

Abstract
The resolution of inflammation is an active process driven by specialized pro-resolving lipid mediators, such as 15-epi-LXA4 and resolvin D1 (RvD1), that promote tissue regeneration. Macrophages regulate the innate immune response being key players during the resolution phase to avoid chronic inflammatory pathologies. Their half-life is tightly regulated to accomplish its phagocytic function, allowing the complete cleaning of the affected area. The balance between apoptosis and autophagy appears to be essential to control the survival of these immune cells within the inflammatory context. In the present work, we demonstrate that 15-epi-LXA4 and RvD1 at nanomolar concentrations promote autophagy in murine and human macrophages. Both compounds induced the MAP1LC3-I to MAP1LC3-II processing and the degradation of SQSTM1 as well as the formation of MAP1LC3+ autophagosomes, a typical signature of autophagy. Furthermore, 15-epi-LXA4 and RvD1 treatment favored the fusion of the autophagosomes with lysosomes, allowing the final processing of the autophagic vesicles. This autophagic response involves the activation of MAPK1 and NFE2L2 pathways, but by an MTOR-independent mechanism. Moreover, these pro-resolving lipids improved the phagocytic activity of macrophages via NFE2L2. Therefore, 15-epi-LXA4 and RvD1 improved both survival and functionality of macrophages, which likely supports the recovery of tissue homeostasis and avoiding chronic inflammatory diseases.
Keywords: inflammation, macrophages resolution, resolvin-D1, 15-epi-lipoxin-A4
Abbreviations
- AA
arachidonic acid
- AKT
v-akt murine thymoma viral oncogene homolog
- ATG5
autophagy-related 5
- CAMK2
calcium/calmodulin-dependent protein kinase II
- CQ
chloroquine
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- EPI
15-epi-LXA4
- MAPK1/ERK2
mitogen-activated protein kinase 1
- MAPK8/JNK1
mitogen-activated protein kinase 8
- KEAP1
kelch-like ECH-associated protein 1
- KO
knockout
- LAMP1
lysosomal-associated membrane protein 1
- MAP1LC3A/LC3A
microtubule-associated protein 1 light chain 3 alpha
- LXA4
lipoxin-A4
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NFE2L2/NRF2
nuclear factor erythroid 2-like 2
- PCC
Pearson correlation coefficient
- PDPK1
3-phosphoinositide dependent protein kinase 1
- RvD1
resolvin-D1
- SPMs
specialized pro-resolving lipid mediators
- ULK1
unc-51 like autophagy activating kinase 1
- WT
wild type.
Introduction
Inflammation is a pathophysiological response of the organism against infection or tissue damage. To neutralize the causing agent, the innate immune system launches a coordinate and multifaceted inflammatory program that is initiated by the recruitment of neutrophils to the affected area. After performing their action, these cells undergo apoptosis and are phagocytized by macrophages allowing the recovery of tissue homeostasis.1,2 The resolution of inflammation is an active process driven by specialized pro-resolving lipid mediators (SPMs)3 that are endogenously generated during the course of the inflammatory process favoring tissue regeneration.4 Among these, the best studied are lipoxin-A4 (LXA4) and the aspirin-triggered derived epimeric 15-epi-LXA4, both derived from the ω-6 polyunsaturated fatty acid, arachidonic acid. These transiently generated and locally acting eicosanoids limit neutrophil tissue infiltration, promote apoptotic events in neutrophils which have reached an inflammatory lesion, induce monocyte recruitment and macrophage actions, stimulate efferocytosis (phagocytosis of dead neutrophils and other cell debris) and promote resolution of the inflammation and tissue repair (for a review see refs.5,6). In addition to the lipoxins, lipid mediators derived from long chain ω-3 polyunsaturated fatty acids are formed during the inflammatory response and regulate the extent and pace of the resolution process. These SPMs comprise resolvins, protectins, and maresins and their individual contributions and roles in inflammation are under study.7,8 At least 2 series of resolvins have been recognized: D-series resolvins are oxygenation products of docosahexaenoic acid (DHA) and E-series are synthetized from eicosapentaenoic acid (EPA). Resolvin D1 (RvD1) is among the best studied SPMs, promoting resolution after lung or kidney damage,9,10 in periodontal diseases 11 and in allergic airways responses.12 Moreover, RvD1 increases efferocytosis13 by promoting an antiinflammatory/pro-resolving “M2-like” macrophage phenotype.14
Macrophages regulate the innate immune response being key players during the resolution process to avoid chronic inflammatory pathologies.15 The half-life of these cells is relatively short and is tightly regulated to carry out a correct resolution of inflammation mediated by its phagocytic function, allowing the complete elimination of dead neutrophils and tissue debris of the inflamed area. The balance between apoptosis and autophagy has been recognized to be essential in the determination of the life span of macrophages within the inflammatory context.16 In recent years it has become evident that both processes exhibit substantial molecular crosstalk that is not yet fully understood.17 We have demonstrated that LXA4 increases macrophage survival via inhibition of the apoptotic process.18 Nevertheless, whether LXA4, and other SPMs modulate cell fate decisions in macrophages by regulating autophagy had not been addressed.
Autophagy is the major intracellular degradation system by which cytoplasmic materials are engulfed into double-membrane vesicles, known as autophagosomes, and delivered to lysosomes for degradation.19,20 It serves as a dynamic recycling system that produces energy and new precursors for cell renewal and homeostasis.21 Moreover, autophagy plays a multifunctional role in host defense, by promoting pathogen clearance and modulating innate and adaptive immune responses. More than 30 genes regulate autophagy, the majority of them with multiple roles in the different stages of the process of which the AuTophaGy-related (ATG) genes have particularly important functions (for a review see ref.22). The initiation of autophagosome formation is mediated by the formation of the macromolecular complex of proteins known as the class III phosphatidylinositol 3-kinase complex consisting of BECN1 and PIK3C3/Vps34, among others. The activity of the phosphatidylinositol 3-kinase complex can be regulated by the activation of MAPK8/JNK1;23 MAPK8 phosphorylates BCL2, which under basal conditions inhibits autophagy by interacting with BECN1, whereas its phosphorylation inhibits this interaction and stimulates autophagy.24 The initiation step can be also modulated by the kinase ULK1, whose activation induces autophagosome formation.25 During the autophagosome elongation stage there is an activation of the MAP1LC3 (microtubule-associated protein 1 light chain 3) system that is involved in the maturation of the autophagosome and its fusion with the lysosome where dysfunctional or unnecessary cellular components are degraded. MAP1LC3 is present in the cytoplasm as MAP1LC3-I but upon autophagy induction, MAP1LC3-I conjugates with phosphatidylethanolamine to form MAP1LC3-II, which specifically associates with phagophores (the precursor to the autophagosome), making it an excellent marker of autophagy.26 ATG5 plays an essential role in this step since it may form a complex with other ATG proteins to allow MAP1LC3-II formation.27 Another important protein is SQSTM1 (sequestosome 1), a ubiquitin-binding receptor protein that binds directly to MAP1LC3.28 SQSTM1 is itself degraded by autophagy and may serve to link ubiquitinated proteins to the autophagic machinery to enable their degradation in the lysosome. Since SQSTM1 accumulates when autophagy is inhibited, and decreased levels can be observed when autophagy is induced, SQSTM1 may also be used as a marker to study autophagic flux.29
Several signaling pathways have been reported to regulate autophagy in mammalian cells. The classical and most studied pathway involves the MTOR (mechanistic target of rapamycin [serine/threonine kinase]), which integrates the stress signals from the initiation complexes.30 This protein can form 2 complexes (MTORC1 and MTORC2) that in turn, can activate or inhibit autophagy depending on the cellular environment. The major signaling cascade controlling MTORC1 is the phosphoinositide 3-kinase pathway that once activated recruits PDPK1 and AKT to the plasma membrane modulating autophagy.31 Apart from this classic pathway, autophagy can be also mediated by several MTOR-independent pathways. Among these, a phosphoinositol (IP3) signaling pathway that directly regulates the BECN1 complex32 and the Ca2+-CAPN-Gs ɑ pathway in which elevated cytoplasmic Ca2+ levels activate calpain that inhibit autophagy.33
More recently it has become apparent that NFE2L2, an antioxidant transcription factor, can be also implied in autophagy regulation.34 Under quiescent conditions, NFE2L2 interacts with KEAP1 (kelch-like ECH-associated protein 1), which continuously targets it for proteasomal degradation, maintaining a low basal expression of NFE2L2-regulated genes. In the presence of oxidative signals, NFE2L2 is released from KEAP1 and translocates to the nucleus transactivating the expression of cytoprotective genes. In this cell context, SQSTM1 can interact with the NFE2L2-binding site on KEAP1 and thus disrupts the KEAP1 inhibitory effect on NFE2L2.35 Therefore, while a KEAP1-SQSTM1 complex is driven to autophagy, at the same time NFE2L2 escapes KEAP1-dependent degradation, accumulates in the nucleus and activates the expression of a large number of cytoprotective targets.
Autophagy is now recognized to regulate both survival and cell death with the outcome depending on cues from the extracellular environment. Indeed, autophagy exerts an important role in the control of inflammatory signaling.36 Since the local microinflammatory milieu determines macrophage function and life span, in the present study we sought to determine whether SPMs modulate autophagy. Here we describe that 2 pro-resolving lipid mediators, 15-epi-LXA4 and RvD1, promote autophagy in macrophages, revealing a new role for SPMs in regulating macrophage life span. The study addresses the involvement of previously recognized signaling pathways that operate within the autophagic process. Induction of autophagy by lipid mediator autacoids likely constitutes an important regulatory process within the control of the inflammatory response and recovery of tissue homeostasis.
Results
15-epi-LXA4 and RvD1 activate autophagy in macrophages through promotion of MAP1LC3 processing and autophagosome formation
We evaluated if SPMs formed in the course of resolution of inflammation, might play a role in macrophage autophagy. Thus, 2 SPMs 15-epi-LXA4 and RvD1 were employed to determine their activity in modulating autophagy in macrophages. First, we incubated murine peritoneal macrophages with different concentrations of these pro-resolving lipids at concentration ranges at which they have previously been shown to activate significant pro-resolving actions12,14,37 to determine the potential effects on MAP1LC3 processing (Fig. 1A). 15-epi-LXA4 at 100 nM and RvD1 at 50 nM were found to be the lower effective concentrations at which a measurable increase in the formation of MAP1LC3-II was observed, indicative of an autophagy-promoting action of both lipid mediators. These concentrations were chosen for all further experiments in this study. Moreover, we discard that 15-epi-LXA4 nor RvD1 had any toxicity in macrophages at the selected concentrations but also significantly reduced staurosporine-induced apoptosis to levels of cell death observed in control incubations as indicated by MTT reduction (Fig. S1A) and flow cytometric assays (Fig. S1B). Therefore, both lipids mediators are protective for macrophages prolonging their survival similar to native LXA4 and appear to activate autophagy.
Figure 1.
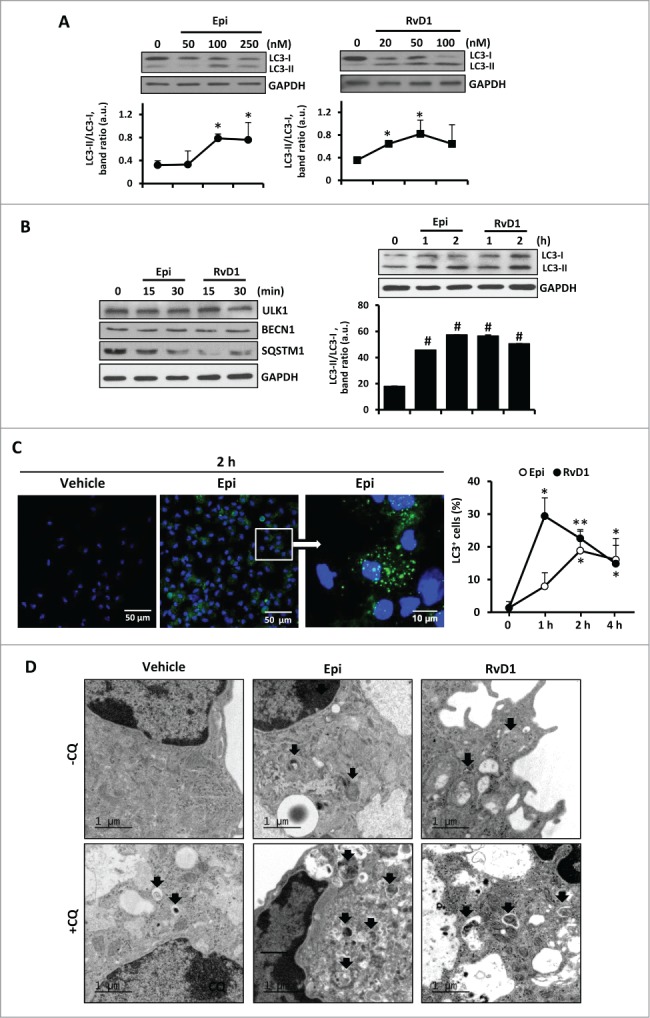
The autophagic pathway is enhanced in 15-epi-LXA4 and RvD1-treated murine macrophages. (A) Representative images of MAP1LC3 showing the 2 protein bands (LC3-I and LC3-II) of murine macrophages treated with different concentrations of 15-epi-LXA4 and RvD1 for 2 h. Immunoblots were normalized by GAPDH levels. The corresponding graph represents the mean ±SD values of band intensity ratios (LC3-II/LC3-I) of 3 different experiments. *P ≤ 0.05 vs. control. (B) Left panel, representative immunoblots of ULK1, BECN1 and SQSTM1 of peritoneal macrophages incubated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for 15 or 30 min. Right panel, representative blots of MAP1LC3 (LC3) in macrophages treated with 15-epi-LXA4 or RvD1 for 1 or 2 h. GAPDH was used for normalization in both cases. The corresponding histogram represents the mean ±SD values of band intensity ratios (LC3-II/LC3-I) of 3 different experiments. #P ≤ 0.001 vs. control. (C) Representative images (left panel) of immunostaining of MAP1LC3+ (green) and DAPI (blue) in macrophages exposed to 15-epi-LXA4 for 2 h. The graph (right panel) shows the mean ±SD values of the percentage of cells with MAP1LC3+ dots of 3 different experiments. *P ≤ 0.05; **P ≤ 0.01 vs. control. (D) Representative electron microscopy images of macrophages treated with vehicle, 15-epi-LXA4 or RvD1 for 2 h in the presence or absence of CQ as indicated. Black arrows show autophagosomes.
Treatment of peritoneal murine macrophages with these SPMs for 15 to 30 min activated the classic autophagic response, characterized mainly by the degradation of SQSTM1 and the accumulation of MAP1LC3-II, with the maximal effect occurring at 1 to 2 h (Fig. 1B). No significant changes were detected in ULK1 or BECN1 protein levels. Immunostaining of MAP1LC3 (green) in macrophages exposed to 15-epi-LXA4 or RvD1 for 1 to 4 h resulted in the accumulation of cells with MAP1LC3+ dots in a time-dependent manner, whereas control cells exhibited a lower and diffuse staining (Fig. 1C). Furthermore, to confirm that 15-epi-LXA4 and RvD1 indeed trigger autophagosome formation in macrophages, we performed transmission electron microscopy analysis in cells treated with these SPMs and, as Fig. 1D shows, formation of typical double-membrane vesicles corresponding to autophagosomes was observed. In these experiments we identified different degrees of autophagosome maturation and autolysosome formation (see Fig. S1C). Moreover, the addition of chloroquine (CQ), a known inhibitor of autolysosome formation,38 in the presence of 15-epi-LXA4 or RvD1 inhibited the fusion of autophagosomes and lysosomes leading to a significant accumulation of immature vesicles in the cytoplasm of the macrophages (Fig. 1D).
BECN1 is an essential autophagic protein implied in the formation of the autophagosome initiation complex but also at later stages of autophagosome maturation interacting with several proteins. Although the treatments with 15-epi-LXA4 or RvD1 did not modify BECN1 protein levels in our model (see Fig. 1B), we analyzed its involvement in the autophagy mediated by these SPMs, using specific siRNA (reducing ≥80% to 90% of mRNA levels; Fig. S2A). In the absence of BECN1, there was a significant reduction in SQSTM1 degradation and MAP1LC3 processing induced by 15-epi-LXA4 or RvD1 (Fig. 2A). These results highlight the important role of BECN1 in the autophagic process. Moreover, in the initial steps of autophagy, the interaction between BCL2 and BECN1 is suppressed after BCL2 phosphorylation. Hence, we analyzed the interaction between both proteins and, as Fig. 2B shows, the coimmunoprecipitation of BCL2 with BECN1 antibody observed in vehicle-treated cells diminished significantly after 1 h of 15-epi-LXA4 or RvD1 exposure.
Figure 2.
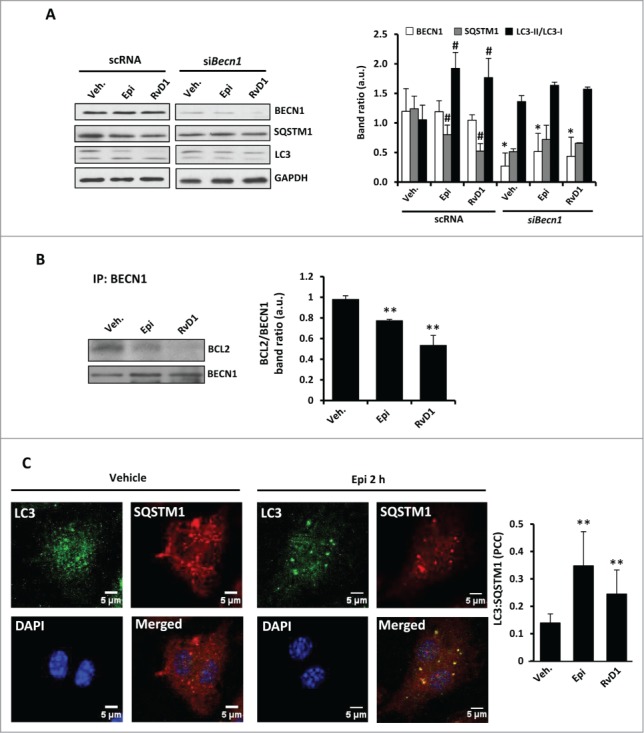
15-epi-LXA4 and RvD1 treatment in murine macrophages induce BECN1-BCL2 dissociation and increase MAP1LC3:SQSTM1 colocalization. (A) Representative blots (left side) of BECN1, SQSTM1, MAP1LC3 (LC3) and GAPDH (as normalization) of peritoneal macrophages transfected with scRNA or siBecn1 and treated with 100 nM of 15-epi-LXA4, 50 nM of RvD1or the vehicle for 2 h. The corresponding histogram (right side) represents the mean ±SD of band intensity ratios of 3 different experiments. *P≤0.05 vs. the corresponding condition in scRNA transfected cells; #P ≤ 0.05 vs. vehicle condition. (B) Representative blots (left side) of BECN1 and BCL2 corresponding to coimmunoprecipitation experiments in macrophages exposed to 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for 1 h. The histogram (right side) shows the mean of BCL2 vs. the corresponding BECN1 band ratio ±SD of 3 different experiments. **P ≤ 0.01 vs. vehicle condition. (C) Left panel shows representative immunofluorescence images of MAP1LC3 (green), SQSTM1 (red) and DAPI (blue) and the corresponding merged image of peritoneal macrophages treated with vehicle or 15-epi-LXA4 for 2 h. The corresponding histogram (right panel) indicates the mean ±SD of the Pearson correlation coefficient (PCC) between MAP1LC3 (LC3) and SQSTM1 proteins in macrophages treated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for 2 h in 3 different experiments. **P ≤ 0.01 vs. vehicle condition.
Regarding the elongation stage, we examined by immunofluorescence the localization of the autophagy receptor protein, SQSTM1, which specifically binds to MAP1LC3 to facilitate the degradation of ubiquitinated protein aggregates by autophagy.28 After 2 h of 15-epi-LXA4 or RvD1 treatment, the colocalization of both proteins was significantly increased compared to vehicle-exposed cells (Fig 2C). Moreover, to demonstrate if 15-epi-LXA4 and RvD1 induce a dynamic autophagic flux in the cells, we examined the effects of LY294002 (LY), a pan-inhibitor of PI3K which prevents the initial steps in the formation of autophagosomes, and of CQ, an inhibitor of the later steps of autophagy.39 The presence of LY failed to significantly change ULK1, BCL2, SQSTM1 or MAP1LC3 protein levels in cells treated with 15-epi-LXA4 or RvD1 (Fig. 3A). By contrast, cotreatment with CQ increased the ratio MAP1LC3-II/MAP1LC3-I and enhanced degradation of SQSTM1, indicating a higher autophagic flux in 15-epi-LXA4 and RvD1-treated macrophages (Fig. 3B). We next analyzed by immunofluorescence the colocalization of MAP1LC3 and LAMP1, a lysosomal marker. In macrophages exposed to 15-epi-LXA4 or RvD1 increased the presence of MAP1LC3 in lysosomes which was maximal at 6 h (Fig. 3C). These results suggest that these SPMs can modulate the autophagosome elongation and the final fusion of the autophagosomes with lysosomes.
Figure 3.
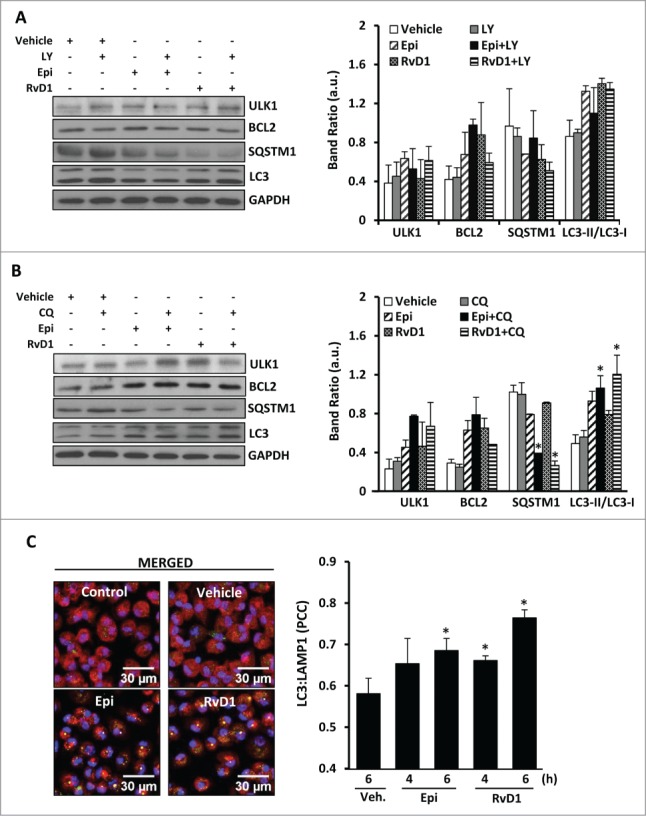
15-epi-LXA4 and RvD1 induce mainly the final stages of autophagy allowing the formation of autolysosomes. (A, B) Representative blots (left panel) of ULK1, BCL2, SQSTM1 and MAP1LC3-II/MAP1LC3-I (LC3-II/LC3-I) of macrophages treated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) or the vehicle for 2 h in the presence or absence of a pretreatment with LY in A (10 µM 30 min) or CQ in B (20 µM 18 h). GAPDH was used for normalization in both cases. The corresponding histograms (right panel) represent the mean ±SD of band ratio of 3 different experiments *P ≤ 0.05 vs. the corresponding condition in the absence of the inhibitor. (C) Left panel shows representative immunofluorescence images of MAP1LC3 (green), LAMP1 (red) and DAPI (blue) in macrophages treated with 15-epi-LXA4 (100 nM), RvD1 (50 nM) or the vehicle for 6 h where MAP1LC3 (LC3) and LAMP1 colocalization appears in yellow color. Right panel presents the histogram quantification representing the mean ±SD of the Pearson correlation coefficient (PCC) between MAP1LC3 and LAMP1 proteins in 3 different experiments. *P ≤ 0.05 vs. vehicle condition.
15-epi-LXA4 and RvD1 activate autophagy via the MAPK1 pathway and its interaction with MAP1LC3
We have previously described that the antiapoptotic effects of LXA4 in macrophages are mainly mediated by AKT and MAPK1 activation.18 To determine if the proautophagic action of SPMs also involves these pathways, the expression and phosphorylation status of individual kinase members of early-response pathways was evaluated by western blot Figure 4. Treatment of macrophages with 15-epi-LXA4 or RvD1 promoted a rapid increase in the levels of P-AKT Figure 4A. However, since LY did not affect the autophagic response (see Fig. 3A), AKT phosphorylation is not likely to be essential for autophagy induction, but might be involved in downstream kinases activation. Also, after 15 to 30 min of 15-epi-LXA4 or RvD1 administration the levels of P-MAPK1 increased significantly and to a lesser extent those of P-MAPK8 Figure 4B. To confirm the selectivity of these changes in the autophagic response, we used specific inhibitors of MAPK1 and MAPK8 phosphorylation 30 min before the incubation with 15-epi-LXA4 or RvD1 for 1 h. In the presence of PD98059 (PD), a specific MAPK1 inhibitor, SQSTM1 degradation was inhibited and there were significantly higher levels of MAP1LC3-I, indicating an important role for MAPK1 pathway in the 15-epi-LXA4 and RvD1-dependent autophagy Figure 4C. However, MAPK8 inhibition did not alter the autophagic response mediated by these SPMs (data not shown). MAPK1 can interact directly with the MAP1LC3 present in the autophagic structures leading to increased MAPK1 phosphorylation.40 Indeed, 15-epi-LXA4 and RvD1 favored the interaction between MAP1LC3 and MAPK1 in colocalization assays Figure 4D. In agreement with these results we confirmed that MAPK1 and MAP1LC3 coimmunoprecipitated in macrophages exposed to 15-epi-LXA4 and RvD1 for 1 h (Fig. S3A). To further support these results, we carried out “FRET-like” experiments, using specific antibodies for MAP1LC3 (Alexa Fluor 488) and MAPK1 (Alexa Fluor 546) to detect by confocal microscopy the endogenous interaction between both proteins in macrophages. We observed that both 15-epi-LXA4 and RvD1 treatment (1 or 2 h) effectively induced the interaction between MAP1LC3 and MAPK1 proteins (Fig. S3B). Moreover, some studies suggest that MAPK1 can regulate the maturation of autophagic vesicles, allowing their fusion with lysosomes. 41 Accordingly we have determined that after MAPK1 inhibition (by PD pretreatment), there was a significant reduction in the number of autolysosomes induced by 15-epi-LXA4 and RvD1 exposure, showed by lower colocalization levels between MAP1LC3 and LAMP1 (Fig. 4E). Altogether our experiments indicate that MAPK1 plays an important role in SPM-mediated autophagy activation by modulating the maturation of the autophagosomes.
Figure 4 (See previous page).

The effects in autophagy observed in macrophages after 15-epi-LXA4 or RvD1 treatments are mediated by the MAPK1 pathway and its interaction with MAP1LC3 protein. (A, B) Representative blots (left side) of P-AKT, AKT (A) and P-MAPK1, MAPK1, P-MAPK8 and MAPK8 (B) of murine macrophages exposed to different concentrations of 15-epi-LXA4 or RvD1 for the indicated periods of time. Immunoblots were normalized by GAPDH levels. The corresponding histograms (right side) represent the mean ±SD of band ratio quantification of 3 independent experiments. *P ≤ 0.05; **P ≤ 0.01 vs. the corresponding control condition. (C) Representative immunoblots (upper panel) of SQSTM1 and MAP1LC3 (LC3) of peritoneal macrophages incubated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for 2 h. PD (1 µM) was added 30 min prior to the lipids where indicated. Immunoblots were normalized by GAPDH levels. Histogram (lower panel) represents the mean ±SD of band ratio quantification of 3 independent experiments. *P ≤ 0.05; #P ≤ 0.01 vs. the corresponding condition in the absence of PD. (D) Left panel shows representative images of immunofluorescence of MAP1LC3 (green), MAPK1 (red) and DAPI (blue) of peritoneal macrophages exposed to 15-epi-LXA4 (100 nM), RvD1 (50 nM) or vehicle for 1 h. Yellow color represents colocalization areas. The histogram (right panel) represents the mean ±SD of the MAPK1-MAP1LC3 (LC3) Pearson correlation coefficient (PCC) of 3 independent experiments. *P ≤ 0.05 vs. control condition. (E) Left panel shows representative images of immunofluorescence of MAP1LC3 (green), LAMP1 (red) and DAPI (blue) of peritoneal macrophages exposed to 15-epi-LXA4 (100 nM), RvD1 (50 nM) or vehicle for 6 h in the presence or absence of a pretreatment with PD (1 µM, 30 min) as indicated. Yellow color represents colocalization areas. The histogram (right panel) represents the mean ±SD of the MAP1LC3 (LC3)-LAMP1 Pearson correlation coefficient (PCC) of 3 independent experiments. *P ≤ 0.05 **P ≤ 0.01 vs. vehicle condition.
Autophagic induction by 15-epi-LXA4 and RvD1 is MTOR independent
One of the main regulators of autophagy is the kinase MTOR that coordinately regulates the balance between cell growth and autophagy in response to the nutritional status, growth factors, and stress signals. Macrophages stimulated with 15-epi-LXA4 or RvD1 exhibited an increase of phosphorylated MTOR levels at 15 and 30 min (Fig. 5A), which disappeared at longer exposure times (Fig. 5B). Interestingly, 30 min pretreatment with rapamycin, a classic inhibitor of the MTOR activity, did not modify the increased percentage of MAP1LC3+ cells induced after 2 h of 15-epi-LXA4 or RvD1 treatment (Fig. 5C). To corroborate the effects on the MTOR pathway we also performed 30 min pretreatment with 20 nM of rapamycin or 10 nM of XL-388, which blocks the kinase activity of MTOR. Both inhibitors independently induced SQSTM1 degradation and had a synergistic action in the presence of 15-epi-LXA4 or RvD1, with higher levels of autophagy induction (Fig. 5D). These results suggest that 15-epi-LXA4 or RvD1 induced autophagy by an MTOR-independent mechanism.
Figure 5 (See previous page).

15-epi-LXA4 and RvD1 induce autophagy in macrophages through an MTOR-independent mechanism. (A) Representative blots (left side) of P-MTOR and MTOR of peritoneal macrophages treated with 15-epi-LXA4 or RvD1 at the different concentrations for the indicated periods of time. The corresponding histogram (right side) shows the mean ±SD of P-MTOR/MTOR band ratio of peritoneal macrophages exposed for 15 or 30 min to15-epi-LXA4 (100 nM) or RvD1 (50 nM) of 3 different experiments. *P ≤ 0.05 vs. control. (B) Histogram represents the mean ±SD of P-MTOR/MTOR band ratio of macrophages treated with 100 nM of 15-epi-LXA4 for the indicated times of 3 different experiments. (C) Left panel shows representative images of immunofluorescence of MAP1LC3 (green) and DAPI (blue) of peritoneal macrophages exposed to 15-epi-LXA4 for 2 h vs. vehicle-treated cells. The histogram (right panel) represents the mean ±SD of the percentage of MAP1LC3 (LC3)+ cells in 3 independent experiments. (D) The histogram represents the mean ±SD of theSQSTM1/GAPDH band ratio of peritoneal macrophages treated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for 2 h in the presence or absence of rapamycin (20 nM) or XL-388 (10 nM) as indicated. *P ≤ 0.05; **P ≤ 0.01 and #P ≤ 0.001 vs. the control condition.
One of the main models of MTOR-independent autophagy is mediated by a reduction in the cytoplasmic calcium levels.33 It has recently been described that RvD1 suppresses the activation of the calcium-sensing kinase CAMK2 (calcium/calmodulin-dependent protein kinase II), limiting LTB4 biosynthesis and facilitating a rapid transition to resolution of an inflammatory process.42 In this work, RvD1 suppresses the phosphorylation of CAMK2 induced by arachidonic acid (AA), which rapidly increases intracellular calcium. Accordingly, we corroborated that stimulation of macrophages with AA for 2 h induced higher levels of P-CAMK2 increasing in parallel to cytoplasmic calcium measured with FLUO4 probe (Fig. 6). Then, we determined that 15-epi-LXA4 reduced the phosphorylation of CAMK2 compared to AA treatment (Fig. 6A) and, moreover, both 15-epi-LXA4 and RvD1 reduced cytoplasmic calcium levels (Fig. 6B). According to these experiments, 15-epi-LXA4 and RvD1 diminished calcium levels in the cytoplasm, permitting further progress of the autophagic process. These results support our hypothesis that 15-epi-LXA4 and RvD1 induce autophagy also involving an MTOR-independent mechanism.
Figure 6.
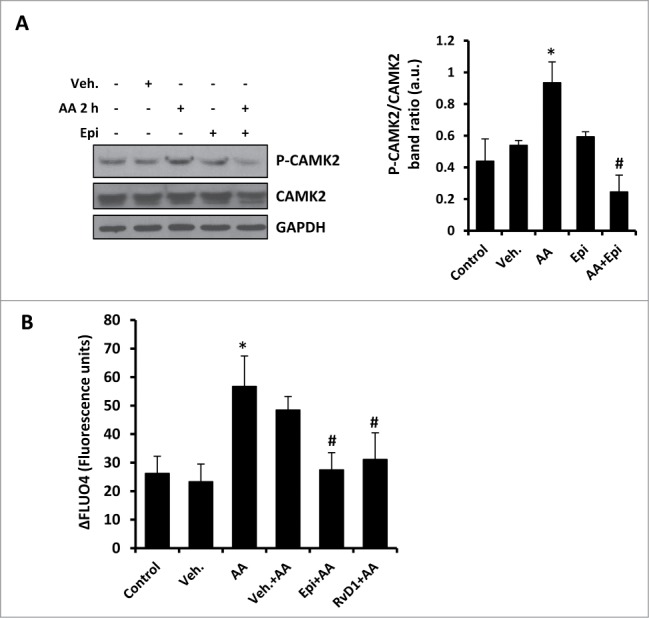
15-epi-LXA4 and RvD1 decreased the cytoplasmic calcium levels in macrophages, reducing the activation of CAMK2. (A). Representative blots (left panel) of P-CAMK2 and CAMK2 of peritoneal macrophages treated with 15-epi-LXA4 (100 nM) or the vehicle for 2 h in the presence or absence of a pretreatment with arachidonic acid (AA; 10 µM) for 2 h as indicated. Immunoblots were normalized to GAPDH levels. The corresponding histogram (right panel) represents the mean ±SD of P-CAMK2/CAMK2 band ratio quantification in 3 different experiments. *P ≤ 0.05 vs. control; #P ≤ 0.01 vs. the AA condition. (B) Histogram represents the mean ±SD of the percentage of increase of specific fluorescence corresponding to FLUO-4 calcium assay of at least 10 different cells per condition vs. their specific background in macrophages treated with 15-epi-LXA4 (100 nM), RvD1 (50 nM) or the vehicle for 2 h in the presence or absence of a pretreatment with arachidonic acid (AA; 10 µM) for 2 h as indicated. *P ≤ 0.05 vs. control; #P ≤ 0.001 vs. the AA condition.
15-epi-LXA4 and RvD1-dependent autophagy in murine macrophages is modulated by NFE2L2
We have previously shown that LXA4 activates an antioxidant mechanism in murine macrophages18 and 15-epi-LXA4 and RvD1 presented a similar action, indicated by a significantly reduced capability of murine macrophages to oxidize DCFH after 1 and 2 h (Fig. S4A). It was of interest to assess if these antioxidative actions occurring during autophagy were NFE2L2-dependent. Using macrophages from WT and Nfe2l2-deficient (knockout, KO) mice, the autophagic effects of 15-epi-LXA4 and RvD1 were maintained in the WT cells (showing increased levels of MAP1LC3-II as well as lower levels of SQSTM1) but blunted in the Nfe2l2 KO counterparts (Fig. 7A). Indeed, 15-epi-LXA4 and RvD1-treated Nfe2l2 KO macrophages showed a significantly lower number of cells containing MAP1LC3+ dots than the WT (Fig. 7B), as well as an impaired colocalization ratio of MAP1LC3 and SQSTM1 (Fig. 7C). To confirm these results, we silenced Nfe2l2 with specific siRNA in peritoneal macrophages (60 to 80% knockdown; Fig. S4B). In the presence of siNfe2l2 there was a decrease in the percentage of MAP1LC3+ cells (Fig. 7D) after 15-epi-LXA4 or RvD1 treatment compared to the negative control (scRNA). These results suggest that autophagy in macrophages is, at least in part, dependent on NFE2L2 activity. Since SQSTM1 can interact with the Nfe2l2-binding site on KEAP1 and thus disrupts its inhibitory effect on Nfe2l2,35 we performed coimmunoprecipitation assays of SQSTM1 and KEAP1 to determine if SPMs modify this interaction. As Figure 7E shows, treatment with 15-epi-LXA4 or RvD1 increased the interaction between both proteins, indicating a direct relationship between the NFE2L2 pathway and the autophagy induced by these SPMs.
Figure 7.
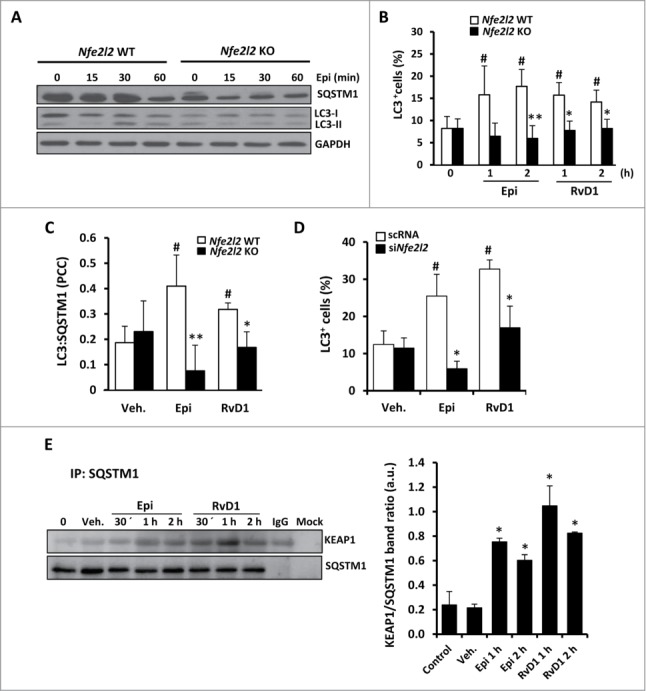
The autophagic induction by 15-epi-LXA4 and RvD1 in murine macrophages is mediated by NFE2L2 activation and the interaction between SQSTM1 and KEAP1. (A) Representative blots of SQSTM1 and MAP1LC3 (LC3) of peritoneal macrophages obtained from Nfe2l2 WT and Nfe2l2 KO mice treated with 100 nM of 15-epi-LXA4 for the indicated periods of time. Immunoblots were normalized by GAPDH levels. (B) Graph represents the mean ±SD of the percentage of cells with MAP1LC3+ dots in Nfe2l2 WT and KO macrophages treated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for one or 2 h of 3 different experiments. #P ≤ 0.05 vs. control; *P ≤ 0.01; **P ≤ 0.001 vs. the corresponding condition in Nfe2l2 WT macrophages. (C) Histogram represents the mean±SD of the Pearson correlation coefficient (PCC) between MAP1LC3 and SQSTM1 in Nfe2l2 WT or KO murine macrophages treated as indicated in the figure in 3 different experiments. #P ≤ 0.05 vs. vehicle; *P ≤ 0.05 and **P ≤ 0.01 vs. the corresponding condition in Nfe2l2 WT macrophages. (D) The histogram represents the mean ±SD of the percentage of MAP1LC3+ cells in peritoneal macrophages transfected with scRNA or siNfe2l2 and treated with 100 nM of 15-epi-LXA4, 50 nM of RvD1or the vehicle for 2 h of 3 different experiments. #P ≤ 0.05 vs. vehicle; *P ≤ 0.05 vs. the corresponding condition in scRNA transfected cells. (E) Representative blots (left side) of KEAP1 and SQSTM1 corresponding to coimmunoprecipitation experiments in macrophages exposed to 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for the indicated periods of time. The histogram (right side) shows the mean of KEAP1 vs. the corresponding SQSTM1 band ratio ±SD of 3 different experiments. *P ≤ 0.01 vs. control condition.
15-epi-LXA4 and RvD1 improve macrophage phagocytic function through the NFE2L2 pathway
It has been described that RvD1 is a locally acting signal that stimulates robust antiinflammatory actions in macrophages promoting phagocytosis, a critical step for the resolution of the inflammatory process.14 Also, LXA4 and 15-epi-LXA4 are known to promote macrophage phagocytosis.13,43-45 Here we confirmed that 15-epi-LXA4 or RvD1-treated cells for 1 or 2 h showed significantly increased phagocytic activity measured as zymosan-Alexa Fluor 488 particles internalization (Fig. 8A). The phagocytosis-stimulating activity of 15-epi-LXA4 was significantly impaired in Nfe2l2 KO macrophages, which already had a weakened basal phagocytic function (Fig. 8B). These results indicate that phagocytosis, and the stimulating actions of these SPMs on phagocytosis, are mediated at least partially by NFE2L2. In this sense, recent evidence suggest that NFE2L2 can be also involved in phagocytosis by modulating the expression of scavenger receptors such as MARCO (macrophage receptor with collagenous structure).46 We analyzed the expression of this receptor by PCR and observed that both 15-epi-LXA4 and RvD1 treatments significantly increased Marco expression (Fig. S4C). These results go deeply into the role of these pro-resolutive lipids in phagocytosis, since they do not only promote interaction between SQSTM1 and KEAP1-NFE2L2 pathway activation, but also involve the activation of the expression of receptors involved in phagocytosis.
Figure 8.
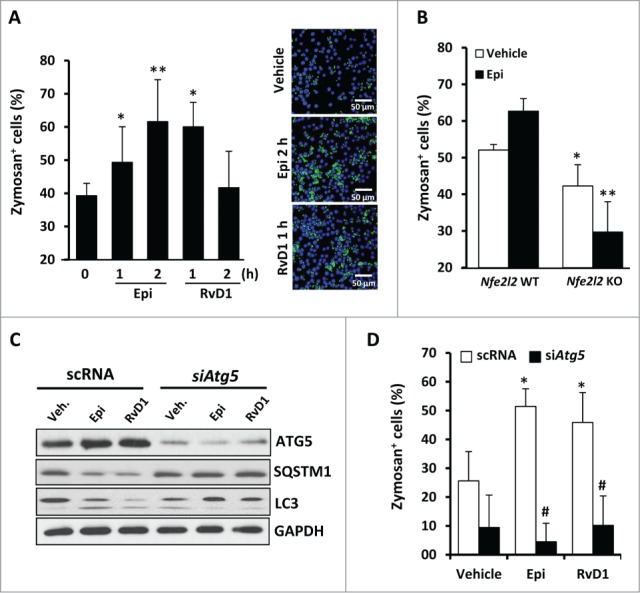
Phagocytosis in 15-epi-LXA4 or RvD1-treated macrophages is tightly regulated by the ATG5 protein and the NFE2L2 antioxidant factor. (A) The histogram (left panel) represents the mean ±SD of the percentage of zymosan+ cells in murine macrophages treated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for the indicated periods of time of 3 different experiments. *P ≤ 0.05 and **P ≤ 0.01 vs. the control. Right panel shows representative images of an immunofluorescence of zymosan Alexa Fluor 488 (green) and DAPI (blue) of vehicle-, 15-epi-LXA4- (2 h) or RvD1 (1 h)-treated cells. (B) The histogram represents the mean ±SD of the percentage of zymosan+ cells in peritoneal macrophages derived from Nfe2l2 WT or KO mice and treated with 15-epi-LXA4 (100 nM) or the vehicle for 2 h of 2 different experiments. *P ≤ 0.05 vs. vehicle and **P ≤ 0.01 vs. the corresponding condition in Nfe2l2 WT macrophages. (C) Representative blots of ATG5, SQSTM1, MAP1LC3 (LC3) and GAPDH (as normalization) of peritoneal macrophages transfected with scRNA or siAtg5 and treated with 100 nM of 15-epi-LXA4, 50 nM of RvD1 or the vehicle for 2 h in 2 different experiments. (D) The histogram represents the mean ±SD of the percentage of zymosan+ cells in peritoneal macrophages transfected with scRNA or siAtg5 and treated with 100 nM of 15-epi-LXA4, 50 nM of RvD1 or the vehicle for 2 h in 2 different experiments. *P ≤ 0.05 vs. the corresponding vehicle; #P ≤ 0.01 vs. the corresponding scRNA condition.
SPM-stimulated phagocytosis is tightly regulated by autophagy in peritoneal murine macrophages
Since autophagy and phagocytosis are conserved and tightly related cell functions in macrophages, we also analyzed their relationship in our model. To address this, we silenced Atg5 (an essential autophagic protein), reducing in ≥80% to 90% both mRNA and protein levels (Fig. S2B). In these experiments, siAtg5 abolished SQSTM1 degradation and MAP1LC3 processing induced by 15-epi-LXA4 and RvD1 treatments (Fig. 8C). Moreover, we have determined that, in the absence of this protein, the macrophages presented a significant decrease in their capacity to phagocyte zymosan particles (Fig. 8D). These results demonstrate the tight relationship between autophagy and phagocytosis in this model.
15-epi-LXA4 and RvD1 promote autophagy in human monocytes
Given the results obtained in murine macrophages, next we determined if 15-epi-LXA4 and RvD1 also induce autophagy in human cells. Human monocytes obtained from healthy donors were exposed to 15-epi-LXA4 or RvD1 for 1 or 2 h and the expression of various components of the autophagic signaling pathway was analyzed by western blot (Fig. 9A). Both SPMs induced a significant increase of MAP1LC3-II/MAP1LC3-I ratio and SQSTM1 degradation without affecting BECN1 levels. Moreover, colocalization of BCL2 and BECN1 was diminished in cells treated for 2 h (Fig. 9B), suggesting an early autophagy activation by these SPMs. Finally, both lipid mediators induced a significantly increased colocalization of MAP1LC3 and SQSTM1 measured by immunofluorescence (Fig. 9C). Taken together, autophagy induction by 15-epi-LXA4 and RvD1 in human monocyte-derived macrophages is similar to the events described in murine peritoneal macrophages.
Figure 9.
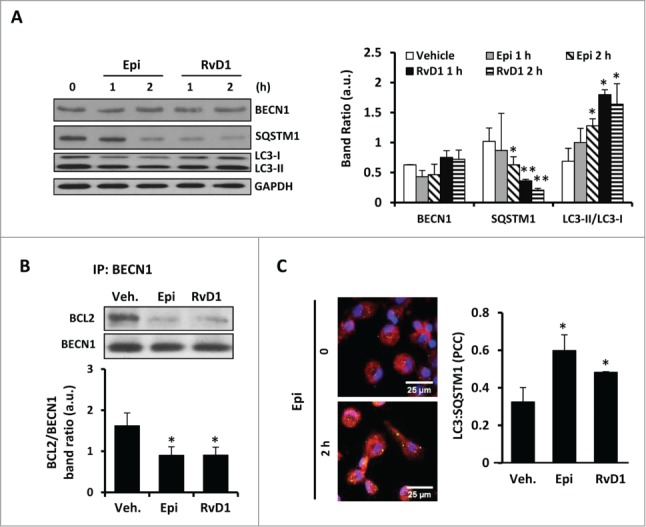
Human monocytes also augmented their autophagy after treatment with 15-epi-LXA4 or RvD1. (A) Left panel shows representative blots of BECN1, SQSTM1 and MAP1LC3 (LC3) of human monocytes treated with 15-epi-LXA4 (100 nM) or RvD1 (50 nM) for one or 2 h. The histogram (right panel) presents the mean ±SD of the band ratio of each protein vs. GAPDH of 3 different experiments. *P ≤ 0.05 and **P ≤ 0.01 vs. the corresponding vehicle. (B) Representative blots (upper panel) correspond to coimmunoprecipitation experiments between BECN1 and BCL2 in human monocytes treated with 15-epi-LXA4 or RvD1 for 2 h. Histogram (lower panel) shows the quantification of the mean ± SD of BCL2/BECN1 band ratio of 3 different experiments. *P ≤ 0.05 vs. vehicle. (C) Left panel shows representative images of immunofluorescence of MAP1LC3 (green), SQSTM1 (red) and DAPI (blue) of 15-epi-LXA4 (2 h)-treated human monocytes vs. control. The histogram (right panel) represents the mean ±SD of the Pearson correlation coefficient (PCC) between MAP1LC3 (LC3) and SQSTM1 of human monocytes exposed to 15-epi-LXA4 or RvD1 for 2 h vs. vehicle-treated cells of 3 different experiments. *P ≤ 0.05 vs. vehicle condition.
Discussion
The present study provides evidence for a new role of 15-epi-LXA4 and RvD1 in the immune system, the activation of autophagy in macrophages. This process, combined with the inhibition of apoptosis by LXA4,18 suggests that pro-resolving lipids play a physiological role in macrophage biology prolonging their survival by regulating apoptosis and autophagy. The concerted regulation of both fundamental cellular processes likely contributes to a well-coordinated and correct resolution of the inflammatory process. A controlled and sustained macrophage survival within an inflamed tissue is required to allow appropriate time for complete efferocytosis, avoiding excessive accumulation of cellular debris within injured tissues and permitting progress to resolution and tissue regeneration. Proper regulation of macrophage half-life by activation of autophagy may contribute to permitting timely macrophage egress from inflamed tissues into draining lymph toward lymph nodes and presentation of antigen to the adaptive arm of the immune system, before macrophage apoptosis can ensue. Thus, our results support a new role of SPMs in the inflammatory response.
The interplay between autophagy and apoptosis is a complex multifactorial network, which controls the cellular balance between survival and death. In each specific cellular context, a small prevalence of one of them could tip the balance one way or the other. We have described that some SPMs, such as LXA4 impair apoptosis of macrophages, but the possible implication of these compounds in autophagy regulation remained unknown. Autophagy is generally activated by conditions of nutrient deprivation but it has also been associated with physiological and pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.22 Autophagy and apoptosis are connected both positively and negatively, and extensive crosstalk exists between the 2 pathways. During nutrient deficiency, autophagy functions as a prosurvival mechanism; however, excessive autophagy may lead to cell death. In fact, there are several apoptotic proteins that also play an important role in autophagy, including BCL2, BCL2L1, or MCL1 which inhibit BECN1-dependent autophagy, thereby acting as prosurvival and as antiautophagic regulators.24,47 In this sense, it has been recently reported that these antiapoptotic proteins could regulate autophagy by inhibiting the activity of the proapoptotic proteins BAK1 and BAX.48
To date, MAP1LC3-II is considered one of the main autophagosome markers. Thus, tracking the conversion of MAP1LC3-I to MAP1LC3-II is indicative of autophagic activity, but needs to be corroborated by the formation of MAP1LC3+ aggregates (“dots” or puncta) within the cells, as well as its colocalization with lysosomal markers, such as LAMP1. We have demonstrated that both 15-epi-LXA4 and RvD1 at nanomolar concentrations induced higher levels of MAP1LC3-II in macrophages, as well as the presence of MAP1LC3+ aggregates in the macrophages, interacting with the lysosomes. In fact, the formation of autophagosomes in different stages of integration was confirmed by electron microscopy. These results were also corroborated in human monocyte-derived macrophages extending the results obtained in the mouse model.
One of the well-known mechanisms reported for regulation in autophagy is the AKT-MTOR pathway. Although it has been widely described that AKT activates MTOR suppressing autophagy, there is also evidence of several autophagic pathways that are MTOR-independent.49 A genome-wide screen has identified various MTOR-independent signaling components that contribute to basal autophagy, such as the MAPK8-BECN1 and MAPK1 pathways, although the precise mechanisms whereby these pathways regulate autophagy remain to be completely elucidated.50 We show here that 15-epi-LXA4 and RvD1 induced a rapid activation of AKT and MTOR pathways, but LY, rapamycin or the MTOR kinase inhibition with XL388 did not influence autophagy in these cells, suggesting that neither AKT nor MTOR play an important role in 15-epi-LXA4 or RvD1-activated autophagy, although they might participate in determining the basal level of autophagic activity. Moreover, both 15-epi-LXA4 and RvD1 prevented the activation of CAMK2 and reduced the cytoplasmic calcium levels in macrophages, also pointing to an activation of autophagy by an MTOR-independent mechanism. In accordance, these SPMs induced the activation of MAPK8 during autophagy resulting in the phosphorylation of BCL2 and releasing its binding to BECN1, permitting autophagosome formation.51 Interestingly, our data indicate that MAPK1 plays a key role in SPM-mediated autophagy as deduced by the increased phosphorylation of MAPK1 in 15-epi-LXA4 and RvD1-treated cells, and corroborated by the use of specific inhibitors of this pathway. MAPK1 is important in cell survival, and its implication in the modulation of autophagy has also become increasingly relevant. For example, MAPK1 activation has been implicated in the induction of autophagy in response to different antitumor and cytotoxic compounds in several human cancer cells,52-54 as well as in neuronal55 or endothelial cells.56 It has also been described that direct MAPK1 activation by overexpression of constitutively active MAP2K1 promote autophagy without any other stimulus41 and MAPK1 inhibition has been associated to an impaired autophagy.52 Moreover, MAPK1 can also interact directly with MAP1LC3 in the autophagic structures.40 Our results shows that the levels of interaction between MAPK1 and MAP1LC3 increased in macrophages treated with 15-epi-LXA4 and RvD1 as concluded by colocalization and coimmunoprecipitation assays. These data were also supported by “FRET-like” approach. Moreover, some studies suggest that MAPK1 can regulate the maturation of autophagic vesicles, allowing their fusion with lysosomes.41 Accordingly we have determined that after MAPK1 inhibition there was a significant reduction in the number of autolysosomes induced by 15-epi-LXA4 and RvD1. Thus, autophagy in macrophages activated by SPMs appears to be mediated by an MTOR-independent pathway, through a reduction in the cytoplasmic calcium levels and activation of MAPK1 that promotes the maturation of autophagosomes.
In addition to the involvement of kinase signaling pathways, 15-epi-LXA4 and RvD1 also require NFE2L2, a component of an intracellular antioxidant pathway, in the activation of autophagy in macrophages. Using KO mice and siRNA assays, our data proved that NFE2L2 can be essential for the activation of several components of the autophagic process suggesting its involvement in the early steps of autophagy initiation or control over those steps. This transcription factor has been implicated in autophagy through the receptor protein SQSTM1, which activates and stabilizes this stress responsive transcription factor through the inactivation of its main inhibitor, KEAP1.34,35 Indeed, our results show that both 15-epi-LXA4 and RvD1 treatments in macrophages promoted the KEAP1-SQSTM1 interaction, releasing the protective factor NFE2L2. Thus, this transcription factor appears to have a central role in the SPMs mediated autophagy. Indeed, phagocytosis, an essential function of macrophages activated by SPMs also proved to be compromised in the absence of a functional NFE2L2 protein, being significantly impaired after 15-Epi-LXA4 treatment in KO mice.57 SPMs also increased the expression of MARCO receptors, that modulate phagocytosis in macrophages and whose expression can be regulated by NFE2L2. Looking to these data, NFE2L2 is another key factor modulating SPMs mediated autophagy in macrophages and as well as regulating the phagocytic function of these immune cells (see Fig. S5).
Autophagy and phagocytosis are basic cellular functions playing important roles in innate immunity whose crosstalk appears to be dependent on the specific immune scenario. Although some studies indicate that bone-marrow derived macrophages deficient for several ATGs are capable to phagocytose dead cells58 and bacteria46 at a rate similar to that of wild-type macrophages, other groups have demonstrated that disruption of the same ATGs impairs the capacity of macrophages to phagocytose fungi59 and also bacteria.60 Here we show that knocking down Atg5 in murine peritoneal macrophages induced a decreased phagocytosis concomitant with the absence of autophagy, suggesting a close relationship between both basic cellular processes. These discrepancies can be explained by the application of different experimental approaches, mainly dependent on the cellular context and even on the type of “particles” to phagocytose (related to the class of receptor involved in their internalization). Therefore, we considered our results as an alternative model that can provide additional knowledge of the relationship between autophagy and phagocytosis in another immune scenario, improving our understanding of macrophage physiology and function.
Over the last few years it has become apparent that autophagy may be a primordial component of the eukaryotic innate immune response against invading microorganism, being activated in response to virus, bacteria, and inflammatory cytokines among others.61 It has been described that both toll-like receptors58,62 and NOD-like receptors63,64 can interact with the autophagic machinery. Recently, defects in autophagy have been associated with several human disorders such as cardiovascular diseases,65-67 obesity and diabetes,68 Alzheimer disease69,70 and even cancer,71-73 all of them share a low-grade chronic inflammatory background. In this line, therapeutic approaches to the majority of these diseases include some antiinflammatory compounds with important limitations linked to their secondary effects. With the recent discovery of endogenous SPMs, the attention has changed in favor of understanding the actions of these endogenous molecules, including the lipoxins, resolvins, protectins, and maresins, which actively orchestrate and mediate the resolution of inflammation. Possibly, a deficit in the synthesis of SPMs such as lipoxins and resolvins may contribute to poor resolution, involving inadequate activation of macrophage autophagy. The knowledge of the mechanism of action of these autacoids is highly relevant since it is believed that they promote resolution inducing neither immunosuppression nor undesirable side-effects, opening a door to potential new approaches in pharmacological treatments for chronic inflammatory diseases.
Materials and Methods
Materials
15-epi-LXA4 (90415) and RvD1 (13060) were from Cayman; PD98059 and arachidonic acid were from Sigma (P215, A3555); rapamycin and LY294002 were from Calbiochem (553210, 440202); thioglycollate broth was from Becton Dickinson (BD 225650) and chloroquine was from Abcam (ab142116); XL388 was from Selleckchem (S7035). Antibodies were from Santa Cruz Biotechnology, Sigma-Aldrich, Abgent, MBL, Life Technologies and Cell Signaling Technology (see Table 1). Zymosan A Bioparticles® Alexa Fluor 488 (Z23373), FLUO4 direct™ calcium assay reagent (F10471) and fluorescent secondary antibodies were from Molecular Probes (Alexa Fluor anti-mouse 488, A11029 and Alexa Fluor anti-rabbit 546, A11035). Tissue culture dishes were from Falcon (353004) and tissue culture media (RPMI, 21875-034 and DMEM, 41966-029) were from Gibco-Invitrogen. Stock solutions and dilutions of lipid mediators were prepared in ethanol, maintained out of direct light and handled under cold conditions at all steps. Vehicle-treated cells received 0.01% (vol/vol) of ethanol for the indicated periods of time in all the experiments.
Animals
The study was conducted in mice following the guidelines of the Spanish Animal Care and Use Committee and the European Union (2010/63/EU). All procedures were approved by the Bioethics Committee of our institution. Male Balb/c mice (Charles River Laboratories), male wild-type C57BL/6 mice and their Nfe2l2-deficient (KO) littermates were housed at RT under 12 h light/dark cycle and food and water was provided ad libitum. Mice were euthanized by CO2 inhalation immediately prior to extraction of elicited-peritoneal macrophages.
Peritoneal macrophage isolation
Mice aged 8 to 12 wk were used as follows: Four d prior to harvesting the peritoneal macrophages, 2.5 ml of 3% thioglycollate broth (BD, 225650) were intraperitoneally injected. Elicited peritoneal macrophages were prepared from mice as previously described.74 Cells were seeded in RPMI 1640 medium (Gibco, 21875) supplemented with 10% heat-inactivated fetal bovine serum (Sigma, F7524) and antibiotics (100 IU/ml penicillin and 100 μg/ml streptomycin; Sigma, P4458).
Human monocyte isolation
The study protocol adhered to the ethical guidelines of the 1975 Declaration of Helsinki and received approval from the Ethics Committee of La Paz Hospital in Madrid. All participants provided their written informed consent to participate in the study and the Ethic Committee approved this consent procedure. Peripheral blood mononuclear cells were isolated from the blood of healthy donors by centrifugation on Ficoll-paque Plus (Fisher, 17-1440-03). The purity of the monocyte cultures was tested by flow cytometry after CD14 labeling (>89% positive cells). Peripheral blood mononuclear cells were cultured for 2 h at 106 cells/ml in DMEM (Gibco, 41966) supplemented with antibiotics. After this period, the supernatant fraction was removed and the adherent cells were cultured in the same medium supplemented with antibiotics and 10% fetal bovine serum.18
Preparation of total cell extracts
Cells were homogenized at 4°C in a medium containing 10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 10% glycerol, 0.5% CHAPS (Sigma, C3023) and protease and phosphatase inhibitor cocktails (Sigma, P8340, P5726, P0044). The extracts were vortexed for 30 min at 4°C and after centrifuging for 15 min at 13,000 g, the supernatant fractions were stored at −20°C. Protein levels were determined with Bradford reagent (Bio-Rad, 500-0006).
Western blot analysis
Equal amounts of protein (20 to 40 μg) were loaded onto and size-fractionated by 8-12% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, 170-4157). After blocking with 5% nonfat dry milk and incubation with the corresponding antibodies the blots were developed by ECL protocol. Optimization of exposition times was performed for each blot to ensure the linearity of band intensities. Values of densitometry were determined using ImageJ software (http://imagej.nih.gov).
Immunofluorescence microscopy
Cells were seeded 16 h before treatment into sterile 8-well chamber slides (Falcon, 354108). After treatments, cells were fixed with 2% paraformaldehyde for 10 min, permeabilized in iced methanol and incubated with 3% bovine serum albumin (Sigma, A2153) for 30 min. After incubating with antibody against MAP1LC3 (MBL, M115-3), SQSTM1 (MBL, PM045), LAMP1 (Abgent, AP1823) or MAPK1 (Cell Signaling Technology, 9102) at 4°C for 18 h, cells were washed with phosphate-buffered saline (PBS; Lonza, BE17-515F) followed by incubation with the corresponding secondary antibody (1:500) and DAPI (1:1000) for 20 min. Coverslips were mounted in ProLong® Gold Antifade reagent (Life technologies, P36930) and examined using a confocal microscope (Leica PCS SP5, Illinois, USA). Values of fluorescence intensity and Pearson colocalization coefficient (PCC) quantification were performed with ImageJ software. PCC values range from 1 for 2 images whose fluorescence intensities are perfectly, linearly related, to −1 for 2 images whose fluorescence intensities are perfectly, but inversely, related to one another. Values near zero reflect probe distributions that are uncorrelated with one another.75
Protein coimmunoprecipitation
After treatment with the indicated lipid mediators, cell proteins were extracted with RIPA buffer and 500 to 800 µg of total protein extract was incubated with the corresponding primary antibody for 18 h at 4°C on a rotating platform. Then, 20 µl of Protein A/G PLUS-agarose (Santa Cruz Biotechnology, sc2003) were added to all samples and incubated for 3 h at 4°C. After centrifugation, the pellets were washed 3 times with PBS and finally resuspended in 4X concentrated Laemmli sample buffer and the presence of the corresponding protein was determined by western blot. Blots were incubated with a specific secondary antibody from Cell Signaling Technology (5127) that only binds to native IgG eliminating the problem of comigrating bands corresponding to IgG heavy and light chains.
siRNA assays
Specific siRNAs or the equivalent scrambled sequences (scRNA; Ambion) were used to knock down the gene expression of Nfe2l2 (s70522), Becn1 (s80166) and Atg5 (s62453). Peritoneal murine macrophages were transfected with 50 nM of siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778). The degree of gene knockdown after 48 h of transfection was determined by PCR, obtaining a transfection efficiency of siRNA of 60 to 90%. Transfection with scrambled RNAs did not modify the basal levels of mRNA.
Phagocytosis assay
Cells were seeded into sterile 8-well chamber slides. After treatments, zymosan A BioParticles® Alexa Fluor 488 (Molecular Probes, Z23373) was incubated with cells for 2 h at 37°C following the manufacturer´s instructions. Then, cells were fixed with 2% paraformaldehyde for 10 min, permeabilized in ice-cold methanol and incubated with DAPI 1:1000 for 20 min at room temperature for nuclear labeling. Coverslips were mounted in ProLong® Gold Antifade reagent and examined using a confocal microscope Leica PCS SP5. Quantification of phagocytic cells (with green particles in their cytoplasm) vs. total cells was performed with ImageJ software.
Fluo-4 Direct™ calcium assay
Cytoplasmic calcium levels were analyzed in peritoneal macrophages seeded into specific 8-chamber slides with a glass bottom (Ibidi, 80827) and stimulated as indicated. Cells were exposed to FLUO4 reagent for 30 min at 37°C following the manufacturer´s instructions and probe specific fluorescence (Ex-494nm/Em-516nm) was measured using a confocal microscope LSM710 (Zeiss, New York, USA).
Electron microscopy
For electron microscopy, peritoneal macrophages were seeded into sterile 8-well Permanox®-treated chamber slides (Thermo Scientific, 177445) and incubated with the indicated stimulus. Then, cells were washed with PBS and fixed in glutaraldehyde (2%) plus paraformaldehyde (2%) for 60 min. Samples were postfixed in 1% osmium tetroxide for 60 min at 25°C, stained with uranyl acetate (5 mg/ml) for 1 h at 25°C, dehydrated in acetone and embedded in Epon 812 (Electron Microscopy Science, EMbed 812). Ultrathin sections, unstained or poststained with uranyl acetate and lead hydroxide, were examined under a Morgagni 268D transmission electron microscope (FEI, Hillsboro, OR) equipped with a Mega View II charge-coupled device camera (SIS, Soft Imaging System GmbH, Munster, Germany) and analyzed with AnalySIS software (SIS).
Statistical analysis
The values shown in graphs correspond to the means ±SD. The statistical significance of differences between mean sample values was estimated with the Student t test for unpaired observations. All experiments were carried out at least in triplicate. A P ≤ 0.05 value was considered as statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Thanks to Dolores Morales (SIDI-UAM) for the excellent technical assistance in the Confocal Microscopy Images capture and analysis.
Funding
This work was supported by grants BFU2011/24760, SAF2013-43271-R, SAF2013-43713-R, SAF2014-52492 and IPT-2012-1331-060000 from MINECO; RD12/0042/0019 from Instituto de Salud Carlos III (ISCIII), Spain, and S2010/BMD2378 from Comunidad de Madrid.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Bannenberg G, Moussignac RL, Gronert K, Devchand PR, Schmidt BA, Guilford WJ, Bauman JG, Subramanyam B, Perez HD, Parkinson JF, et al.. Lipoxins and novel 15-epi-lipoxin analogs display potent anti-inflammatory actions after oral administration. Brit J Pharmacol 2004; 143:43-52; PMID:15302682; http://dx.doi.org/ 10.1038/sj.bjp.0705912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11:519-31; PMID:21785456; http://dx.doi.org/ 10.1038/nri3024 [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014; 510:92-101; PMID:24899309; http://dx.doi.org/ 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell CD, Schwarze J. The role of pro-resolution lipid mediators in infectious disease. Immunology 2014; 141:166-73; PMID:24400794; http://dx.doi.org/ 10.1111/imm.12206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serhan CN, Krishnamoorthy S, Recchiuti A, Chiang N. Novel anti-inflammatory-pro-resolving mediators and their receptors. Curr Topics Med Chem 2011; 11:629-47; PMID:21261595; http://dx.doi.org/ 10.2174/1568026611109060629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med 2013; 5:661-74; PMID:23592557; http://dx.doi.org/ 10.1002/emmm.201202382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol 2007; 28:176-83; PMID:17337246; http://dx.doi.org/ 10.1016/j.it.2007.02.007 [DOI] [PubMed] [Google Scholar]
- 8.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochimica et Biophysica Acta 2010; 1801:1260-73; PMID:20708099; http://dx.doi.org/ 10.1016/j.bbalip.2010.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Zheng X, Cheng Y, Zhang YL, Wen HX, Tao Z, Li H, Hao Y, Gao Y, Yang LM, et al.. Resolvin D1 stimulates alveolar fluid clearance through alveolar epithelial sodium channel, Na,K-ATPase via ALX/cAMP/PI3K pathway in lipopolysaccharide-induced acute lung injury. J Immunol 2014; 192:3765-77; PMID:24646745; http://dx.doi.org/ 10.4049/jimmunol.1302421 [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Shetty S, Zhang P, Gao R, Hu Y, Wang S, Li Z, Fu J. Aspirin-triggered resolvin D1 down-regulates inflammatory responses and protects against endotoxin-induced acute kidney injury. Toxicol Appl Pharmacol 2014; 277:118-23; PMID:24709673; http://dx.doi.org/ 10.1016/j.taap.2014.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mustafa M, Zarrough A, Bolstad AI, Lygre H, Mustafa K, Hasturk H, Serhan C, Kantarci A, Van Dyke TE. Resolvin D1 protects periodontal ligament. Am J Physiol Cell Physiol 2013; 305:C673-9; PMID:23864609; http://dx.doi.org/ 10.1152/ajpcell.00242.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, Pfeffer MA, Priluck R, Serhan CN, Levy BD. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol 2012; 189:1983-91; PMID:22802419; http://dx.doi.org/ 10.4049/jimmunol.1101665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012; 120:e60-72; PMID:22904297; http://dx.doi.org/ 10.1182/blood-2012-04-423525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, Arroyo V, Claria J. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol 2011; 187:5408-18; PMID:22013115; http://dx.doi.org/ 10.4049/jimmunol.1100225 [DOI] [PubMed] [Google Scholar]
- 15.Chazaud B. Macrophages: supportive cells for tissue repair and regeneration. Immunobiology 2014; 219:172-8; PMID:24080029; http://dx.doi.org/ 10.1016/j.imbio.2013.09.001 [DOI] [PubMed] [Google Scholar]
- 16.Nakahira K, Cloonan SM, Mizumura K, Choi AM, Ryter SW. Autophagy: a crucial moderator of redox balance, inflammation, and apoptosis in lung disease. Antioxidants Redox Signaling 2014; 20:474-94; PMID:23879400; http://dx.doi.org/ 10.1089/ars.2013.5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delgado ME, Dyck L, Laussmann MA, Rehm M. Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis 2014; 5:e1011; PMID:24457955; http://dx.doi.org/ 10.1038/cddis.2013.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prieto P, Cuenca J, Traves PG, Fernandez-Velasco M, Martin-Sanz P, Bosca L. Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ 2010; 17:1179-88; PMID:20094061; http://dx.doi.org/ 10.1038/cdd.2009.220 [DOI] [PubMed] [Google Scholar]
- 19.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/ 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 22.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al.. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- 23.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008; 4:949-51; PMID:18769111; http://dx.doi.org/ 10.4161/auto.6788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 25.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/ 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pyo JO, Nah J, Jung YK. Molecules and their functions in autophagy. Exp Mol Med 2012; 44:73-80; PMID:22257882; http://dx.doi.org/ 10.3858/emm.2012.44.2.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 29.Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methodsn Enzymol 2009; 452:181-97; PMID:19200883; http://dx.doi.org/ 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- 30.Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin cell Dev Biol 2014; 36:121-9; PMID:25158238 [DOI] [PubMed] [Google Scholar]
- 31.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296:1655-7; PMID:12040186; http://dx.doi.org/ 10.1126/science.296.5573.1655 [DOI] [PubMed] [Google Scholar]
- 32.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101-11; PMID:16186256; http://dx.doi.org/ 10.1083/jcb.200504035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al.. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. N Chem Biol 2008; 4:295-305; PMID:18391949; http://dx.doi.org/ 10.1038/nchembio.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al.. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51:618-31; PMID:24011591; http://dx.doi.org/ 10.1016/j.molcel.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 35.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al.. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213-23; PMID:20173742 [DOI] [PubMed] [Google Scholar]
- 36.Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol 2010; 189:925-35; PMID:20548099; http://dx.doi.org/ 10.1083/jcb.201002021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Wu Y, Wang Y, Wu J, Song L, Xian W, Yuan S, Pei L, Shang Y. Resolvin D1 promotes the interleukin-4-induced alternative activation in BV-2 microglial cells. J Neuroinflammation 2014; 11:72; PMID:24708771; http://dx.doi.org/ 10.1186/1742-2094-11-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990-5; PMID:15528435; http://dx.doi.org/ 10.1126/science.1099993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Euro J Biochem/FEBS 1997; 243:240-6; PMID:9030745; http://dx.doi.org/ 10.1111/j.1432-1033.1997.0240a.x [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun 2013; 4:2799; PMID:24240988; http://dx.doi.org/ 10.1038/ncomms3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, Fenichel P, Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res 2006; 66:6861-70; PMID:16818664; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3557 [DOI] [PubMed] [Google Scholar]
- 42.Fredman G, Ozcan L, Spolitu S, Hellmann J, Spite M, Backs J, Tabas I. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc Natl Acad Sci U S A 2014; 111:14530-5; PMID:25246560; http://dx.doi.org/ 10.1073/pnas.1410851111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, Serhan CN. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem 2010; 285:3451-61; PMID:19906641; http://dx.doi.org/ 10.1074/jbc.M109.044131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, Godson C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J: Off Pub Federat Am Soc Exp Biol 2010; 24:4240-9; PMID:20570963; http://dx.doi.org/ 10.1096/fj.10-159913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maderna P, Godson C. Lipoxins: resolutionary road. Brit J Pharmacol 2009; 158:947-59; PMID:19785661; http://dx.doi.org/ 10.1111/j.1476-5381.2009.00386.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonilla DL, Bhattacharya A, Sha Y, Xu Y, Xiang Q, Kan A, Jagannath C, Komatsu M, Eissa NT. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 2013; 39:537-47; PMID:24035364; http://dx.doi.org/ 10.1016/j.immuni.2013.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6:1221-8; PMID:15558033; http://dx.doi.org/ 10.1038/ncb1192 [DOI] [PubMed] [Google Scholar]
- 48.Lindqvist LM, Heinlein M, Huang DC, Vaux DL. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci U S A 2014; 111:8512-7; PMID:24912196; http://dx.doi.org/ 10.1073/pnas.1406425111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 2013; 41:1103-30; PMID:24059496; http://dx.doi.org/ 10.1042/BST20130134 [DOI] [PubMed] [Google Scholar]
- 50.Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al.. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010; 18:1041-52; PMID:20627085; http://dx.doi.org/ 10.1016/j.devcel.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivaprasad U, Basu A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-alpha-induced cell death in MCF-7 cells. J Cell Mol Med 2008; 12:1265-71; PMID:18266953; http://dx.doi.org/ 10.1111/j.1582-4934.2008.00282.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem 2000; 275:39090-5; PMID:10993892; http://dx.doi.org/ 10.1074/jbc.M006198200 [DOI] [PubMed] [Google Scholar]
- 54.Gong K, Zhang Z, Chen Y, Shu HB, Li W. Extracellular signal-regulated kinase, receptor interacting protein, and reactive oxygen species regulate shikonin-induced autophagy in human hepatocellular carcinoma. Euro J Pharmacol 2014; 738C:142-52; PMID:24886888; http://dx.doi.org/ 10.1016/j.ejphar.2014.05.034 [DOI] [PubMed] [Google Scholar]
- 55.Jo C, Kim S, Cho SJ, Choi KJ, Yun SM, Koh YH, Johnson GV, Park SI. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett 2014; 588(17):3081-8; PMID:24952354 [DOI] [PubMed] [Google Scholar]
- 56.Cui H, Li X, Li N, Qi K, Li Q, Jin C, Zhang Q, Jiang L, Yang Y. Induction of autophagy by Tongxinluo via the MEK/ERK pathway protects human cardiac microvascular endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc Pharmacol 2014; 64(2):180-90; PMID:24705173 [DOI] [PubMed] [Google Scholar]
- 57.Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, Feller-Kopman D, Wise R, Biswal S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Trans Med 2011; 3:78ra32; PMID:21490276; http://dx.doi.org/ 10.1126/scitranslmed.3002042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al.. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID:18097414; http://dx.doi.org/ 10.1038/nature06421 [DOI] [PubMed] [Google Scholar]
- 59.Nicola AM, Albuquerque P, Martinez LR, Dal-Rosso RA, Saylor C, De Jesus M, Nosanchuk JD, Casadevall A. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect Immun 2012; 80:3065-76; PMID:22710871; http://dx.doi.org/ 10.1128/IAI.00358-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, Lemichez E, Luciano F, Solary E, Auberger P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012; 119:4527-31; PMID:22452982; http://dx.doi.org/ 10.1182/blood-2011-11-392167 [DOI] [PubMed] [Google Scholar]
- 61.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/ 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J 2008; 27:1110-21; PMID:18337753; http://dx.doi.org/ 10.1038/emboj.2008.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al.. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11:55-62; PMID:19898471; http://dx.doi.org/ 10.1038/ni.1823 [DOI] [PubMed] [Google Scholar]
- 64.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 2010; 16:90-7; PMID:19966812; http://dx.doi.org/ 10.1038/nm.2069 [DOI] [PubMed] [Google Scholar]
- 65.Ma S, Wang Y, Chen Y, Cao F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochimica et Biophysica Acta 2014; 1(4):338-48; PMID:2559358325593583 [Google Scholar]
- 66.Mei Y, Thompson MD, Cohen RA, Tong X. Autophagy and oxidative stress in cardiovascular diseases. Biochimica et Biophysica Acta 2014; 1852(2):243-51; PMID:24834848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sergin I, Razani B. Self-eating in the plaque: what macrophage autophagy reveals about atherosclerosis. Trends Endocrinol Metab: TEM 2014; 25:225-34; PMID:24746519; http://dx.doi.org/ 10.1016/j.tem.2014.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stienstra R, Haim Y, Riahi Y, Netea M, Rudich A, Leibowitz G. Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 2014; 57(8):1505-16; PMID:24795087 [DOI] [PubMed] [Google Scholar]
- 69.Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, Hiltunen M. Impaired autophagy and APP processing in Alzheimer's disease: the potential role of Beclin 1 interactome. Prog Neurobiol 2013; 106-107:33-54; PMID:23827971; http://dx.doi.org/ 10.1016/j.pneurobio.2013.06.002 [DOI] [PubMed] [Google Scholar]
- 70.Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Euro J Neurosci 2013; 37:1949-61; PMID:23773064; http://dx.doi.org/ 10.1111/ejn.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic autophagy in cancer therapy. Int J Mol Sci 2014; 15:10034-51; PMID:24905404; http://dx.doi.org/ 10.3390/ijms150610034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol 2014; 85:830-8; PMID:24574520; http://dx.doi.org/ 10.1124/mol.114.091850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res 2014; 74:647-51; PMID:24459182; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2966 [DOI] [PubMed] [Google Scholar]
- 74.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection. J Exp Med 2001; 194:1231-42; PMID:11696589; http://dx.doi.org/ 10.1084/jem.194.9.1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 2011; 300:C723-42; PMID:21209361; http://dx.doi.org/ 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.