

Abstract
Here, we present a summary of our recent findings on the (patho-)physiological relevance of PINK1-phosphorylated ubiquitin (p-S65-Ub). Using novel polyclonal antibodies, we find that p-S65-Ub specifically accumulates on damaged mitochondria. Phosphorylation of ubiquitin on serine 65 depends on the activity of PINK1 and the signal is vastly amplified by the activity of the E3 ubiquitin ligase PARK2/Parkin in a feed-forward loop. The induction of p-S65-Ub in primary cells suggests a significant role of p-S65-Ub also in neurons. Consistent with a marker for damaged mitochondria that are undergoing mitophagy, we find anti-p-S65-Ub immunoreactive granules that partially colocalize with mitochondria, lysosomes and ubiquitin in human post-mortem brain. The number of p-S65-Ub positive granules increases with age and with PD, highlighting the relevance of p-S65-Ub as a potential biomarker and therapeutic target.
Keywords: autophagy; mitochondria; mitochondrial quality control; mitophagy; PARK2/Parkin; Parkinson disease, phosphorylated ubiquitin; PINK1
The kinase PINK1 and the E3 ubiquitin (Ub) ligase PARK2, of which either loss causes early-onset Parkinson disease (PD), together orchestrate a cytoprotective mitochondrial quality control. Upon mitochondrial stress, PINK1 is stabilized on the outer membrane of damaged organelles, phosphorylates the small modifier protein Ub and the Ub-like domain of PARK2 at the conserved serine 65 (S65) residue. As a result, PARK2 releases its auto-inhibited structure, translocates from the cytosol to mitochondria and is fully activated. The phosphorylated Ub (p-S65-Ub) signal is then amplified by the concerted enzymatic action of PINK1 kinase and PARK2 Ub ligase. Eventually, mitochondrial components are selectively targeted for degradation via the proteasome and the autophagy-lysosome system (via mitophagy). Structural and functional effects of Ub phosphorylation for (dis-)assembly of poly-Ub chains in general and PINK1-PARK2-mediated mitochondrial quality control in particular have been suggested from studies in vitro and in cells. However, due to the lack of appropriately sensitive tools and methods, the physiological significance and disease relevance of this mitochondrial quality control remained uncertain. Using novel antibodies, we could provide evidence for the existence of p-S65-Ub under endogenous conditions in primary cells including neurons, and human post-mortem brain, but not in samples from PD patient with PINK1 mutations.
We developed polyclonal antibodies against p-S65-Ub and validated their performance across a spectrum of applications in vitro, and in cells and tissues, as illustrated in Figure 1. Two rabbits were immunized with phosphorylated peptides surrounding S65 of Ub. Sera were affinity purified and depleted against nonphosphorylated peptides. The antibodies were first tested with PINK1-phosphorylated recombinant Ub monomers and poly-Ub chains. We noted a slight preference of the p-S65-Ub antibodies for phosphorylated K48- over K63-linked poly-Ub chains, but the latter linkage could equally affect PINK1 phosphorylation and antibody binding. The PINK1-PARK2 pathway is repressed under normal conditions and p-S65-Ub is indeed almost undetectable, but strongly induced upon mitochondrial stress in cell lines and primary neurons as an early marker of an apparently fundamental cell biological process. We further confirmed lack of stress-induced p-S65-Ub signal in primary fibroblasts, iNeurons (induced neurons) and brain samples of PD patients carrying a homozygous PINK1 loss-of-function mutation to fully establish the specificity of the p-S65-Ub antibodies.
FIGURE 1.
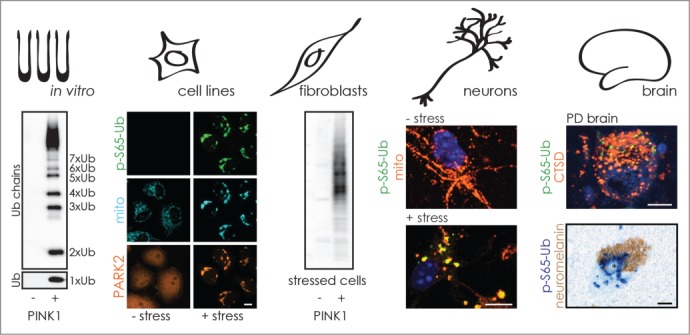
p-S65-Ub is a specific marker for mitochondrial stress. The performance of novel anti-p-S65-Ub antibodies is illustrated across various applications and highlights the potential for biomarker and therapeutic development. The antibodies were validated for their specificity to p-S65-Ub in vitro and in cell lines as well as in primary cells and post-mortem brains from PD patients with PINK1 kinase mutations. p-S65-Ub is almost undetectable in cells under basal conditions, but is induced by mitochondrial stress also in primary mouse neurons. In accordance with a mitophagy label, p-S65-Ub partially colocalizes with total Ub, mitochondria, and lysosomal markers. In sporadic PD brains, p-S65-Ub reactivity can be found in the vicinity of, but not within, Lewy bodies. Strikingly, p-S65-Ub-positive granules increase with age and disease, possibly reflecting enhanced mitochondrial damage and/or declined degradative capacities.
In line with a dual role for p-S65-Ub as a signaling molecule for PARK2 activation and as a receptor for its recruitment to mitochondria, mono- and dimeric forms of p-S65-Ub are found in the cytosol, whereas phosphorylated poly-Ub chains are mostly detected on damaged organelles. Shortly after translocation of PARK2 to mitochondria, we could confirm a strong amplification of mitochondrial p-S65-Ub signal (mostly poly-Ub chains) in the presence of both functional enzymes, PINK1 and PARK2. When functional PARK2 is missing, we find enhanced levels of cytosolic p-S65-Ub monomers. It remains unclear if more of these are released from mitochondria in the absence of functional PARK2 or if momomeric cytosolic p-S65-Ub moieties are utilized by PARK2 for the formation of phosphorylated poly-Ub chains on mitochondria.
Upon withdrawal of mitochondrial stress, the p-S65-Ub signal swiftly declines and is followed by reduction of total poly-Ub chains shortly after. Together with effects of Ub phosphorylation on (dis-)assembly of phosphorylated poly-Ub chains and their recognition by Ub binding proteins that decode the signals, this further highlights the complexity of PINK1-PARK2 regulation. Several E2 Ub-conjugating and opposing de-ubiquitinating enzymes regulate PARK2 and mitochondrial quality control at distinct steps of a sequential process. Now, it will be important to identify the p-S65-Ub binding proteins and phosphatases as additional drug targets and for a complete understanding of phospho-Ub signaling.
In human brain sections, similar to cultured cells, we found small anti-p-S65-Ub immunoreactive cytoplasmic granules that are partially colocalized with markers for mitochondria, lysosomes, and total ubiquitin. In appearance, the p-S65-Ub-positive granules resemble bodies observed in granulovacuolar degeneration that have been suggested to present late-stage autophagic vacuoles. This is consistent with a role of p-S65-Ub as a mitophagy label, but the exact cellular functions during the entire course of mitochondrial quality control remain unclear. In addition, accumulation of p-S65-Ub could be a sign of increased mitochondrial damage and of defective clearance via mitophagy. Strikingly, p-S65-Ub granules appear to increase with age and sporadic PD in our small brain cohort, but are absent in PINK1 mutant brain. Of note, p-S65-Ub granules are not found within Lewy bodies, the neuropathological hallmarks of PD, but frequently surrounding these cytoplasmic inclusions. It will be essential to clarify the nature of p-S65-Ub granules and to quantify p-S65-Ub levels across brain regions during aging and PD as well as in a spectrum of other neurodegenerative disorders in which alterations in PINK1-PARK2-dependent mitochondrial quality control have been implicated.
In summary, our findings highlight the potential of p-S65-Ub for biomarker and therapeutic development as it accumulates with stress, age, and disease. While the exact functions of p-S65-Ub remain unknown, our study suggests that the PINK1-PARK2-dependent mitochondrial quality control pathway is activated in vivo in human brain. Though PINK1 has just recently been discovered as the first Ub kinase, several other post-translational modifications of Ub have already been identified. This further expands the Ub code beyond ‘simple’ signaling from mono-Ub or topologically and functionally diverse poly-Ub chains. At the same time this provides unprecedented specificity for a ‘ubiquitous’ label that will be key to resolve downstream biological signaling mechanisms and to further unravel the fundamental roles of Ub in health and disease.
Funding
WS is partially supported by NIH/NINDS R01NS085070, the Michael J. Fox Foundation for Parkinson Research and the Foundation for Mitochondrial Medicine, Mayo Clinic Foundation and the Center for Individualized Medicine, the Marriott Family Foundation, and a Gerstner Family Career Development Award. FCF is partially supported by a fellowship from the American Parkinson Disease Association (APDA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.