured for several days with IL2, TH1 cells presented higher levels of autophagy than freshly isolated naive CD4+ T cells (data not shown). Moreover, our data showed that costimulation synergized with TCR to enhance activation-induced autophagy and CD28 engagement is also associated with increased IL2 expression, as we confirmed by increased phosphorylation levels of STAT5 when T cells were activated with anti-CD3 and anti-CD28 (data not shown). Finally, other cytokines, such as IFNG or IL4, had been previously shown to modulate autophagy in macrophages, suggesting that cytokines could also be involved in the regulation of autophagy in T cells.
We first determined the ability of IL2 to modulate autophagy in T cells by challenging TH1 cells with different doses of IL2 and observed that autophagy flux, asses as LC3 turnover, was promoted in a dose-dependent manner (Fig. 3A). However, we did not detect a corresponding increase in SQSTM1/p62 flux (Fig. S1E), suggesting that this receptor protein may not participate in cargo recognition in activated CD4+ effector T-helper cells. In order to validate that the increase of LC3-II turnover in IL2-stimulated TH1 cells reflected an increase in autophagy activity as previously shown for activated T cells,21,22 we first analyzed the formation and turnover of LC3+ puncta, representing autophagic vesicles, by immunofluorescence. We found that following IL2 stimulation, turnover of LC3 puncta was highly increased, while no significant changes were observed in resting cells (Fig. 3B). Secondly, we also compared the levels of autophagic vesicles by electron microscopy in TH1 cells that were either left resting or stimulated with IL2. Supporting increased autophagy in CD4+ T cells in response to IL2, we found that numbers of autophagic vacuoles were significantly increased in IL2-treated cells compared to resting cells (Fig. 3C and S2). This increase was mainly due to a higher number of autolysosomes while the number of autophagosomes remained comparable to resting cells, further supporting an increase in the autophagic flux in IL2-treated cells.
Figure 3 (See previous page).

Common γ-chain cytokines induce autophagy in T cells. (A) TH1 effector T helper cells were incubated without or with increasing doses of IL2 for 20 h. NL were added for the last 3 h. LC3-II turnover was assessed by immunoblot. Y694 phosphorylation of STAT5 was used as control of the response to cytokine stimulation. (B) TH1 cells were incubated without or with IL2 (10 ng/mL) for 16 to 18 h. NL were added for the last 3 h. LC3 puncta (FITC) were analyzed by immunofluorescence using anti-LC3 and DAPI for nucleus staining. Some of the puncta are indicated by a white arrow. Bar graph represents mean + SEM from the analysis of 200 cells per condition from 3 independents experiments. Data was analyzed with a Student t test: *, P < 0.05; ns, not statistically different. (C) TH1 cells were left resting or stimulated with IL2 (10 ng/mL) for 16 to 18 h and cells were processed for electron microscopy. Left side shows representative resting (magnification x8000) and IL2-stimulated (magnification x6000) TH1 cells (additional examples of both type of cells are shown in Fig. S2A). Right side insets show examples of autophagosomes and autolysosomes at higher magnification. Bar graph (mean+SEM) indicates the number of autophagosomes (APG), autolysosomes (AUTL) and total autophagic vesicles (AV) per cell. t test, n = 20 cells per condition, ***, P < 0.001. ((D)to F) Naive CD4+, TH1 or TH2 effector T helper cells were incubated without or with 4 increasing doses of the indicated cytokines for 20 h. NL were added for the last 3 h. LC3-II turnover was assessed by immunoblot. Y694 phosphorylation of STAT5 was used as control of the response to cytokine stimulation. (G) Bar graph indicates autophagy flux normalized to the autophagy flux measured in unstimulated conditions, and show mean + SEM of 3 independent experiments per cytokine. ANOVA followed by the Tukey post-hoc test; *, P < 0.05. (H) Th2 cells were treated with different doses of IFNG or 10 ng/mL of IL4 (last 2 lanes) for 20 h. NL were added for the last 3 h. Whole-cell protein extracts were subject to immunoblot. Y701 Phosphorylation of STAT1 and Y641 Phosphorylation of STAT6 were used as controls of IFNG and IL4-induced stimulation, respectively. IL4 stimulation was used as a positive control of autophagy induction in TH2 cells.
IL2 is a member of the common γ-chain family cytokines, which share signaling pathways activated by their receptor's γ-chain and display redundancy in some of their functions. We therefore analyzed whether other common γ-chain cytokines would also be able to modulate autophagy in T cells. To this goal, we extended dose-response assays to 3 other common γ-chain cytokines in 3 different CD4+ T cell populations, which are preferentially targeted by those common γ-chain cytokines. Thus, we tested IL15 on TH1 cells, IL4 on TH2 cells and IL7 on naive CD4+ T cells. As for IL2, we observed for all cytokines tested a dose-dependent induction of autophagy (Fig. 3D to G). We next examined whether this effect was restricted to common γ-chain cytokines. Since IFNG has been shown to activate autophagy in macrophages, this cytokine might constitute a good candidate to analyze the specificity, or lack of thereof, of autophagy induction in T cells on common γ-chain cytokine signaling. During TH1 differentiation, the IFNG-receptor is downregulated, however TH2 cells that were never challenged with IFNG still express its receptor and can respond to this cytokine.28,29 We used this feature of TH2 cells to test the effect of IFNG on autophagy in CD4+ T cells. Interestingly, we did not observe any significant changes in autophagy flux on TH2 cells after stimulation with different doses of IFNG, even though these cells clearly responded to this cytokine with a marked increase in STAT1 phosphorylation (Fig. 3H).
Having established the potential role of common γ-chain cytokines in the regulation of autophagy in CD4+ T cells, we next investigated whether the secretion of common γ-chain cytokines by activated T cells could be ultimately, responsible for activation-induced autophagy. For this purpose we tested the role of IL2 and IL4 secreted upon TCR and CD28 stimulation by TH1 and TH2 cells, respectively on the levels of activation induced autophagy in those T-helper cell populations. When neutralizing antibodies against IL2 and IL4 were used to block these cytokines during TH1 and TH2 cell activation, respectively, a consistent reduction of autophagy flux was observed in both conditions, with at least 50% of the activation-induced autophagy being sensitive to IL2 or IL4 blockade (Fig. 4A to C). Altogether, our data demonstrate that IL2 and IL4 are not only inducers of autophagy on T cells but are also essential for full induction of autophagy in response to TCR and CD28 engagement.
Figure 4.
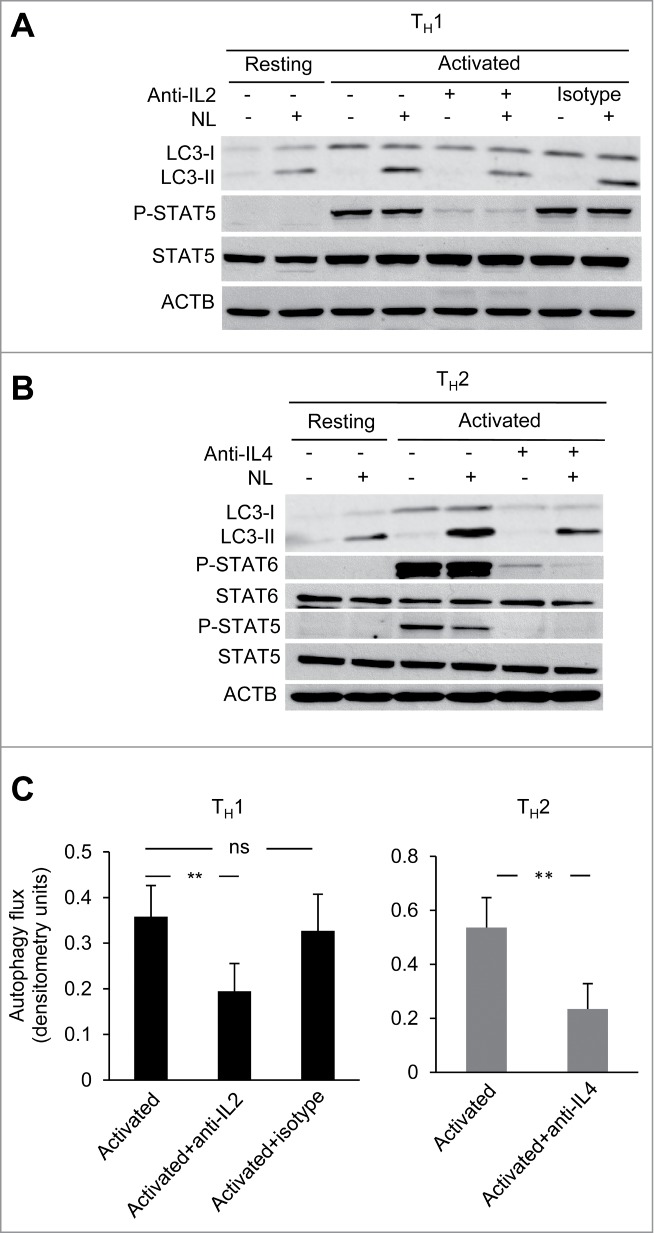
Common γ-chain cytokine signaling contributes to activation-induced autophagy in T-helper cells. (A and B) TH1 and TH2 cells were activated with plate-bound anti-CD3 (0.4 µg/ml) and soluble anti-CD28 (0.5 µg/ml) antibodies for 16 to 18 h in the presence or absence of cytokine-neutralizing monoclonal antibodies for IL2 (30 µg/mL) or IL4 (100 µg/mL), respectively. Autophagy flux was assessed on whole cell lysates by immunoblot. Inhibition of Y694 phosphorylation of STAT5 or Y641 phosphorylation of STAT6 were used to control for the neutralizing activity of the anti-IL2 and anti-IL4 antibodies. (C) Bar graph indicates mean+SEM autophagy flux induction of 4 (TH1) or 3 (TH2) independent experiments (ANOVA with Tukey post-hoc test; **, P < 0.01; ns, not statistically significant).
JAK signaling participates in TCR- and cytokine-induced autophagy in CD4+ T cells
The JAK3 kinase is associated with the γ-chain of the common γ-chain cytokine receptors, whereas JAK1 is commonly associated with other cytokine-specific chains present in those receptors. To determine if JAK signaling was responsible for the induction of autophagy caused by those cytokines, we initially used the JAK inhibitor I (JAKi-I), which displays potent inhibitory activity against JAK1, JAK2 and JAK3. As expected, when TH1 cells were stimulated with IL2 in the presence of this JAK inhibitor, upregulation of Y694 phosphorylation of STAT5 was abrogated (Fig. 5A). When we measured autophagy flux under those conditions, a significant reduction of autophagy induction was observed when JAK activity was blocked (Fig. 5A). To further characterize this effect, we used specific inhibitors of JAK1 (ruxolitinib), or JAK3 (tofacitinib). Similar to the results obtained with JAKi-I, STAT5 phosphorylation and autophagy flux induction in response to IL2 stimulation were markedly diminished when the specific JAK inhibitors were used (Fig. 5B). Furthermore, TH2 cells stimulated with IL4 in the presence of those inhibitors consistently displayed an equally marked abrogation of autophagy flux (Fig. 5C). Interestingly, both JAK inhibitors also downregulated autophagy flux induced by TCR activation, suggesting that an autocrine or paracrine loop involving engagement of the IL2 or IL4 receptor by cytokines secreted by activated T cells, had an important role in the upregulation of autophagy during T-cell activation (Fig. 5D and E).
Figure 5.
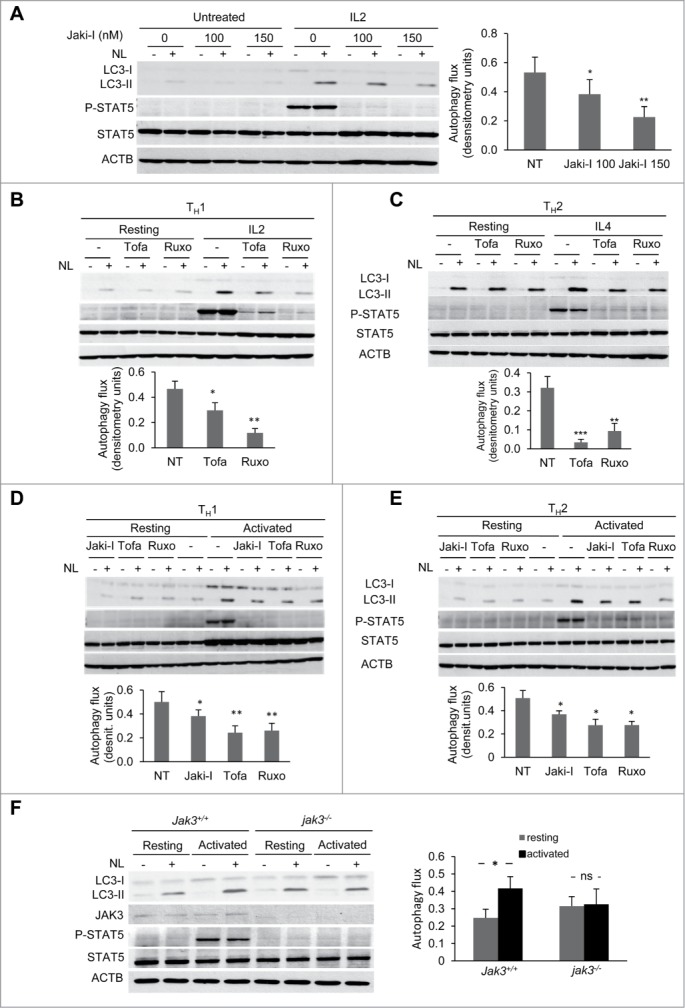
JAK activity is required for cytokine- and activation-induced autophagy in T-helper cells. (A-C) TH1 and TH2 cells were left resting or stimulated with IL2 (10 ng/mL) or IL4 (10 ng/ml), respectively, for 16 to 18 h, treated or untreated (NT) with several JAK inhibitors. (A) 100 and 150 nM of JAKi-I, (B) 150 nM and (C) 100 nM of tofacitinib (Tofa) or ruxolitinib (Ruxo). (D and E) TH1 and TH2 cells were left resting or activated in the presence of 100 nM of different JAK inhibitors as indicated. In all conditions, NL were added for the last 3 h and autophagy flux was assessed by immunoblot. P-STAT5 (Y694) was used as a control of stimulation and inhibition of JAK activity. Graphs show of mean + SEM of induced autophagy flux from 4 to 6 independent experiments. (ANOVA with Tukey post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001). (F) Freshly isolated CD4+ T cells from Jak3+/+ and jak3-/- mice were incubated in media alone or activated with plate-bound anti-CD3 and soluble anti-CD28 antibodies for 10 h. NL were added during the last 3 h. Autophagy flux was assessed by immunoblot, and JAK3 protein as well as STAT5 phosphorylation (Y694) were used to control JAK3 deletion and activity. Pool of CD4+ T cells from 2 to 3 mice was used for each experiment. Bar graph represents mean + SEM of autophagy flux from 5 independent experiments (paired 2-tailed t test; *, P < 0.05; ns, non-statistically different).
To further ascertain whether JAK activity was responsible for cytokine-dependent induction of autophagy, we used CD4+ T cells isolated from Jak3-deficient (jak3-/-) mice.30 As T cells lacking Jak3 exhibit altered activation and proliferation, we used freshly isolated naive CD4+ T cells and activated them for shorter time periods (9 to 10 h) that still allowed for a robust and reproducible early induction of autophagy in wild-type cells. In accordance with the results obtained with the JAK inhibitors, we found that TCR and CD28-stimulation of jak3-/- CD4+ T cells failed to induce autophagy (Fig. 5F). Our results support the conclusion, thus, that JAK1/3 activity is required for full activation of autophagy in response to both common γ-chain cytokines or TCR and CD28 stimulation in T-helper cells.
Discussion
Autophagy has emerged in the past few years as an important mechanism that contributes to T-cell homeostasis and activation, although the mechanisms that are involved in the regulation of autophagy in T cells remained unknown. This study unveils a new mechanism of autophagy induction and maintenance following T-cell activation that involves common γ-chain cytokine signaling pathways. Moreover, we provide evidence that JAK1 and JAK3 activity, which is a key early event downstream of the IL2-receptor, is required for the induction of autophagy in activated T cells.
As has been described for different cell types and tissues, autophagy plays an important role in T-cell homeostasis. Turnover of mitochondria and the endoplasmic reticulum maintain redox and calcium homeostasis in T cells.26,31 Furthermore, autophagy promotes T-cell survival through the degradation of caspases and proapoptotic BCL2 family proteins.24 As we and others have reported, autophagy is also induced in response to T-cell activation and, when inhibited, T cells respond poorly to TCR engagement, with decreased proliferation and cytokine expression.22,23 In this situation, autophagy allows T cells to adapt to the increase metabolic demand that follows activation, although specific regulation of signaling pathways through targeted degradation of signaling intermediates has also been recently proposed.22,32 The mechanisms that are responsible for the induction of autophagy in activated T cells remain, however, not yet characterized.
Our data support the idea that signaling through cytokine receptors that contain the common γ chain induces autophagy in CD4+ T cells. Cytokines have been implicated in modulation of autophagy not only in immune cells but also in several other cell types.33 However, the emerging characterization of pathways implicated in cytokine modulation of autophagy has revealed that a uniform model cannot be established for all cell types and conditions. For instance, IL4 and IL13 inhibit starvation-induced autophagy in macrophages through an AKT-dependent pathway, whereas STAT6, but not AKT, is involved in IL4- and IL13-mediated inhibition of IFNG-induced autophagy in the same cells.20 As opposed to Th2 cytokines, in macrophages IFNG promotes autophagy through a mechanism that involves lRGM activation.34 However, given the pleiotropic nature of cytokines it is quite possible that the mechanisms of cytokine-mediated regulation of autophagy might be cell-type specific. Our data supports this notion, as we found that IFNG was not sufficient to induce autophagy in T cells. It was instead cytokines that signal through common γ-chain receptors, including IL2, IL4, IL7 and IL15, which were able to activate autophagy in different subsets of T helper cells. Interestingly, administration of IL2 has been proposed to induce systemic activation of autophagy, and IL2-mediated activation of autophagy is required to sustain IL2-induced fibroblast growth,35,36 suggesting that γ chain signaling-mediated regulation of autophagy might not be restricted to the T cell compartment. Given the role of common γ chain cytokines in promoting T-cell survival and the defined role of autophagy as a survival mechanism in several T cell populations it is likely that autophagy represents one of the cytokine-activated mechanisms that underlie their prosurvival activity.23,24,37 Recently, it has also been shown that memory CD8+ T-cell generation requires autophagy.37,38 Given that common γ chain cytokines play essential roles in ensuring the generation and maintenance of efficient T-cell memory, it is tempting to speculate that these cytokines might be responsible for the induction of autophagy in T cells during the generation of memory CD8+ T cell populations. Whether a similar process may occur during the generation of CD4+ T cell memory remains to be determined.
Signaling through the IL2 receptor leads to the activation of different signaling pathways including JAK1 and JAK3.39 By using genetic deletion and chemical inhibition, we show that the activation of JAK1 and JAK3 is required for the induction of autophagy in T cells in response to IL2 or IL4. A screen of kinase inhibitor libraries also found a JAK3 inhibitor to be an efficient autophagy inhibitor in MCF7 cells.40 However, in this study the readout was MTOR-dependent autophagy and based on additional experiments the authors suggested that the inhibitor was most likely acting through an off-target effect. Our data using more specific inhibitors and JAK3-deficient T cells suggest that JAK signing is directly involved in the regulation of activation-induced macroautophagy in T helper cells, although it is possible that this pathway may also be active in other cell types in response to specific stimuli. Our results not only support an important role of common γ-chain cytokines in the induction of autophagy in CD4+ T cells, but also indicate that auto- or paracrine signaling loops mediated by those cytokines have a crucial role in maintaining the overall induction of autophagy during T cell activation in response to TCR and CD28 engagement. It is likely that the regulation of autophagy during T cell activation may get input from other signaling pathways to allow T cells to fine-tune autophagic activity in response to different stimulatory contexts. For instance, JNK has been proposed to participate in the activation of autophagy in T cells,21 possibly through regulation of the BECN1-BCL2 complex, as has been described in other cell types.41 Recently, the glycolytic enzyme PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3) has also been shown to positively regulate autophagy in T cells, although the mechanism responsible for this effect remains unknown.42 Moreover, autophagy activity in T cells is also modulated by FADD and CASP8, likely to prevent excessive autophagy activation in T cells following stimulation that could otherwise result in cell death.43 However, the importance of IL2-receptor signaling in mediating activation-induced autophagy in T cells is underscored by the fact that inhibition of JAK kinases has a marked inhibitory effect on the levels on autophagy induced by TCR engagement.
Our data show that autophagy is induced concomitantly with MTOR activation. MTOR has been extensively studied in the regulation of starvation-induced autophagy and defined as major autophagy inhibitor that, by sensing nutrient levels, acts on the Atg1/ULK complex to prevent autophagosome formation.10,44 Our observations were not completely surprising since MTOR is activated upon TCR engagement and downstream of the IL2-receptor. Moreover, we have also found that inhibiting MTOR with rapamycin during T-cell activation did not significantly modulate autophagy in our experimental conditions, supporting that activation-induced autophagy in CD4+ T cells does not require MTOR inhibition. How autophagy avoids or counteracts the inhibitory effect of MTOR in T cells is not yet known. Nevertheless, not only MTOR activity but also its subcellular localization are important for exerting its regulatory role on autophagy.45,46 In nutrient-rich conditions MTOR localizes to the lysosome, where it can regulate autophagy.47 Therefore, exclusion of the MTORC1 complex from a specific subcellular compartment could prevent it from exerting its inhibitory activity on autophagosome formation. Recent studies in mammalian autophagy have already defined other examples of potential ULK1-independent or MTOR-independent forms of autophagy.48-50 Our study also confirms the role of BECN1 in activation-induced macroautophagy in T cells. Indeed, previous studies have reported the requirement for T cell homeostasis of BECN1 as well as of PIK3C3/VPS34, 2 major components of the class III PtdIns3K complex, and a direct link with autophagy in T cells has been shown for PIK3C3.24,51,52
In further support of the unique nature of autophagy regulation in activated T cells, we have also identified in this study that the increase in autophagy flux upon T cell activation is accompanied by postranscriptional regulation of LC3 expression. Autophagosome formation requires LC3. Thus, it is crucial for the cell to continuously adapt LC3 expression to the rate of autophagy. Following T cell activation or IL2 stimulation, we found that LC3 was actively synthesized without transcriptional upregulation or increased stabilization of Lc3 mRNA, which suggests a post-transcriptional regulation of LC3 expression. Previous studies have reported an upregulation of LC3 mRNA in response to several cellular stresses that activate autophagy, such as nutrient starvation or hypoxia, and identified specific transcription factors that can regulate LC3 expression.53-57 Nevertheless, in response to those stresses, upregulation of LC3 mRNA was generally observed after 8 to 10 h and then downregulated, hence, it might not completely account for the long-term maintenance of autophagy and the requirement of LC3 expression for longer time periods.55-57 Indeed, a study that extends the analysis to the regulation of translation has found an enrichment of LC3 mRNA in polysome fractions in response to hypoxia, supporting an increase in the translation rate of LC3.56 Furthermore, several studies have recently unveiled a prominent role for specific miRNAs in the regulation of the total levels of several autophagy-related proteins, including LC3, indicating that post-transcriptional regulation of LC3 expression may constitute a mechanism that contributes to the activation of autophagy under specific conditions.58,59 The fast rate of autophagosome turnover that we have detected in activated CD4+ T cells would make it necessary to increase the cellular availability of LC3 to maintain the levels of autophagic activity required to support T-cell activation. Our observations also highlight the fact that, given the marked increased in LC3-I expression that occurs in activated T cells, the LC3-I to LC3-II ratio has little meaning as a method to assess autophagic activity, whereas quantification of autophagy flux by assessing LC3-II turnover represents a much more accurate measurement of autophagy in T cells.6,60,61 All together these results indicate that high autophagy flux correlates with high rate of synthesis and degradation of LC3 following T cell activation.
In conclusion, our results have unveiled a link between common γ-chain cytokine and JAK signaling and activation of autophagy in T cells. Moreover, our data provide new evidence to support the conclusion that LC3 protein synthesis is upregulated by a post-transcriptional mechanism that is likely required to maintain the high rate of autophagy observed in T cells following TCR+CD28 engagement.
Materials and Methods
Mice
Six- to 12-wk-old C57BL/6J female mice were purchased from the Jackson Laboratory. JAK3-deficient mice (jak3−/−) were also obtained from the Jackson Laboratory and bred using Jak3+/− breeding pairs under specific pathogen-free conditions. Six- to 7-wk-old jak3−/− mice and Jak3+/+ littermate controls were used in the experiments. All mice were housed in a pathogen-free barrier facility. All procedures were reviewed and approved by The Albert Einstein College of Medicine's Institutional Animal Care and Use Committee, conforming to accepted standards of humane animal care.
T-cell culture
Naive CD4+ T cell were isolated from lymph nodes and spleens using Dynabeads® Mouse CD4 (L3T4) positive selection and following the manufacturer's protocol (Life Technologies, 11445D). For T helper (TH) cell differentiation, naive CD4+ T cells were activated with plate-bound with anti-CD3E/CD3ε (10 µg/ml; eBioscience, clone 145–2C11) and soluble anti-CD28 (0.5 µg/ml; eBioscience, clone 37.51) in T cell media (DMEM supplemented with, 10% fetal bovine serum, 5 U/mL penicillin, 5 µg/mL streptomycin, 4 mM glutamine, 1x nonessential amino acids (Lonza, 13–114E), 1x MEM Eagle vitamin mixture (Lonza, 13–607C), 50 µM β-mercaptoethanol, 660 µM L-arginine, 270 µM L-aspartic acid, 6 µM folic acid, 10 mM HEPES, pH 7.2 to 7.5, 1 mM sodium pyruvate); in the presence of 10 ng/mL of mouse IL12 p70 (eBioscience, 34–8121) and 10 µg/mL anti-mouse IL4 (clone 11B11) for TH1; or 10 ng/mL of mouse IL4 (BD Biosciences, 550067) and 2 µg/mL anti-mouse IFNG (eBioscience, clone R4–6A2) for TH2 cells. After 24 h of activation human IL2 (10 U/mL; NCI BRB preclinical repository) was added. TH cells were differentiated for 6 d. Unless otherwise indicated, differentiated TH cells were restimulated on plate bound with anti-CD3E (0.6 µg/ml) and soluble anti-CD28 (0.5 µg/ml) or alternatively using mouse T-activator CD3-CD28 beads (Invitrogen, 11452D) at ratio 0.7 beads per cell. Other cytokines used in specific experiments include: mouse IFNG (Cell Sciences, CRI001B), mouse IL7 (Cell Sciences, CRI131B) and mouse IL15-IL15R complex (eBioscience, 14–8152), mouse IL2 (eBioscience, 14–8021). Where indicated, anti-mouse IL2 (Biolegend, clone JES6–5H4) and its corresponding isotype control (Biolegend, clone RTK4530) were added to cultures to block IL2-induced signaling. For signaling inhibition, unless otherwise indicated, T cells were incubated for 1.5 to 2 h prior to stimulation with any of the following inhibitors at the concentration indicated in the figure legend: rapamycin (Sigma, r8781), JAKi-I (EMD, 420099), tofacitinib (LC Laboratories, T-1377), ruxolitinib (LC Laboratories, R-6600). Other inhibitors used in T cell cultures include: MG-115 (Enzo, BML-ZW8445), lactacystin (Enzo, BML-PI104), cycloheximide (Sigma, C4859) and actinomycin D (Sigma, 114666) at the concentrations indicated in each experiment.
Immunoblots
Cells were harvested and washed once in cold phosphate-buffered saline (PBS; HyClone, SH30028.02) and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.7, 150 mM NaCl, 1% [v/v] NP40 [Sigma, 74385] 0.5% sodium deoxycholate [Sigma, D6750], 0.1% SDS [American Bioanalytical, AB01920]) and freshly added cOmplete Protease Inhibitor cocktail 1x (Roche, 11697498001), 2 mM PMSF, 1 mM DTT, phosphatase inhibitor cocktail (Roche, 04906845001) for 30 min on ice. Protein concentration of precleared (20,000 g, 4°C, 30' min) extracts was quantified using the Bradford assay (Bio-Rad, 500–0006). Ten to 12 µg of protein was separated by SDS-PAGE (13% acrylamide) and transferred onto nitrocellulose membrane. Specific proteins were detected by immunoblot using the following primary antibodies: rabbit anti-LC3 (MBL, PM036), mouse anti-STAT5 (Invitrogen, 33–5900), rabbit anti-phospho-STAT5 (Y694; Invitrogen, 716900), mouse anti-STAT6 (BD bioscience, 611290), rabbit anti-phospho-STAT6 (Y641; Invitrogen, 700247), rabbit anti-STAT1 (Cell Signaling Technology, 9172), rabbit anti-phospho-STAT1 (Y701; Invitrogen, 44376G), mouse anti-HSP70–72 (Enzo, ADI-SPA-810), rabbit anti-JAK3 (Cell Signaling Technology, 8863), mouse anti-GFP (Acris Antibody, AM20840PU-N), rabbit anti-RPS6KB (Cell Signaling Technology, 9202), rabbit anti-phospho-RPS6KB (T389; Cell Signaling Technology, 9205), mouse anti-BECN1 (Novus, NBP1–00084), rabbit anti-SQSTM1/p62 (Enzo BML-PW9860), mouse anti-TUBA/αTubulin (Cell Signaling Technology, 3873), and mouse anti-ACTB/β-actin (Abcam, Ab6276). Where multiple proteins were analyzed on the same gel, membranes were either directly probed for the first protein then reprobed for the next protein or stripped before reprobing. Band intensities were quantified using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence
Cells were harvested and resuspended in cold PBS and an aliquot of 100 µL containing 1 to 1.5×105 cells were loaded into cytology funnel chambers, spun for 5 min at 220 g in a cytospin centrifuge to deposit a layer of cells on a microscope slide. From here, all steps were performed at room temperature and solutions discarded by aspiration. Cells were fixed in paraformaldehyde 4%-PBS for 10 min and permeabilized in digitonin (Sigma, D141) 0.01%-PBS for 12 min. Excess digitonin was quenched by incubation in 50 mM NH4Cl−PBS for 3 to 5 min, and rinsed 3 times in PBS. Cells were incubated with anti-LC3 for 30 min, rinsed in PBS for 30 min and incubated with Alexa Fluor 488-conjugated anti-rabbit (Invitrogen, A11008) for 30 min. After 30 min of rinse, 50 µL of mounting solution containing DAPI (Southernbiotech, 0100–20) was loaded on the cells and a cover slip was lower slowly and sealed on it. Images were acquired using a fluorescence microscope with apotome technology (Carl Zeiss, Jena, Gernany).
Autophagy flux and LC3 puncta assessment
For any experimental condition, to measure autophagy flux lysosomal turnover of LC3-II was quantified on immunoblot by comparing the levels of LC3-II in untreated T cells and in T cells that were treated with 20 mM NH4Cl and 100 µM leupeptin (Fisher scientific, BP2662) or 100 µM vinblastine (LC labs, V-7300) to block lysosomal proteases or autophagosome-lysosome fusion, respectively, for the last 3 h of culture in each specific condition analyzed. Immunoblot-band intensity was measured using ImageJ and the average quantification of at least 2 different exposures per gel was used for each experiment. ACTB-normalized LC3-II densitometric value in the sample without lysosomal inhibitors was subtracted from the one with inhibitors (i.e. autophagy flux). Induced autophagy was defined as the autophagy flux above the flux measured in basal level (resting cell) induced by a specific stimulation. For immunofluorescence images analysis, the number of LC3 puncta per cell was quantified using ImageJ MaxEntropy auto-threshold method and particles quantification tool. Autophagic flux was defined as the accumulation of puncta in sample treated with lysosome inhibitor vs untreated.
Pulse-chase and LC3 immunoprecipitation
TH1 cells were activated in T-cell media without cysteine or methionine in the presence of 35S-labeled cysteine and methionine (Perkin Elmer, Easy tag 35S mix) at 0.15 mCi/ml. After 3.5 h, half of the cells were harvested and whole protein extract was prepared as described for immunoblot. An excess of cold cysteine and methionine (1 mM) were added in the other half for 3.5 additional h and whole protein extracts were prepared. For immunoprecipitation, 0.75 µg of anti-LC3 (MBL, PM036) or isotype control antibodies (Cell Signaling Technology, 2729) were immobilized on 22 µL of protein-G beads (Invitrogen, 10004D). 80 µg of protein diluted in 50 mM Tris-HCl, pH 7.7, 150 mM NaCl, 5 mM EDTA, 0.1% NP40 with protease inhibitors were incubated for 1 h at 4°C with the antibody-conjugated beads. After 3 washes immunoprecipitated products were run on SDS-PAGE and transfer to nitrocellulose membrane. The membrane was first used to expose BioMax MR Film (Carestream) for the 35S signal and then used to blot for LC3 and ACTB.
Real-time quantitative PCR
Total RNA was extracted and purified using TRIzol reagent (Life Technologies, 10296–010). Purified RNA (1 to 2 μg) was treated with RNase-free DNase (Roche, 04716728001) then reverse transcribed using SuperScript III First-Strand Synthesis System (Life Technologies, 18080–051) and random hexanucleotides primers. cDNAs was then amplified by real-time PCR using power Sybr green (Applied Biosystems, 4309155) with the following primers: mouse Lc3b (Map1lc3b) mRNA: forward 5'-ACAAAGAGTGGAAGATGTCCGGCT-3', Reverse 5'-TGCAAGCGCCGTCTGATTATCTTG-3'; mouse Map1lc3b pre-mRNA: forward 5'-ATAGGCTGTGAACGCTGTGGATGT-3', reverse 5'-AAGGCTTGGTTAGCATTGAGCTGC-3'; mouse ACTB (Actb) forward 5'-GGCTGTATTCCCCTCCATCG-3' reverse 5'-CCAGTTGGTAACAATGCCATGT-3'. In the case of pre-mRNA quantification, RNA controls for each sample were processed in same conditions without the reverse transcriptase and quantify by real-time quantitative PCR to control insignificant genomic DNA contamination.
Plasmid constructs and generation of lentiviral vector particles for GFP-LC3 or shRNA transduction in T cells
BglII-SmaI HsC3wt and HsLC3-GΔ open reading frame (ORF) was extracted from pEX-CFP-HsLC3 (Addgene, 24985 and 24986; Isei Tanida laboratory plasmids) and inserted in frame with GFP into pEGFP-C1 (Clonetech, 6084–1). Then, the GFP-LC3 (WT or GΔ) ORF was extracted from these new constructs and inserted into the lentiviral vector pCCL.sin.PPT.promPGK.GFP replacing the original GFP cDNA present in the lentiviral vector. Lentiviral particles were produced using a third generation lentiviral system with 3 packaging-plasmids (pMDLg-pRRE, pRSV-Rev and pMD2.VSVG) and the lentiviral vector expressing GFP alone or GGP-LC3 fusions (WT or GΔ) transfected into HEK293T-cells. Virus-containing supernatants were collected at 36 h, 48 h and 72 h post-transfection and concentrated using an ultracentrifugation procedure. Lentiviruses were added to T cells after 24 h of activation. 48 h postinfection GFP positive T cells were sorted and expanded in culture until d 6 as described above For BECN1 downregulation pLKO.1 shRNA expression vectors from the TRC library were purchased from Sigma. Target sequences for mouse Becn1 used are: for BEC88 5'-CCACATGTTTACAATACCAAA-3' and for BEC92 5'-CGGACAGTTTGGCACAATCAA-3'. Nontarget shRNA control was purchased from Sigma (SCH002). Lentiviral particles were produced as described above using the shRNA plasmids as backbone. Lentiviruses were added to T cells after 24 h of activation. 48 h postinfection puromycin (4 µg/mL) was added to the media to select for positively infected cells and cells were expanded until d 6.
Transmission electron microscopy and morphometric analysis
Cells were fixed with 2.5% glutaraldehyde, and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer pH 7.4 for 45 min at room temperature. Cells were postfixed with 1% osmium tetroxide, dehydrated with a graded series of ethanol, and then infiltrated and embedded using EMbed 812 (Electron Microscopy Sciences, 14900). Ultrathin sections (7080 nm) were cut using a Leica Ultramicrotome (Leica, Buffalo Grove, IL, USA) and stained using uranyl acetate and contrasted with lead citrate. Samples were viewed under a Jeol JEM-1200EX transmission electron microscope (Jeol, Peabody, MA, USA) at 80 kV. Morphometric analysis was done using ImageJ (National Institutes of Health, Bethesda, MD, USA). Briefly, cytosolic area and individual autophagic vesicle areas were calculated after tracing the membrane profiles using the measure function of the ImageJ software. Classification of autophagic vacuoles according to their luminal content was done by a blinded independent observers using one single-category allocation for each vesicle. Autophagic vacuoles (vesicles <0.5 μm) were classified as autophagosomes when they met 2 or more of the following criteria: double membranes (complete or at least partially visible), absence of ribosomes attached to the cytosolic side of the membrane, luminal density similar to cytosol, and identifiable organelles or regions of organelles in their lumen. Vesicles of similar size but with a single membrane (or less than 40% of the membrane visible as double), luminal density lower than the surrounding cytosol, multiple single membrane-limited vesicles containing light or dense amorphous material were classified as autolysosomes.
Statistical analysis
Statistical analysis were performed using a 2-tailed Student t test or an ANOVA test with a Tukey post-hoc test. Significance was defined as P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We want to thank Drs. Ana Maria Cuervo, Eric Batsche, Eric Allemand and Genevieve Marcelin for valuable discussions. The authors acknowledge the Einstein Flow Cytometry Facility for sorting and the Analytical Imaging Facility for the processing of samples for electron microscopy experiments.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by NIH grants AG031782, the Glenn Foundation and an Irma T Hirschl award (to FM). The use of specific research cores was supported by the Einstein Nathan Shock Center in Basic Research in Aging (NIH grant AG038072).
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 2.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inoue Y, Klionsky DJ. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin Cell Dev Biol 2010; 21:664-70; PMID:20359542; http://dx.doi.org/ 10.1016/j.semcdb.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- 5.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol 2008; 445:77-88; PMID:18425443; http://dx.doi.org/ 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. . Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. . A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488-92; PMID:11100732; http://dx.doi.org/ 10.1038/35044114 [DOI] [PubMed] [Google Scholar]
- 8.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008; 90:313-23; PMID:17928127; http://dx.doi.org/ 10.1016/j.biochi.2007.08.014 [DOI] [PubMed] [Google Scholar]
- 9.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507-13; PMID:10995454; http://dx.doi.org/ 10.1083/jcb.150.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. . Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 2001; 20:5971-81; PMID:11689437; http://dx.doi.org/ 10.1093/emboj/20.21.5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes–Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell 2006; 17:1527-39; PMID:16421251; http://dx.doi.org/ 10.1091/mbc.E05-09-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/ 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- 14.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360-72; PMID:18843052; http://dx.doi.org/ 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. . Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol 2009; 335:71-84; PMID:19802560 [DOI] [PubMed] [Google Scholar]
- 17.Valdor R, Macian F. Autophagy and the regulation of the immune response. Pharmacol Res 2012; 66:475-83; PMID:23063674; http://dx.doi.org/ 10.1016/j.phrs.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307:593-6; PMID:15591165; http://dx.doi.org/ 10.1126/science.1104904 [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753-66; PMID:15607973; http://dx.doi.org/ 10.1016/j.cell.2004.11.038 [DOI] [PubMed] [Google Scholar]
- 20.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 2007; 27:505-17; PMID:17892853; http://dx.doi.org/ 10.1016/j.immuni.2007.07.022 [DOI] [PubMed] [Google Scholar]
- 21.Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol 2006; 177:5163-8; PMID:17015701; http://dx.doi.org/ 10.4049/jimmunol.177.8.5163 [DOI] [PubMed] [Google Scholar]
- 22.Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy Regulates Energy Metabolism during Effector T Cell Activation. J Immunol 2010; 185:7349-57; PMID:21059894; http://dx.doi.org/ 10.4049/jimmunol.1000576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204:25-31; PMID:17190837; http://dx.doi.org/ 10.1084/jem.20061303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, Roodman DG, Windle JJ, Zhang X, Lu B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ 2012; 19:144-52; PMID:21660048; http://dx.doi.org/ 10.1038/cdd.2011.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arsov I, Adebayo A, Kucerova-Levisohn M, Haye J, MacNeil M, Papavasiliou FN, Yue Z, Ortiz BD. A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol 2011; 186:2201-9; PMID:21239722; http://dx.doi.org/ 10.4049/jimmunol.1002223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 2009; 182:4046-55; PMID:19299702; http://dx.doi.org/ 10.4049/jimmunol.0801143 [DOI] [PubMed] [Google Scholar]
- 27.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84-91; PMID:16874052; http://dx.doi.org/ 10.4161/auto.1.2.1697 [DOI] [PubMed] [Google Scholar]
- 28.Bach EA, Szabo SJ, Dighe AS, Ashkenazi A, Aguet M, Murphy KM, Schreiber RD. Ligand-induced autoregulation of IFN-gamma receptor β chain expression in T helper cell subsets. Science 1995; 270:1215-8; PMID:7502050; http://dx.doi.org/ 10.1126/science.270.5239.1215 [DOI] [PubMed] [Google Scholar]
- 29.Pernis A, Gupta S, Gollob KJ, Garfein E, Coffman RL, Schindler C, Rothman P. Lack of interferon gamma receptor β chain and the prevention of interferon gamma signaling in TH1 cells. Science 1995; 269:245-7; PMID:7618088; http://dx.doi.org/ 10.1126/science.7618088 [DOI] [PubMed] [Google Scholar]
- 30.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science 1995; 270:794-7; PMID:7481767; http://dx.doi.org/ 10.1126/science.270.5237.794 [DOI] [PubMed] [Google Scholar]
- 31.Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol 2011; 186:1564-74; PMID:21191072; http://dx.doi.org/ 10.4049/jimmunol.1001822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity 2012; 36:947-58; PMID:22658522; http://dx.doi.org/ 10.1016/j.immuni.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris J. Autophagy and cytokines. Cytokine 2011; 56:140-4; PMID:21889357; http://dx.doi.org/ 10.1016/j.cyto.2011.08.022 [DOI] [PubMed] [Google Scholar]
- 34.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006; 313:1438-41; PMID:16888103; http://dx.doi.org/ 10.1126/science.1129577 [DOI] [PubMed] [Google Scholar]
- 35.Liang X, De Vera ME, Buchser WJ, Romo de Vivar Chavez A, Loughran P, Beer Stolz D, Basse P, Wang T, Van Houten B, Zeh HJ 3rd, et al. . Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res 2012; 72:2791-801; PMID:22472122; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang R, Tang D, Lotze MT, Zeh Iii HJ. Autophagy is required for IL-2-mediated fibroblast growth. Exp Cell Res 2013; 319:556-65; PMID:23195496; http://dx.doi.org/ 10.1016/j.yexcr.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al. . Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol 2014; 15:1152-61; PMID:25362489; http://dx.doi.org/ 10.1038/ni.3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, et al. . Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 2014; 3:e03703; PMID:25385531; http://dx.doi.org/ 10.7554/eLife.03706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013; 38:13-25; PMID:23352221; http://dx.doi.org/ 10.1016/j.immuni.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farkas T, Daugaard M, Jaattela M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem 2011; 286:38904-12; PMID:21930714; http://dx.doi.org/ 10.1074/jbc.M111.269134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 2013; 210:2119-34; PMID:24043759; http://dx.doi.org/ 10.1084/jem.20130252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, Morrissette NS, Walsh CM. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A 2008; 105:16677-82; PMID:18946037; http://dx.doi.org/ 10.1073/pnas.0808597105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korolchuk VI, Rubinsztein DC. Regulation of autophagy by lysosomal positioning. Autophagy 2011; 7:927-8; PMID:21521941; http://dx.doi.org/ 10.4161/auto.7.8.15862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol 2013; 203:563-74; PMID:24385483; http://dx.doi.org/ 10.1083/jcb.201306041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290-303; PMID:20381137; http://dx.doi.org/ 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A 2011; 108:11121-6; PMID:21690395; http://dx.doi.org/ 10.1073/pnas.1107969108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 2010; 3:ra31; PMID:20424262 [DOI] [PubMed] [Google Scholar]
- 50.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101-11; PMID:16186256; http://dx.doi.org/ 10.1083/jcb.200504035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A 2012; 109:8670-5; PMID:22592798; http://dx.doi.org/ 10.1073/pnas.1205305109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parekh VV, Wu L, Boyd KL, Williams JA, Gaddy JA, Olivares-Villagomez D, Cover TL, Zong WX, Zhang J, Van Kaer L. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol 2013; 190:5086-101; PMID:23596309; http://dx.doi.org/ 10.4049/jimmunol.1202071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 2014; 15:65-74; PMID:24326622; http://dx.doi.org/ 10.1038/nrm3716 [DOI] [PubMed] [Google Scholar]
- 54.Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 2013; 27:1016-31; PMID:23651856; http://dx.doi.org/ 10.1101/gad.212282.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. . FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458-71; PMID:18054315; http://dx.doi.org/ 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 56.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al. . The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127-41; PMID:20038797; http://dx.doi.org/ 10.1172/JCI40027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A 2010; 107:18511-6; PMID:20937856; http://dx.doi.org/ 10.1073/pnas.1006124107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meenhuis A, van Veelen PA, de Looper H, van Boxtel N, van den Berge IJ, Sun SM, Taskesen E, Stern P, de Ru AH, van Adrichem AJ, et al. . MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood 2011; 118:916-25; PMID:21628417; http://dx.doi.org/ 10.1182/blood-2011-02-336487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, et al. . VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012; 21:532-46; PMID:22516261; http://dx.doi.org/ 10.1016/j.ccr.2012.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol 2010; 648:193-214; PMID:20700714; http://dx.doi.org/ 10.1007/978-1-60761-756-3_13 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.