

Abstract
This study confirms that autophagy is activated concomitantly with KSHV lytic cycle induction, and that autophagy inhibition by BECN1 knockdown reduces viral lytic gene expression. In addition, we extend previous observations and show that autophagy is blocked at late steps, during viral replication. This is indicated by the lack of colocalization of autophagosomes and lysosomes and by the LC3-II level that does not increase in the presence of bafilomycin A1 in primary effusion lymphoma (PEL) cells induced to enter the lytic cycle, either by TPA/sodium butyrate (BC3 and BCBL1) or by doxycycline (TRExBCBL1-Rta). The autophagic block correlates with the downregulation of RAB7, whose silencing with specific siRNA results in an autophagic block in the same cells. Finally, by electron microscopy analysis, we observed viral particles inside autophagic vesicles in the cytoplasm of PEL cells undergoing viral replication, suggesting that they may be involved in viral transport.
Keywords: autophagy, herpesvirus, KSHV, lytic cycle, PEL, RAB7
Abbreviations
- ACTB/β-actin
actin beta
- ATG
autophagy related
- Baf
bafilomycin
- BECN1
Beclin 1 autophagy related
- Doxy
doxycycline
- EBV
Epstein-Barr virus
- EM
electron microscopy
- KSHV
Kaposi's sarcoma-associated herpesvirus
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- PBS
phosphate-buffered saline
- PEL
primary effusion lymphoma
- RAB7
RAB7 member RAS oncogene family
- SD
standard deviation
- T/B
TPA/sodium butyrate
- TPA
2-O-tetradecanoylphorbol-13-acetate
- TUBA1A/α-tubulin
tubulin alpha 1a
- VZV
varicella zoster virus.
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV) belongs to the human gamma-herpesvirus subfamily, Rhadinoviridae genus.1 It is associated with Kaposi's sarcoma and some hematological malignancies, such as primary effusion lymphoma and multicentric Castleman disease.2 KSHV infection of tumor cells is mostly latent, but it can be switched into lytic cycle either in vivo or in vitro upon appropriate stimuli such as exposure to 12-O-tetradecanoylphorbol-13-acetate (TPA) alone or in combination with sodium butyrate (T/B),3-5 Bortezomib,6 ORF50 expression7 and doxycycline in TRExBCBL1-Rta cells.8 Upon the activation of the lytic cycle, different sets of KSHV genes can be expressed:9 immediate early (e.g., K-bZIP),10,11 early (e.g., ORF69)12 and late (e.g., K8.1).13 Autophagy is a process of self-degradation of cellular components, upregulated in tumor cells and in stressful conditions. This is a multistep process regulated by the autophagy-related (ATG) genes.14 The vesicle nucleation, that represents the first step of autophagy, is dependent on the class III phosphatidylinositol 3-kinase complex formed by PIK3C3/Vps34 (mammalian/yeast protein), PIK3R4/Vps15, BECN1/Vps30/Atg6, and ATG14/Atg14. Among them BECN1/Beclin 1, has a central role in this process, since it interacts with molecules that regulate autophagy at several steps.15 A marker of the autophagic vacuoles is MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), an ortholog of yeast Atg8, that is expressed as a full-length cytosolic protein and upon autophagy induction is cleaved by ATG4 to form LC3-I, which is then conjugated to phosphatidylethanolamine generating LC3-II. In the last autophagic phases, autophagosomes, transporting intracellular proteins, fuse with lysosomes. Autophagosomes can also transport intracellular microorganisms to the lysosomes, contributing to the clearance of the microbial infections.16 For that reason viruses, especially those that persist in the infected host, subvert autophagy to avoid their own elimination.17 However, exceptions have been described and, for example, among herpesviruses, varicella zoster virus (VZV) is able to establish a successful infection without affecting the autophagic process in the host cells.18 In contrast, KSHV encodes several proteins that block autophagy at multiple levels19-21 even if autophagy has been shown to be activated and to promote the KSHV lytic cycle.22,23 In this study, besides confirming that autophagy is activated concomitantly with the KSHV lytic cycle in 2 different PEL cell systems and that autophagy promoted viral lytic gene expression, we found that the late autophagic steps were blocked, as indicated by several lines of experimental evidence. Thus, the autophagic block might represent a mechanism that allows KSHV to avoid reaching the lysosomes and be degraded by the lysosomal proteases. Meanwhile, this strategy enables KSHV to hijack the autophagic machinery to travel toward the cell surface. Based on the results obtained in this study, we suggest that autophagy manipulation could be exploited to reduce viral replication and consequently to improve the outcome of therapies against KSHV-associated malignancies, whose pathogenesis correlates with continuous viral release.
Results and Discussion
BC3 and BCBL1 PEL cells were treated with TPA (20 ng/ml) and sodium butyrate (0.3 mM) combined (T/B) for 36 h, to induce the KSHV lytic cycle. We found that at this time, when the KSHV lytic cycle was activated (as indicated by K-bZIP lytic antigen expression), the level of the lipidated form of LC3 (LC3-II) increased in comparison to the onset of viral replication (time 0) and to untreated controls (-) in both PEL cell lines (Fig. 1A). Of note, in BCBL1 cells a certain amount of spontaneous viral replication occurred (indicated by the low expression of K-bZIP in the untreated cells) and concomitantly a higher basal level of LC3-II was also detected. Then, BC3 and BCBL1 cells were transfected with a pEGFP-LC3 plasmid and treated with T/B.24 We found that autophagosomes, revealed by GFP-LC3 puncta, accumulated in PEL cells undergoing KSHV replication and expressing K8.1 membrane antigen (Fig. 1B). The majority of replicating cells (about 70%) showed GFP-LC3 puncta; conversely, only 10% of the cells not undergoing KSHV replication, showed GFP-LC3 puncta (data not shown), suggesting that autophagy was induced during KSHV lytic cycle activation. We then stably transfected BC3 cells with a pEGFP-LC3 plasmid, to avoid artifactual results14,25 and quantified the number of LC3-positive BC3 cells, induced or not to enter the KSHV lytic cycle by T/B for 0, 18, 36 and 60 h. The results shown in Fig. 1C indicate that the number of GFP-LC3-positive cells (containing at least 5 GFP-LC3 puncta)26 increased up to 36 h of T/B treatment, whereas it was slightly changed in the untreated control cells. Similar results were obtained with transiently pEGFP-LC3 plasmid-transfected BC3 cells (Fig. 1D). For these quantitative experiments we counted about 200 cells induced or not to enter the lytic cycle, for each time point. The accumulation of LC3-II as well as of autophagosomes suggest the activation of the autophagic process, but they do not make it possible to assess the completeness of the autophagic flux, since both LC3-II and autophagosomes are formed and degraded during autophagy induction.14
Figure 1.

T/B activates autophagy concomitant with KSHV lytic cycle induction in PEL cells. (A) Western blot analysis showing the accumulation of the lipidated form of LC3 (LC3-II) concomitant with the expression of K-bZIP lytic cycle antigen induced by TPA at 20 ng/ml and sodium butyrate at 0.3 mM (T/B) upon 36 h of treatment in BC3 and BCBL1 cells, in comparison to the untreated control. LC3-I/-II as well as K-bZIP at the onset of the viral lytic cycle are also shown. TUBA1A/α-tubulin was used as loading control and a representative experiment out of 3 is shown. The histograms represent the mean plus standard deviation (SD) of the densitometric analysis of the ratio of K-bZIP or LC3-II to TUBA1A of 3 different experiments. (B) PEL cells were transfected with pEGFP-LC3 plasmid before inducing the lytic cycle with T/B. Autophagy activation was revealed by the appearance of GFP-LC3 puncta (green) and the cells undergoing KSHV lytic cycle were indicated by the expression of KSHV K8.1, a membrane lytic protein, by using a specific monoclonal antibody (red staining). Bar: 5 micron. (C) BC3 cells were stably transfected with pEGFP-LC3 plasmid and quantification of cells expressing more than 5 GFP-LC3 puncta per cell (as evaluated by immune fluorescence microscopy) was performed in both unreactivated and T/B reactivated PEL cells at 4 time points: 0, 18, 36 and 60 h. (D) BC3 cells were transiently transfected with pEGFP-LC3 plasmid and quantification of cells expressing more than 5 GFP-LC3 puncta per cell (as evaluated by immune fluorescence microscopy) was performed in both unreactivated and T/B reactivated PEL cells at 4 time points: 0, 18, 36 and 60 h. About 200 cells were counted for each time point.
To shed more light on this process, we added bafilomycin A1 (Baf; 20 nM) to PEL cells, during the last 3 h of T/B treatment. Baf, as an inhibitor of the vacuolar-type H+-ATPase, prevents LC3-II degradation and allows the evaluation of total LC3-II formation. We found that the LC3-II level did not increase after 36 h of T/B treatment in combination with Baf (Fig. 2A), a time at which the KSHV lytic cycle was induced (evidenced by the expression of the KSHV lytic antigen K-bZIP), suggesting the occurrence of an autophagic block during KSHV replication. In contrast, a complete autophagic flux was observed in BJAB, a KSHV-negative lymphoma cell line, undergoing 36 h of T/B treatment (Fig. 2B). Concomitant with the lytic cycle activation and the autophagic block, we found that expression of RAB7 (a member of the RAS oncogene family) was reduced in BC3 cells (Fig. 2A). RAB7,27 besides regulating lysosome biogenesis,28 plays an important role in the fusion of autophagosomes with lysosomes, a step required for a complete autophagic flux.29 Next, a kinetic evaluation of the expression of K-bZIP, RAB7 and LC3-I/-II was performed after 0, 9, 18 and 36 h of T/B treatment in the presence or in the absence of Baf. As shown in Figure 2C, when the KSHV lytic cycle was induced, RAB7 expression was reduced and an autophagic block occurred, based on the change in the level of LC3-II in the presence or in the absence of Baf.
Figure 2.

The autophagic flux was blocked in BC3 cells undergoing KSHV lytic cycle activation. (A) Analysis of K-bZIP lytic antigen, RAB7 and LC3-I/-II expression in the presence or in the absence of Baf (used for the last 3 h at 20 nM) in BC3 cells induced to enter the lytic cycle by T/B treatment for 36 h or (B) in the KSHV-negative BJAB cells undergoing the same treatment. (C) Kinetic analysis of the expression of K-bZIP, RAB7 and LC3-II and effects of Baf on the expression of K-bZIP, RAB7 and LC3-II in BC3 cells induced to enter the lytic cycle by T/B at the indicated time points. ACTB was used as loading control and a representative experiment out of 3 is shown. The histograms represent the mean plus SD of the densitometric analysis of the ratio of specific proteins to ACTB of 3 different experiments. (D) BC3 cells were transfected with pEGFP-LC3 plasmid and induced to enter the lytic cycle by T/B treatment for 0, 18 and 36 h. Lysosomes were stained in red with LysoTracker while autophagosomes were indicated by GFP-LC3 puncta. Bar: 5 micron. (E) Gallery of BC3 cells transfected with pDest-mCherry-EGFP-LC3B plasmid and treated with T/B for 36 h or starved for 2 h. The yellow puncta indicate autophagosomes while red puncta indicate autolysosomes. A representative experiment out of 3 is shown. Bar: 5 micron.
Then, to further analyze the autophagic flux, we investigated the localization of autophagosomes and lysosomes and observed that in the majority of PEL cells treated with T/B for 36 h, the GFP-LC3 puncta (stained in green and indicating autophagosomes) did not colocalize with the lysosomes (stained in red with LysoTracker Red). Conversely, at earlier time points some of the autophgosomes and lysosomes colocalized, as indicated by the appearance of yellow puncta (Fig. 2D). These results are in agreement with those obtained for LC3-II in the presence of Baf by western blot analysis (Fig. 2C) and suggest that the autophagic flux was complete during the first 18 h of T/B treatment, whereas it was blocked when the KSHV lytic antigens start to be expressed. Finally, by transfecting BC3 cells with the pDest-mCherry-EGFP-LC3B plasmid,30 we found that in T/B-treated cells most of the autophagic structures displayed yellow fluorescence, due to the concomitant expression of mCherry and GFP, corresponding to autophagosomes. In contrast, the GFP component of the fluorescence was decreased by the complete autophagic flux induced by starvation in the same cells (due to instability of GFP in the lysosome) and consequently most of the autophagic structures appeared red, corresponding to autolysosomes (Fig. 2E). These results further suggest that the KSHV lytic cycle activation might impair the fusion between autophagosomes and lysosomes in PEL cells.
We then investigated the completeness of the autophagic flux in a different cell system, the TRExBCBL1-Rta cells. We observed that an autophagic block occurred also in these cells induced to enter the lytic cycle by doxycycline treatment for 48 h while a complete autophagy was observed in TRExBCBL1-vector control cells undergoing doxycycline treatment for the same time (Fig. 3A). The efficiency of lytic cycle induction by doxycycline in TRExBCBL1-Rta cells was analyzed in the same cells either by western blot (Fig. 3B) or by immunofluorescence (Fig. 3C), to further confirm that a block of the autophagic flux was occurring concomitantly with KSHV lytic cycle activation. Upon lytic cycle activation, we found that RAB7 was downregulated also in these cells (Fig. 3B), suggesting that it could be one of the mechanisms underlying the block of autophagy at the final steps during KSHV replication. Then, to demonstrate that the reduction of RAB7 would lead to a block of autophagy in PEL cells, we silenced this molecule by specific siRNA (Fig. 4A) and found that indeed LC3-II did not further increase in the presence of Baf in RAB7 knockdown cells (Fig. 4B). A reduction of RAB7 expression was also previously observed during the replication of Epstein-Barr virus (EBV),17 another member of the gamma-herpesviruses subfamily, Lymphocryptoviridae genus, suggesting that it represents a common feature during gamma-herpesvirus replication. Moreover, the results obtained in this study are in agreement with a recent paper reporting that the transfection of the KSHV K7 lytic protein impaired the fusion of autophagosomes with lysosomes in HeLa cells, in which autophagy was induced by rapamycin. 31
Figure 3.

The autophagic flux was blocked in TRExBCBL1-Rta cells undergoing KSHV lytic cycle activation by doxycycline treatment. (A) Evaluation of the autophagic flux based on LC3-II accumulation in the presence or in the absence of Baf (used for the last 3 h at 20 nM) in TRExBCBL1-Rta cells induced to enter the lytic cycle by treatment with doxycycline for 48 h. TRExBCBL1-vector cells were used as control. (B) The expression of KSHV lytic antigen K8.1 and RAB7 was analyzed by western blot. TUBA1A was used as loading control and a representative experiment out of 3 is shown. The histograms represent the mean plus SD of the densitometric analysis of the ratio of LC3-II, K8.1 or RAB7 to TUBA1A of 3 different experiments. (C) Percentage of cells expressing K8.1 (red) lytic antigen analyzed by immunofluorescence. Nuclei were stained by DAPI (blue).
Figure 4.
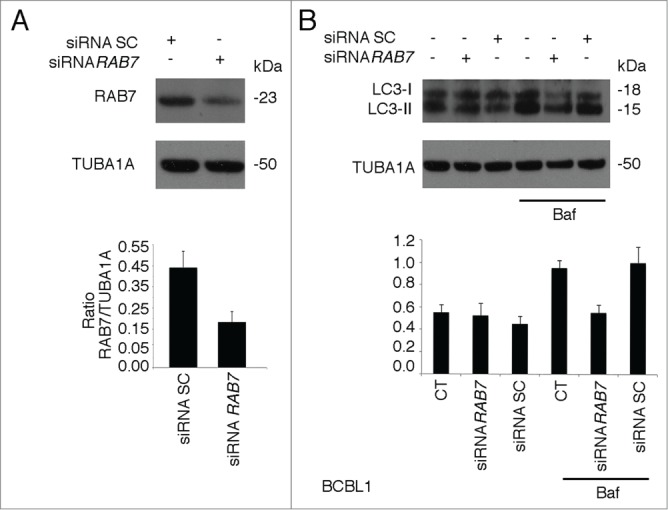
RAB7 knockdown leads to an autophagic block in PEL cells. BCBL1 cells were knocked down for RAB7 or scramble (SC) treated and (A) RAB7 and (B) LC3-II expression level was evaluated in the presence or in the absence of Baf. TUBA1A was used as loading control and a representative experiment out of 3 is shown. The histograms represent the mean plus SD of the densitometric analysis of the ratio of LC3-II to TUBA1A of 3 different experiments.
Next, to investigate the role of autophagy in the KSHV lytic cycle, we knocked down BECN1, a gene essential for the autophagic process, before inducing the lytic cycle with T/B. We found that KSHV K-bZIP lytic protein expression was reduced in BECN1-silenced BC3 cells in comparison to the scramble control (Fig. 5A). Accordingly, the number of cells expressing K-bZIP was strongly reduced by BECN1 knockdown (Fig. 5B). Similar results were obtained by ATG5 silencing (data not shown). These results indicate that autophagy promoted the KSHV lytic cycle, in agreement with a recent study22 and similarly to what we and other authors have previously observed during EBV replication.17,32-34 Finally, by electron microscopy (EM) analysis, autophagic features were observed in the majority of virus-producing cells and about 30% of viral particles were contained within the double-membrane autophagic vesicles present in the cytoplasm of PEL cells induced to enter the KSHV lytic cycle (200 cells were analyzed; Fig. 5C). Based on this observation and on the negative effect of autophagy inhibition on viral lytic expression, we propose that KSHV, similarly to EBV, might exploit the autophagic machinery for its transport, to enhance viral production. The study of the mechanisms that regulate KSHV lytic cycle activation are of fundamental importance since KSHV-associated malignancies, such as Kaposi's sarcoma, are characterized by a continuous release of viral particles that contributes to the disease's maintenance.35 The finding that autophagy is involved in KSHV replication suggests that manipulation of this process could lead to a better control of viral production and could restrain the progression of KSHV-associated malignancy diseases.
Figure 5.
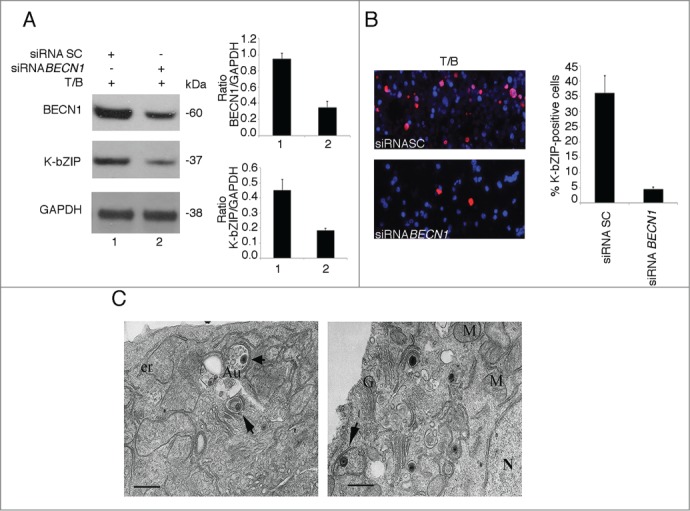
Autophagy enhances the KSHV lytic cycle. (A) K-bZIP expression was evaluated by western blot analysis in BC3 cells transfected with scramble (SC) or BECN1 siRNA for 48 h and then induced to enter the lytic cycle by 36 h of T/B treatment. GAPDH was used as a loading control and a representative experiment out of 3 is shown. The histograms represent the mean plus SD of the densitometric analysis of the ratio of BECN1 or K-bZIP to GAPDH of 3 different experiments. (B) BC3 cells transfected with scramble (SC) or BECN1 siRNA for 48 h and then induced to enter the lytic cycle by 36 h of T/B were analyzed for K-bZIP (red) expression by immunofluorescence. Nuclei were stained by DAPI (blue). The histograms represent the mean plus SD of the percentage of K-bZIP-positive cells of 3 different experiments. (C) Electron microscopy analysis of BC3 cells induced to enter the lytic cycle by T/B. Autophagic vacuoles containing viral particles (arrows) were observed in the cytoplasm of BC3 virus-producing cells. Au, autophagosomes; er, endoplasmic reticulum; G, Golgi apparatus; M, mitochondrion; N, nucleus. Bar: 0.5 μm.
Materials and Methods
Cell culture and reagents
BC3 (ATCC, CRL-2277), BCBL1 (kindly provided by Prof. P. Monini, National AIDS Center, Istituto Superiore di Sanità, Rome, Italy), TRExBCBL1-Rta (kindly provided by Prof. J. Jung, Dept. of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine, Los Angeles, California, USA) and TRExBCBL1-vector (kindly provided by Prof. J. Jung, Dept. of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine, Los Angeles, California, USA)8 are human B-cell lines derived from PEL-carrying latent KSHV. BJAB is an EBV-negative Burkitt lymphoma cell line (kindly provided by Prof. MG Masucci, Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm), Sweden). The cells were cultured in RPMI 1640 (Sigma, R0883), 10% fetal calf serum (Euroclone, ECLS0180L), L-glutamine and streptomycin (100 µg/ml) and penicillin (100 U/ml) (Gibco, 10378-016) in 5% CO2 at 37°C. TRExBCBL1-Rta and TRExBCBL1-vector cell lines (kindly provided by Prof. J. Jung) were cultured using the same medium in the presence of hygromycin (100 µg/ml) (Sigma Aldrich, H0654) and blasticidin (100 µg/ml; Santa Cruz Biotechnology, sc-204655A) in 5% CO2 at 37°C. The KSHV lytic cycle was induced in BC3 and BCBL1 cells by treatment with TPA (20 ng/ml; Sigma Aldrich, P8139) and sodium butyrate (0.3 mM; Sigma Aldrich, B5887) for the indicated times. Otherwise, the viral replication in TRExBCBL1-Rta cells was activated by treatment with doxycycline (1 µg/ml) (Sigma, D1822) for the indicated times. To investigate autophagy, the cells were treated with Baf (20 nM; Santa Cruz Biotechnology, sc-201550) for the last 3 h.25
A stable BC3 cell line expressing GFP-LC3 was grown in complete RPMI medium supplemented with 0.8 mg/ml geneticin/G418 (Life Technologies, 10131-027)
Antibodies
In western blotting analysis, we used in this work the following primary antibodies: mouse monoclonal anti-K-bZIP (1:500; Santa Cruz Biotechnology, sc-69797), mouse monoclonal anti-K8.1 (1:400; ABI Advanced Biotechnologies, 13-213-100) and rabbit polyclonal anti-RAB7 (1:500; Santa Cruz Biotechnology, sc-10767). To study autophagy we used the following primary antibodies: rabbit polyclonal anti-LC3 (1:1000; Novus Biologicals, NB100-2220SS) and rabbit polyclonal anti-BECN1 (1:1000; Cell Signaling Technology, 3738S).
Monoclonal mouse anti-TUBA1A/α-tubulin (1:1000; Sigma Aldrich, T6199), anti-ACTB/β−actin (1:10000; Sigma Aldrich, A5441) and anti-GAPDH (Santa Cruz Biotechnology, sc-13717) were used as loading controls. The goat polyclonal anti-mouse IgG-HRP (Santa Cruz Biotechnology, sc-2005) and anti-rabbit IgG-HRP (Santa Cruz Biotechnology, sc-2004) were used as secondary antibodies. All the primary and secondary antibodies were diluted in phosphate-buffered saline (PBS; Sigma Aldrich, D8537) -0.1% Tween 20 (Sigma Aldrich, P1379) solution containing 3% BSA (Serva, 11943.03)
Western blot analyses
1 × 106 cells were washed twice with 1X PBS-0.1% Tween 20 solution and centrifuged at 212 x g for 5 min. The pellet fraction was lysed in a RIPA buffer containing 150 mM NaCl, 1% NP-40 (Sigma Aldrich, NP40S), 50 mM Tris-HCl, pH 8, 0.5% deoxycholic acid (Sigma Aldrich, D6750), 0.1% SDS (Sigma Aldrich,71736), protease (Sigma Aldrich, S8830) and phosphatase inhibitors (Sodium Orthovanadate; Sigma Aldrich, S6508) (Sodium Fluoride; Sigma Aldrich, S7920). Then, 30 µg of protein lysates were subjected to electrophoresis on 4-12% NuPage Bis-Tris gels (Life Technologies, N00322BOX) according to the manufacturer's instruction. To evaluate the LC3-I/-II, the cell lysates were denatured in 3X Loading Protein Buffer (150 mM Tris-HCl, pH 6.8, 6% SDS, 30% glycerol, 30 mM EDTA, 0.2% Bromophenol Blue) for 5 min at 100°C and run on a 15% gel (30% acrylamide/Bis Solution 29:1; Bio-Rad, 161-0159) in Tris-glycine-SDS buffer (Bio-Rad, 1610772). Then, the gels were transferred to nitrocellulose membranes (Bio-Rad, 162-0115) for 2 h in Tris-glycine buffer. The membranes were blocked in PBS-0.1% Tween 20 solution containing 3% BSA, probed with specific antibodies and developed using ECL Blotting Substrate (Advansta, K-12045-D20).
Electron microscopy analysis
PEL cells were treated with TPA (20 ng/ml) and sodium butyrate (0.3 mM) at the indicated times. Subsequently, cells were fixed in 2% glutaraldehyde (Electron Microscopy Sciences, 111-30-8) in 1X PBS for 24 h at 4°C, post-fixed in 1% OsO4 (Agar Scientific, R1016) for 2 h and stained for 1 h in 1% aqueous uranyl-acetate (Electron Microscopy Sciences, 22400). The samples were then dehydrated with graded acetone and embedded in Epon-812 (Electron Microscopy Sciences, 14120). One-micron thick sections were cut, stained with 1% methylene blue and viewed by light microscopy to select representative areas. Ultrathin sections were cut with a Reichert ultramicrotome, counterstained with uranyl acetate and lead citrate, and examined with a Philips CM10 transmission electron microscope (FEI).
Knockdown by small interfering RNA
The knockdown of BECN1 (Santa Cruz Biotechnology, sc-29797) or RAB7 (Santa Cruz Biotechnology, sc-29460) was performed in PEL cell lines using specific small interfering RNA duplex. The day before transfection, 3 × 105 cells were seeded in 12-well culture plates in RPMI medium without antibiotics. Subsequently, 75 pmol of siRNA duplex and 7.5 µl of Lipofectamine 2000 Transfection Reagent (Life Technologies, 11668027) were diluted in Optimem medium (Life Technologies, 31985062) and added to the cells for 48 h. The transfection efficiency was evaluated by a Fluorescein Conjugate-A siRNA (Santa Cruz Biotechnology, sc-36869), which was also used as a scrambled control.
Transfection and plasmids
We performed a transient transfection using the pEGFP-LC3 plasmid (kindly provided by Prof. G.M. Fimia, National Institute for Infectious Disease ‘L. Spallanzani’ IRCCS, Rome, Italy)24 or pDest-mCherry-EGFP-LC3B plasmid (kindly provided by Prof. T. Johansen, Molecular Cancer Research Group, Institute of Medical Biology, University of Tromsø and The Arctic University of Norway, Tromsø, Norway)30 in PEL cell lines. 5 × 105 cells were seeded into 6-well plates and then transfected with 5 µg of plasmid DNA/well with Lipofectamine 2000 according to the manufacturer's instructions. After 48 h, the cells were treated with TPA (20 ng/ml) and sodium butyrate (3 mM) for the indicated times. In order to activate autophagy, BC3 cells were transfected with pDest-mCherry-EGFP-LC3B for 48 h and then were cultured in Hank's balanced salt solution (Sigma Aldrich, E6267) medium for 3 h. The pDest-mCherry-EGFP-LC3B plasmid makes it possible to visualize the autophagic steps because the LC3 puncta, indicating autophagosomes arising during autophagy induction, change their color depending on the pH; they appear yellow when both green and red colors are expressed, but when autophagosomes fuse with lysosomes the GFP is unstable and autolysosomes become red. The LC3 dots were visualized with an Apotome Axio Observer Z1 inverted microscope (Zeiss, Germany) equipped with an AxioCam MRM (Zeiss, Germany) Rev.3 at 40 × magnification.
We generated a stably transfected BC3 cells with a plasmid expressing GFP-LC3. Briefly, 5 × 105 BC3 cells were cultured into 6-well plates and transfected with 5 µg of pEGFP-LC3 plasmid/well with Lipofectamine 2000 according to the manufacturer's instructions. After 48 h, the cells were resuspended in a medium containing 1.5 mg/ml of G418 and seeded in 96-well plates. After 10-14 d, the GFP-LC3-positive clones were isolated and selected by fluorescence microscopy.
Immunofluorescence
KSHV replication was activated with the lytic cycle inducers in PEL cell lines, as previously described. The cells were harvested, washed twice in 1X PBS and then incubated with mouse monoclonal anti-K8.1 antibody (1:300 in PBS; ABI Advanced Biotechnologies, 13-213-100) for 1 h on ice. Subsequently, they were washed 3 times in cold 1X PBS and incubated with polyclonal Cy3-conjugated sheep-anti mouse (1:100 in 1X PBS; Jackson Immunoresearch Laboratories, 515-165-062) for 30 min on ice. Finally, after several washing, the cells were resuspended in a 1X PBS-glycerol solution (1:1).
After BECN1 silencing, KSHV replication was induced by TPA (20 ng/ml) and sodium butyrate (0.3 mM) at the indicated times in BC3 cells. Cells were then harvested, washed twice in PBS, seeded on glass slides, air dried and permeabilized/fixed in acetone-methanol (1:1) for 10 min. The slides were then incubated with mouse monoclonal anti-K-bZIP antibody for 1 h. They were washed 3 times in 1X PBS and incubated with polyclonal Cy3-conjugated sheep-anti mouse for 30 min. Finally, after several washings, cells were incubated with 4,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei and the coverslips were mounted face down using a PBS-glycerol (1:1) solution. The immunofluorescence was analyzed using an Apotome Axio Observer Z1 inverted microscope, equipped with an AxioCam MRM Rev.3 at 40 × magnification.
LysoTracker Red staining
BC3 cells transfected with pEGFP-LC3 were treated with TPA (20 ng/ml) and sodium butyrate (0.3 mM) for the indicated times, washed in PBS solution, and incubated with LysoTracker Red DND-99 (100 nM; Life Technologies, L7528), a fluorescent acidotropic probe with high selectivity for acidic organelles, for 30 min at room temperature.35 After several washings with PBS solution, the lysosomes were analyzed using an Apotome Axio Observer Z1 inverted microscope equipped with an AxioCam MRM Rev.3 at x40 magnification.
Densitometric analysis
The quantification of proteins bands was performed by densitometric analysis using the ImageJ software, which was downloaded from the NIH web site (http://imagej.nih.gov).
Statistical analysis
All data are represented by the mean ± standard error of at least 3 independent experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sandro Valia for technical assistance. We thank Prof. J. Jung for the kind gift of TRExBCBL-1-vector and TRExBCBL-1-Rta cell lines. We also thank Prof. T. Johansen and Prof. G. M. Fimia to kindly provided the pDest-mCherry-EGFP-LC3B and pEGFP-LC3 plasmids, respectively.
Funding
This work was supported by ASI (Italian Space Agency) (2014-033-R.O.), Associazione Italiana per la Ricerca sul Cancro (AIRC) N° 10265 and MIUR (2012SNMJRL_003).
References
- 1.Davison AJ. Overview of classification In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Chapter 1. Cambridge, 2007. [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss RA. Epidemiology and pathogenesis of Kaposi's sarcoma-associated herpesvirus. Philos Trans Royal Soc London Series B, Biol Sci 2001; 356:517-34; PMID:11313009; http://dx.doi.org/ 10.1098/rstb.2000.0778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 1996; 2:342-6; PMID:8612236; http://dx.doi.org/ 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 4.Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J Virol 1997; 71:314-24; PMID:8985352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol 1999; 80(Pt 1):83-90; PMID:9934688; http://dx.doi.org/ 10.1099/0022-1317-80-1-83 [DOI] [PubMed] [Google Scholar]
- 6.Brown HJ, McBride WH, Zack JA, Sun R. Prostratin and bortezomib are novel inducers of latent Kaposi's sarcoma-associated herpesvirus. Antivir ther 2005; 10:745-51; PMID:16218174 [PubMed] [Google Scholar]
- 7.Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998; 252:304-12; PMID:9878608; http://dx.doi.org/ 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- 8.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 2003; 77:4205-20; PMID:12634378; http://dx.doi.org/ 10.1128/JVI.77.7.4205-4220.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv Cancer Res 2007; 97:81-109; PMID:17419942; http://dx.doi.org/ 10.1016/S0065-230X(06)97004-3 [DOI] [PubMed] [Google Scholar]
- 10.Lefort S, Flamand L. Kaposi's sarcoma-associated herpesvirus K-bZIP protein is necessary for lytic viral gene expression, DNA replication, and virion production in primary effusion lymphoma cell lines. J Virol 2009; 83:5869-80; PMID:19321621; http://dx.doi.org/ 10.1128/JVI.01821-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin SF, Robinson DR, Miller G, Kung HJ. Kaposi's sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J Virol 1999; 73:1909-17; PMID:9971770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santarelli R, Farina A, Granato M, Gonnella R, Raffa S, Leone L, Bei R, Modesti A, Frati L, Torrisi MR, et al. Identification and characterization of the product encoded by ORF69 of Kaposi's sarcoma-associated herpesvirus. J Virol 2008; 82:4562-72; PMID:18305046; http://dx.doi.org/ 10.1128/JVI.02400-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, MacKey J, Czajak SC, Desrosiers RC, Lackner AA, Jung JU. Identification and characterization of Kaposi's sarcoma-associated herpesvirus K8.1 virion glycoprotein. J Virol 1999; 73:1341-9; PMID:9882339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/ 10.1038/cdd.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009; 5:527-49; PMID:19527881; http://dx.doi.org/ 10.1016/j.chom.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granato M, Santarelli R, Farina A, Gonnella R, Lotti LV, Faggioni A, Cirone M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol 2014; 88:12715-26; PMID:25142602; http://dx.doi.org/ 10.1128/JVI.02199-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckingham EM, Carpenter JE, Jackson W, Zerboni L, Arvin AM, Grose C. Autophagic flux without a block differentiates varicella-zoster virus infection from herpes simplex virus infection. Proc Natl Acad Sci U S A 2015; 112:256-61; PMID:25535384; http://dx.doi.org/ 10.1073/pnas.1417878112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santarelli R, Gonnella R, Di Giovenale G, Cuomo L, Capobianchi A, Granato M, Gentile G, Faggioni A, Cirone M. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci Rep 2014; 4:4241; PMID:24577500; http://dx.doi.org/ 10.1038/srep04241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 2009; 11:1355-62; PMID:19838173; http://dx.doi.org/ 10.1038/ncb1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. E X, Hwang S, Oh S, Lee JS, Jeong JH, Gwack Y, Kowalik TF, Sun R, Jung JU, Liang C. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of gammaherpesvirus 68. PLoS Pathogens 2009; 5:e1000609; PMID:19816569; http://dx.doi.org/ 10.1371/journal.ppat.1000609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen HJ, Yang Z, Zhou Y, Wood C. Enhancement of autophagy during lytic replication by the Kaposi's sarcoma-associated herpesvirus replication and transcription activator. J Virol 2010; 84:7448-58; PMID:20484505; http://dx.doi.org/ 10.1128/JVI.00024-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santarelli R, Granato M, Faggioni A, Cirone M. Interference with the autophagic process as a viral strategy to escape from the immune control: lesson from Gamma Herpesviruses. J Immunol Res 2015; 2015:546063; PMID:26090494; http://dx.doi.org/ 10.1155/2015/546063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121-5; PMID:17589504 [DOI] [PubMed] [Google Scholar]
- 25.Sharifi MN, Mowers EE, Drake LE, Macleod KF. Measuring autophagy in stressed cells. Methods Mol Biol 2015; 1292:129-50; PMID:25804753; http://dx.doi.org/ 10.1007/978-1-4939-2522-3_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou WJ, Deng R, Zhang XY, Feng GK, Gu LQ, Zhu XF. G-quadruplex ligand SYUIQ-5 induces autophagy by telomere damage and TRF2 delocalization in cancer cells. Mol Cancer Ther 2009; 8:3203-13; PMID:19996277; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0244 [DOI] [PubMed] [Google Scholar]
- 27.Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci: Off J Soc Neurosci 2010; 30:12535-44; PMID:20844148; http://dx.doi.org/ 10.1523/JNEUROSCI.1920-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell 2000; 11:467-80; PMID:10679007; http://dx.doi.org/ 10.1091/mbc.11.2.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochimica et Biophysica Acta 2013; 1833:503-10; PMID:23220125; http://dx.doi.org/ 10.1016/j.bbamcr.2012.11.018 [DOI] [PubMed] [Google Scholar]
- 30.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID: 17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 31.Liang Q, Chang B, Brulois KF, Castro K, Min CK, Rodgers MA, Shi M, Ge J, Feng P, Oh BH, et al. Kaposi's sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J Virol 2013; 87:12499-503; PMID:24027317; http://dx.doi.org/ 10.1128/JVI.01898-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hung CH, Chen LW, Wang WH, Chang PJ, Chiu YF, Hung CC, Lin YJ, Liou JY, Tsai WJ, Hung CL, et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J Virol 2014; 88:12133-45; PMID:25122800; http://dx.doi.org/ 10.1128/JVI.02033-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonnella R, Granato M, Farina A, Santarelli R, Faggioni A, Cirone M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochimica et Biophysica Acta 2015; 1853:1586-95; PMID:25827954; http://dx.doi.org/ 10.1016/j.bbamcr.2015.03.011 [DOI] [PubMed] [Google Scholar]
- 34.Nowag H, Guhl B, Thriene K, Romao S, Ziegler U, Dengjel J, Münz C. Macroautophagy proteins assist epstein barr virus production and get incorporated into the virus particles. EBioMedicine 2014; 1(2-3):116-125; PMID:26137519; http://dx.doi.org/ 10.1016/j.ebiom.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gantt S, Casper C. Human herpesvirus 8-associated neoplasms: the roles of viral replication and antiviral treatment. Curr Opin Infect Dis 2011; 24:295-301; PMID:21666458; http://dx.doi.org/ 10.1097/QCO.0b013e3283486d04 [DOI] [PMC free article] [PubMed] [Google Scholar]